

Abstract
We prepared pressure-sensitive adhesive (PSA) patches based on styrene–isoprene–styrene (SIS) thermoplastic elastomer using hot-melt coating method. The liquid paraffine is added in the PSA matrices as a plasticizer to moderate the PSA properties. Three drugs, methyl salicylate, capsaicin, and diphenhydramine hydrochloride are selected as model drugs. The Fourier transform infrared spectroscopy, differential scanning calorimetry test, and wide-angle X-ray diffraction test indicate a good compatibility between drugs and matrices. Peppas equation is used to describe drug release profile. Different drug–matrix absorption, as indicative of drug–matrix interaction, accounts for the variation in release profiles of different drugs. Furthermore, atomic force microscopy and rheological studies of the PSA samples are performed to investigate the effect of SIS structure and plasticizer of PSA on drug release behaviors. For methyl salicylate and capsaicin, drug diffusion in the PSA matrices is the main factor controlled by the release kinetic constant k. The high [SI] diblock content and high plasticizer amount in matrix provide the PSA with a homogeneous and soften microstructure, resulting in a high diffusion rate. But for water-soluble drugs such as diphenhydramine hydrochloride, the release rate is governed by water penetration with the competition from diffusion mechanisms.
KEY WORDS: drug delivery, Peppas equation, rheology, thermoplastic elastomer
INTRODUCTION
Pressure-sensitive adhesives (PSA) have been used for decades in transdermal drug delivery systems (TDDS). It is the main component forming the matrices of the patches to control drug release and offer the adhesive strength to skin. Three types of PSA are commonly used in TDDS: poly isobutylene (PIBs), poly siloxanes (silicones), and polyacrylate copolymers (acrylics). Additionally, natural rubber-based patch is another class of PSA used in many over the counter dermal therapeutic systems (1).
Recently, hot melt PSA (HMPSA) becomes more important in transdermal drug delivery devices due to their superior characteristics compared with other PSA (2). Firstly, the use of hot-melt coating technology offers better economics to solvent or water-based coatings. Secondly, HMPSA is particularly suitable for combined adhesive/drug–matrix device as they can be formulated to contain little or no chemical functional group. This reduces the possibility of medication/adhesive interactions and skin irritation. Thirdly, HMPSA is less prone to swelling when in contact with alcohols used in certain medications, which is a particular problem with acrylics. But the high melt temperature (150–200 °C) and high peel force required removing from skin of HMPSA limit its pharmaceutical application.
Styrene-isoprene-styrene (SIS) block copolymer is a thermoplastic elastomer, which has been widely used in HMPSA manufacturing (3). Its blend is made of [SI] diblock and [SIS] triblock. Both of which are made from polyisoprene and polystyrene chains with different ratios. The soft, rubbery polyisoprene phase, which accounts for the elastomeric properties, is trapped in the hard and rigid polystyrene phase which increases the modulus and yields strength of the copolymer system (4). This entanglement network brings on a special microphase-separated morphology and hence provides viscoelastic solid behavior for SIS copolymer. In addition, a tackifying resin is necessary in the PSA formulation to provide adhesiveness (5).
The chemical and physical properties of SIS have been extensively studied and characterized including adhesiveness (6), morphological and rheological properties (7–9), and thermodynamic behaviors (10,11). But in most of these researches, SIS were mainly used or considered as tapes and adhesives. To the best of our knowledge, the usage of SIS as drug delivery devices was rarely reported. Teruaki Hayashi and his coworkers (12) investigated the release properties of indomethacin from PSA patches based on SIS copolymer. In their paper, drug release behaviors were evaluated by means of drug crystal and thickness of the PSA, and a simple liner compartment model was designed to describe release profile. But the effects of SIS structure and PSA formulations on drug release were not taken into account.
In the present article, patches based on SIS copolymer are prepared using hot-melt coating method. Liquid paraffine, a low molecular weight plasticizer, is added in the formulation to reduce the melt temperature and the peel force (3). Three ordinary topical drugs, methyl salicylate (MS), diphenhydramine hydrochloride (DPH), and capsaicin (CS) are selected as model drugs to evaluate the different drug release profiles (13–15). The chemical structures of the drugs are shown in Fig. 1. The drug–matrix interaction and drug physical state in the matrix is examined. Furthermore, the effects of SIS structure and plasticizer amount on drug release behaviors are investigated by correlating the morphological and rheological properties of the matrix to drug diffusion behaviors.
Fig. 1.

Chemical structures of the three model drugs
MATERIALS AND METHODS
Materials
The SIS selected for this study include Kraton D1113, D1163, D1107 (Kraton, USA), and Zeon 3620 (Zeon, Japan). A synthetic hydrocarbon resin, Hikorez A 1100S (C-5, Kolon, Korea) was selected as the tackifying resin, and a liquid paraffine purchased from LinFeng chemical Co. Ltd (Shanghai, China) was selected as the plasticizer. Antioxidant 1010 was purchased from JiYi chemical Co. Ltd (Beijing, China). Methyl salicylate, capsaicin, and diphenhydramine hydrochloride were purchased from BangCheng chemical Co. Ltd (Shanghai, China). Other chemicals and solvents were obtained commercially.
Method of Preparing HMPSA Matrices (6)
The SIS block copolymer, C-5 hydrocarbon resin, and liquid paraffine were blended in a three-neck flask. The antioxidant was used as a thermal stabilizer. The system was heated and maintained at 130 °C during the whole process. The blends were stirred at 500 rpm (IKA agitator) for 30 min until the PAS specimens melted into transparent. A dry nitrogen atmosphere was maintained during the whole producing process. The PSA solution was then quickly moved from the reactor onto an anti-stickiness film (silicone-coated polyethylene terephthalate film, thickness 50 μm, YiDong Co., Shanghai, China) before it solidified at room temperature. In the HMPSA formulations, the amount of SIS block copolymer was set at 6.0 g, the other substances were weighted in ratio according to the schedule listed in Table I.
Table I.
Formulations and Compositions of Samples
| No | SIS copolymer | C5 resin | Liquid paraffine | |
|---|---|---|---|---|
| Trade name | Content (%) | Content (%) | Content (%) | |
| Sample 1 | D1107 | 40.0 | 20.0 | 40.0 |
| Sample 2 | D1163 | 40.0 | 20.0 | 40.0 |
| Sample 3 | D1113 | 40.0 | 20.0 | 40.0 |
| Sample 4 | 3620 | 40.0 | 20.0 | 40.0 |
| Sample 5 | D1163 | 56.7 | 28.3 | 15.0 |
| Sample 6 | D1163 | 50.0 | 25.0 | 25.0 |
| Sample 7 | D1163 | 33.3 | 16.7 | 50.0 |
The antioxidant 1010 was added in the content of 0.5 %
Method of Preparing Drug Solution
To increase the drug solubility in PSA matrix, CS and DPH were prepared in the forms of solution. A certain amount of CS and DPH was accurately weighted and dissolved in ethanol in ratio of 1:2 (w/v), respectively. For MS, to make a good molecular dispersing state in the PSA matrix, the drug was directly added into liquid paraffine up to the ratio of 1:2 (w/v). The drug solutions were then ultrasonic treated for 30 min to make the mixture homogeneous.
Method of Preparing PSA Patches
The patches were prepared by a direct coating method. A certain amount of HMPSA matrices were heated to 90 °C until it melted into liquid. Then a certain volume of drug solution was added into the adhesive solution respectively and mixed thoroughly. The resulting drug–PSA specimen was then quickly melt-coated onto the backing sheet of non-woven fabrics (thickness, 200 μm, medical grade, YiDong Co., Shanghai, China) with an average thickness of 50 μm using a film applicator (HongWei Trading Co., Guangzhou, China). The coating process was performed on a hot plate operating at 90 °C. The anti-stickiness liner (silicone-coated polyethylene terephthalate film, thickness 50 μm, YiDong Co., Shanghai, China) was then placed onto the patch surface after the PSA solution solidified.
Characterizations
Molecular Weight
The weight average molecular weight (Mw), the number average molecular weight (Mn), and the molecular weight distribution (Mw/Mn) of the SIS copolymer were determined by gel permeation chromatography (GPC, Waters Co.).
Atomic Force Microscopy
The samples were hot-melted and coated into a thin film with the thickness of 1 mm and area of 10 × 10 mm approximately before atomic force microscopy (AFM) study (AJ-III Scanning probe microscope, AJ Nano-Science Development, Co., China). The AFM analysis was performed with a tapping-mode at room temperature. The scanning size was set at 1,000 × 1,000 nm.
Fourier Transform Infrared Spectroscopy
The Fourier transform infrared spectroscopies (FITR) for the pure drugs, the adhesive matrix, and the drug-loaded matrix were determined by an attenuated total reflection Fourier transform infrared spectrometer (ATR-FTIR spectrometer, Thermo Nicolet 5700, USA). The pure drugs were measured by the KBr methods, while the matrix and the drug–matrix blends were measured directly using ATR-FTIR spectra.
Differential Scanning Calorimetry
The differential scanning calorimetry (DSC) thermograms of pure drugs, blank matrix, drug–matrix physical mixture, and drug-loaded patches were recorded by a thermo gravimetric analyzer (DSC 200 PC Phox®, German). A certain amount of sample (10–15 mg) was placed in a sealed aluminum pan and heated at a heating rate of 10 °C/min in the range of 25 to 200 °C. A nitrogen atmosphere at a flow rate of 80 ml/min was maintained during the whole process.
X-ray Diffractometry
Crystal states of drugs in patches were observed by wide-angle X-ray diffraction (XRD) using a Rigaku (Japan) D/max 2250 VB/PC X-diffractometer. All the samples were measured at 100 mA and 40 KV with the scan range of 3–50° and the scan interval of 0.02°.
Rheological Test
Rheological measurements of HMPSA samples were performed on physical advanced rheometer (MCR501, AntonPaar Co Ltd, Austria). Three viscoelastic parameters, storage modulus (G′), loss modulus (G″), and dynamic complex viscosity (η′) were measured as functions of frequency in the range of 10−4 to 102 Hz at 32 °C (the temperature of drug release experiments) with a monitored shear strength (1,000 Pa) (16). A 20-mm diameter stainless steel parallel plate with a gap size of 1,000 μm was used.
Water Uptake Experiment
Compound-free HMPSA samples were cut into pieces of 1.5 × 1.5 cm, and placed into plastic flasks filled with pre-heated release medium (10 %(v/v) ethanol–phosphate buffer solution (pH 7.4)), followed by stirring for 24 h at 32 °C with a stirring speed of 200 rpm; (KaKa TD-120, KaKa, Shanghai, China). The samples were weighed before exposure to the medium (dry weight (t = 0), mdry). At predetermined time points, the samples were withdrawn from the media, carefully dried from adhering water, and accurately weighed (mwet(t)) (XS 105 DU, Mettler Toledo, Switzerland). The water uptake (%) at time t was calculated as follows:
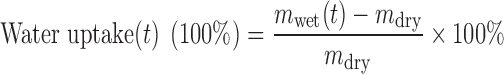 |
1 |
Drug Diffusion Experiments
Drug Absorption Studies
Drug absorption in the PSA matrices was measured by shake-flask method (17). Compound-free samples with a surface area of 2.5 × 2.5 cm were accurately weighed and immersed in 20.0-ml drug saturated solution followed by equilibrating with a gentle shaking at 32 °C in a water bath for 24 h. The amount of drug distributed into the matrix was determined by assaying the drug amount in the solution before and after equilibrium. The solubility of drug in the PSA matrix was determined as follows:
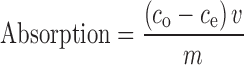 |
2 |
Here, co is the initial concentration of a drug, ce is the equilibrium concentration, v is the volume of drug solution, and m is the weight of the samples.
Determination of Initial Drug Amount in the PSA Patches
The patch was cut into a certain area accurately and immersed into 15-ml methanol with a reflux extraction for 3 h. For patches loaded with MS, a volatile oil extraction device filled with ethyl acetate was used to collect the drug extracted from the matrix. The drug extraction solution was then filtered through a filter membrane with pore size 0.45 μm (Millex ® GV, Millipore, USA) before chromatograph analysis.
Drug Release Experiments
Release profiles were evaluated by means of drug transdermal diffusion apparatus (KaKa TD-120, KaKa, Shanghai, China) equipped with Franz cells. Patches were applied on the receiver compartment with 17.5-ml phosphate buffer solution (pH 7.4) and a diffusion area of 3.14 cm2. Ten percent ethanol (v/v) was added in the buffer solution to increase capsaicin solubility in receiver compartment (15). The receiver compartment was stirred at a constant speed of 200 rpm. The whole release process was performed at 32 °C. At predetermined times, 1.0 ml of the solution was collected from the receive compartment and the same volume of fresh buffer solution was added to keep the volume constant. Sink conditions were maintained throughout the experiment.
Chromatograph Analysis
MS was determined using gas chromatography (HP Series 6890, Agilent Technologies Ltd, USA) with a flame ionization detector (LGH-300, Anpu Sci technologies Ltd, China). MS in the samples was extracted into ethyl acetate, and naphthalene was added as internal standard prior to injection. The capillary column used was a HP-Innowax (30 m × 0.530 mm, Agilent, USA). The injector temperature was set at 23 °C, the oven temperature was 150 °C, and the FID detector temperature was 250 °C. The injection volume was 1.0 μl with a split ratio of 10:1.
CS was identified and quantified using an Agilent 1100 automated chromatograph fitted with a 5-μm C18 250 × 4.6 mm column (Capcell Pak, Shiseido, Japan). The mobile phase comprised of 52 % acetonitrile (HPLC grade, Merck & Co., Inc, USA) and 48 % phosphoric acid solution (0.1 %, v/v). The UV detector was set at 209 nm. The injection volume was 10 μl and the flow rate was 1.0 ml min−1. The retention time for CS was 10.8 min.
DPH was also identified and quantified using an Agilent 1100 automated chromatograph fitted with a 5-μm C18 250 × 4.6 mm column (Capcell Pak, Shiseido, Japan). The mobile phase comprised of 53 % methanol (HPLC grade, Merck & Co., Inc, USA) and 47 % ammonium acetate solution (1 %, w/v). The UV detector was set at 210 nm. The injection volume was 10 μl and the flow rate was 1.0 ml min−1. The retention time for DPH was 9.8 min.
Both the GC and HPLC methods were previously validated, including sensitivity, precision, accuracy, stability with a linear relationship between the concentrations in the range of 10–100 μg/ml of drugs.
RESULT AND DISCUSSION
Characterizations of SIS Polymers
GPC Analysis
The weight average molecular weight (Mw), the number average molecular weight (Mn), and the molecular weight distribution (Mw/Mn) of the SIS copolymers were determined by gel permeation chromatography. As shown in Table II, there are no significant differences among the molecular weight and weight distribution for all the copolymers. The diblock contents increase in the order of D1107 < D1163 < D1113 < 3620.
Table II.
Copolymer Characterization Data
| Trade name | Typea | Styrene contenta (wt.%) | Diblock contenta (wt.%) | M n | M w | M w /M n | Manufacture |
|---|---|---|---|---|---|---|---|
| D1107 | Linear SIS | 15 | 15 | 107,233 | 109,039 | 1.017 | Kraton Polymer |
| D1163 | Linear SIS | 15 | 38 | 105,239 | 107,922 | 1.025 | Kraton Polymer |
| D1113 | Linear SIS | 16 | 56 | 108,582 | 111,748 | 1.029 | Kraton Polymer |
| 3620 | Linear SIS | 15 | 78 | 101,885 | 103,531 | 1.016 | Zeno Crop |
aData supplied by the manufacturer
AFM Studies of SIS Copolymer
The tapping-mode AFM was performed to observe morphologies of SIS copolymer surfaces. As shown in Fig. 2, the bright domain represents the high phase of the surface, which is assigned to the hard and rigid styrene sequences; while the dark domain represents the low phase, which correlates to the soft isoprene sequences (18,19). An obvious phase separation appears on D1107 surface (Fig. 2a) for its least [SI] diblock content (15 %). With the increasing of diblock ratio, the surface tends to grow from long worm-like to mesh-like microphase domain (20). When the diblock content reaches 78 %, the soft isoprene spots are completely dispersed in the continuous phase composed of hard styrene network (Fig. 2d). Hence, it can be concluded that SIS copolymer with higher [SI] diblock content is more homogeneous at microscale.
Fig. 2.

a–d Set of height images of SIS block copolymers obtained by tapping-mode AFM with height variation in the 0–10 nm range and a scan size of 1,000 × 1,000 nm
Characterizations of Patches
FTIR Spectroscopy
To understand the matrix–drug interactions, FTIR of drugs, matrix, and drug–matrix were performed. All the samples were prepared in the composition of sample 2 (Table I). The drug concentrations were 0.2 %, in accordance with the release experiments. The spectra are shown in Fig. 3.
Fig. 3.

a–c FITR spectra of the drug, matrices, and drug–matrices for model drugs, respectively. All the matrices were prepared in the composition of SIS (D1163)/C5 resin/liquid paraffine at the ratio of 1:0.5:1 (w/w) with the drug concentration of 0.20 %
For MS, the peaks at 3,188, 1,680, and 1,091 cm−1 indicate the stretching vibrations of O–H, C = O, and O–CH3 bonds, respectively (21). The spectrum of the matrix exhibits no characteristic peaks, which confirms the inert characters of the matrix. Comparing with spectrum of pure MS, the characteristic peaks are strongly weakened in matrix–drug blends due to its low concentration in the matrix. No significant shifts in the peaks corresponding to MS or the adhesive matrix appear in the drug–polymer blends, which indicate there are no combinations of MS with the matrix (22).
For CS, the peaks at 3,446, 3,291, 1,644, and 1,032 cm−1 are assigned to the stretching vibrations of N–H, O–H, (NH)C = O, and O–CH3 bonds (23). The spectrum is found to be similar with the spectrum of MS: the characteristic peaks are weakened but no shifts are observed in matrix–drug blends.
For DPH, the peaks in the range of 2,400–2,700 cm−1 are attributed to the stretching vibrations of –N(CH3)2·HCl bond, while the peak at 1,103 cm−1 represents the O–CH3 stretching vibration (24). No peaks in the range of 2,400–2,700 cm−1 are observed in the matrix–drug blends, implying the deformation of ionic bonds between hydrochloric acid and dimethylamino, when DPH be loaded in the matrix.
DSC Test
For DSC test, matrices of all the samples were prepared in the formulation of sample 2. Taking the detection limits of the instrument into consideration, the drug concentrations in the patches were set as 2 %, tenfold higher than which was used in release experiments. The DSC curves of drugs, physical mixture of drugs and matrices, drug-loaded patches, and drug-free matrices are depicted, respectively, in Fig. 4. The drugs show their character peaks in the curves of pure drugs and physical mixture of drug–matrix individually. But no character peaks appear in the curves of drug-loaded patches, suggesting a good compatibility between drugs and matrix. The DSC results indicate that the three drug molecules exist in a dissolved or dispersed state in the adhesive matrix with the concentration up to 2 % (25).
Fig. 4.

a–c DSC analysis of the drug, blank matrix, physical mixture of drug–matrix, and drug-loaded patches. All the matrices were prepared in the composition of SIS (D1163)/C5 resin/liquid paraffine at the ratio of 1:0.5:1 (w/w) with the drug concentration of 2 %
XRD Test
For XRD test, all the matrices samples were prepared in the formulation of sample 2. The concentrations of the three drugs were set as 2 %. No crystalline peaks appear in the drug-loaded patches from the XRD curves (Fig. 5). It suggested that in the PSA matrix no crystallization occurs when the drug concentration reaches 2 % (25).
Fig. 5.

a–c XRD analysis of the drug, blank matrix, and drug-loaded patches. All the matrices were prepared in the composition of SIS (D1163)/C5 resin/liquid paraffine at the ratio of 1:0.5:1 (w/w) with the drug concentration of 2 %
Drug Release Experiment
To investigate the effect of [SI] content and plasticizer amount of PSA on drug release behavior, a series of PSA matrices were prepared respectively. Table I lists the compositions and formulations of samples. Samples 1 to 4 were prepared in the same component ratio but different kinds of SIS copolymer; while samples 2, 5, 6, and 7 were prepared with the same SIS copolymer but different ratio of plasticizer amounts in the formulations. The initial amount of the drug loading in the matrix was set at 0.2 % to minimize the influence of drug loading on PSA properties (26).
We use the semi-empirical Peppas equation (27) to describe drug release behavior from HMPSA patches:
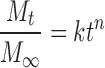 |
3 |
Where Mt/M∞ is the drug release percentage at times t. In our work the initial drug loading in the formulation is considered as M∞. k is a kinetic constant related to drug release rate, and n is the release exponent, which might be indicative of the release character (28).
Statistical analysis was performed using SPSS software (SPSS 11.5, SPSS Science, Chicago, IL, USA). Nonlinear regression was applied to calculate the parameters of k and n.
Figure 6 depicted the drug release profiles from patches with various [SI] content in SIS copolymer. Figure 7 depicted the release profile of drugs from patches made of different content of liquid paraffine. The parameters of k and n are calculated from each release curve and listed in Table III.
Fig. 6.

Release profiles of model drugs from samples 1 to 4. a MS, b CS, and c DHP. Each data represents mean±SD of six determinations (n = 6)
Fig. 7.

Release profiles of model drugs from samples 2, 5, 6, and 7. a MS, b CS, and c DHP. Each data represents mean±SD of six determinations (n = 6)
Table III.
The Parameters of k and n Calculated from Each Release Curve of the Three Drugs (Mean±SD, n = 6)
| MS | CS | DPH | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Kinetic constant(h −n) | Diffusion exponent | R 2 | Kinetic constant(h −n) | Diffusion exponent | R 2 | Kinetic constant(h −n) | Diffusion exponent | R 2 | |
| Sample1 | 0.392 ± 0.013 | 0.228 ± 0.002 | 0.983 | 0.050 ± 0.004 | 0.688 ± 0.032 | 0.996 | 0.119 ± 0.011 | 0.435 ± 0.012 | 0.988 |
| Sample2 | 0.436 ± 0.031 | 0.223 ± 0.012 | 0.982 | 0.068 ± 0.005 | 0.682 ± 0.021 | 0.997 | 0.156 ± 0.013 | 0.424 ± 0.023 | 0.986 |
| Sample3 | 0.475 ± 0.021 | 0.234 ± 0.009 | 0.976 | 0.086 ± 0.006 | 0.681 ± 0.035 | 0.993 | 0.171 ± 0.008 | 0.435 ± 0.003 | 0.986 |
| Sample4 | 0.526 ± 0.038 | 0.226 ± 0.004 | 0.974 | 0.099 ± 0.007 | 0.685 ± 0.024 | 0.995 | 0.187 ± 0.012 | 0.435 ± 0.021 | 0.988 |
| Sample5 | 0.341 ± 0.021 | 0.224 ± 0.011 | 0.980 | 0.033 ± 0.002 | 0.685 ± 0.018 | 0.993 | 0.131 ± 0.013 | 0.433 ± 0.015 | 0.983 |
| Sample6 | 0.387 ± 0.018 | 0.223 ± 0.007 | 0.977 | 0.063 ± 0.009 | 0.679 ± 0.019 | 0.998 | 0.169 ± 0.018 | 0.424 ± 0.013 | 0.989 |
| Sample7 | 0.470 ± 0.031 | 0.224 ± 0.012 | 0.981 | 0.084 ± 0.005 | 0.683 ± 0.013 | 0.998 | 0.106 ±0.012 | 0.434 ± 0.015 | 0.987 |
Different Drug Release Profiles
As discussed above, the FITR results indicate that there are no combinations between the matrix and drugs because of the inherent inert character and lack of functional groups of the adhesives; while the DSC and XRD results show a dispersed or dissolved state of the three drugs in the matrix respectively. Hence the release profiles for drugs depend on their diffusion in the matrix (29). As shown in Figs. 6 and 7, MS exhibits a burst release in the early stage and almost get equilibrium after 12 h. The release exponents n retains at a small but stable value of 0.22 approximately for all the samples and the kinetic constants k are relatively high (Table III). A stable n value of 0.68 and small values of k indicate a slow but sustained release for CS. DPH is released fast in the first 4 h but the release rate slowed down gradually with the value of n stayed at 0.43.
The different release profiles of the three drugs in the same formulation can be attributed to the different absorption abilities of drugs. As shown in Table IV, the level of absorption differs from drug to drug but does not exhibit any dependency on matrix composition. In drug release process, the mass transfer occurs at the matrix–water interface. The higher distribution ability of drug in matrix could improve the drug release by the higher diffusing ability because of the good molecular dispersing state and higher drug concentration gradient between the matrix and the released medium. Hence MS shows small values of n, indicating a burst release profile, and the relatively large values of k are complimentary to the n values (30). When drugs are less absorb in the matrix, such as CS and DPH, the matrix would retard their diffusion, causing an extended and continuous release profile, which is reflected in the large n and small k values. Furthermore, the consistency of n for the same drug in different formulations implies that the matrices maintain a stable structure during the release process comparing with the water-soluble materials (31).
Table IV.
Drug Absorptions in the Matrices (Mean±SD, n = 6)
| Drug | Absorption in the matrix (%, w/w) | ||||||
|---|---|---|---|---|---|---|---|
| Sample 1 | Sample 2 | Sample 3 | Sample 4 | Sample 5 | Sample 6 | Sample 7 | |
| MS | 5.600 ± 0.198 | 5.492 ± 0.247 | 5.150 ± 0.468 | 5.237 ± 0.398 | 5.108 ± 0.364 | 5.249 ± 0.149 | 5.366 ± 0.189 |
| CS | 0.231 ± 0.030 | 0.230 ± 0.051 | 0.228 ± 0.041 | 0.223 ± 0.010 | 0.201 ± 0.032 | 0.243 ± 0.057 | 0.223 ± 0.050 |
| DPH | 0.296 ± 0.007 | 0.272 ± 0.040 | 0.315 ± 0.065 | 0.284 ± 0.043 | 0.345 ± 0.054 | 0.288 ± 0.055 | 0.274 ± 0.037 |
Effect of [SI] Diblock Content on Drug Release
The kinetic constant k is compared among samples 1 to 4 for the three model drugs. As shown in Fig. 8a, the value of k increases as a function of the [SI] content for MS, CS, and DPH, respectively. To further explore the influence of [SI] diblock content on drug releases, the rheological tests of the samples are performed. Figures 9, 10, and 11 depict the rheological curves. The storage modulus, G′ describes the solid-like character, whereas the loss modulus G″ describes the liquid-like character of the samples (16). The rheological curves can be fitted using power law models, which was discussed in detail by Gibert and his coworkers (8).
Fig. 8.

a Effect of [SI] content on drug release; b effect of plasticizer content on drug release (MS black bars, CS gray bars, DPH white bars). Each data represents mean±SD of six determinations (n = 6)
Fig. 9.

a, b Rheological parameters of G′ and G″ versus frequencies of samples 1 to 4
Fig. 10.

a, b Rheological parameters of G′ and G″ versus frequencies of samples 2, 5, 6, and 7
Fig. 11.

a, b Dynamic complex viscosities η′ versus frequencies for samples
As shown in Fig. 9a, the main rheological differences among samples 1 to 4 focus on the terminal region: the storage modulus, G′, tending toward a limiting plateau value at low frequencies, is inversely proportional to the [SI] content in the SIS copolymer. The morphology study of SIS we discussed above may help explain this phenomenon. When the [SI] content is relatively low, a phase separation microstructure can be observed (Fig. 2a). But when [SI] content increases, the isoprene phase is gradually dispersed in the continuous styrene phase, implying a uniform structure of the copolymer (Fig. 2d). The free isoprene ends of the dispersed [SI] diblock soften and relax the rigid polystyrene network of the triblock part, causing a reduction in G′ (8). The rheological behavior is also in accordance with the result of dynamic complex viscosity test (Fig. 11a): sample with higher [SI] content possesses lower entanglement density, resulting in a lower value of dynamic complex viscosity η′ (32). On the other hand, the loss modulus G″ is less influenced by the diblock content of SIS, as shown in Fig. 9b.
Now return to the drug release behavior, we can find that the morphological and rheological properties put a significant impact on the k values. Higher content of [SI] provides the copolymer with a more homogeneous microstructure, producing a lower plateau modulus. The matrix hence possesses a more soften matrix structure, which facilitates the drug diffusion. Additionally, the variations in the kinetic constants k are also attributed to the viscosities of samples. According to the Stokes–Einstein equation, the diffusion coefficient, which controls the release behavior, is inversely proportional to the viscosity of the matrices (33). Hence the lower value of η′ of samples results in a higher diffusion coefficient of drugs.
Effect of Plasticizer Amount on Drug Release
Similarly, the kinetic constant k for the three drugs are compared among samples 2, 5, 6, and 7 to evaluate the effect of plasticizer on drug release (Fig. 8b). For MS and CS release, the k value increases as a function of plasticizer amount in the samples.
Rheological behaviors of samples 2, 5, 6, and 7 are depicted in Figs. 10 and 11b. The lower values of G′ and G″, accounting for a less rigid but more relaxation network of the matrix, can be attributed to the swelling effect of liquid paraffine. Liquid paraffine is a low-molecular-weight material served as a solvent in the matrix, which causes a reduction in polymer–polymer chain and forms secondary bonds with the polymer chains instead (34). Hence the more liquid paraffine included in the formulation can weaken the interactions between molecules and expand the free volume of copolymer, resulting in a lower value of modulus and dynamic viscosity (5). AFM results shown in Fig. 12 further demonstrate this hypothesis: the obvious phase separation structure in Fig. 12a is disappeared when more liquid paraffine is employed in the formulation (Fig. 12b), implying that the isoprene phase is swelled and dissolved with inclusion of liquid paraffine.
Fig. 12.

Set of height images of SIS–Liquid paraffine blends obtained by tapping-mode AFM with height variation in the 0–10 nm range and a scan size of 1,000 × 1,000 nm: a SIS/Liquid paraffine = 2:1; b SIS/Liquid paraffine = 1:1
Taking the results of AFM and rheological test into consideration, the effect of plasticizer on release kinetic constant k can be well explained. According to free volume theory (29), the diffusion coefficient of a drug in polymer relates to the free volume of the polymer, hence the expanding in free volume of matrix results in a higher diffusion coefficient, producing a faster release rate. Additionally, the differences in values of viscosity (Fig. 11b) also help interpret the effect of plasticizer on release behavior as we have discussed previously.
However, for DPH, the k value first increases as increasing of plasticizer amount but then decreases. A maximum value for k is observed at the formulation of sample 6 (Fig. 8b). It seems inconsistent with the explanation above. We performed water uptake experiments to further explore the diffusion mechanism of DPH, as shown in Fig. 13. As evident in this figure, water absorption in samples is not significant but still exists, when comparing with water swellable materials (35). For samples 1 to 4, no significant variation in water absorptions appears (Fig. 13a), implying that water absorption exhibits independency of [SI] content in copolymer. However, samples 2, 5, 6, and 7 present an inverse correlation between water absorption and liquid paraffine amount (Fig. 13b): the water uptake in samples increases as the decreasing of plasticizer content. Nazhat (4) also reported similar findings. The appearance of water forms fluid-filled channels in the entanglement network of the PSA and hence desorbs the water-soluble drug from the matrix to the release medium (36), which strongly facilitates the release behaviors of water-soluble drugs. So it can be concluded that release behavior of DPH is governed by two mechanisms. One is a matrix diffusion controlled mechanism associate with the inherent properties of the matrices such as morphology and rheology (26). The other is a water-content-dependent mechanism relate to the ability of water penetration into the matrix (35). These two mechanisms are under restraint by each other. For samples 1 to 4, the variation in water uptakes is insufficient to influence the drug release rate. The release behavior of DPH is solely controlled by the former mechanism. But for samples 2, 5, 6, and 7, the variation in water absorptions becomes significant and governs the release profile of DPH with the competition from diffusion-controlled mechanism. The effect of water absorption might overwhelm the effect produced by the diffusion-controlled mechanism when the plasticize content exceeds 24 %, thereby making a maximum value of kinetic constant k at sample 6. Comparing with release behavior of DPH, the water penetration, which speeds up the DPH release from matrix, has no effect on MS and CS, for the poor solubility of the two drugs in the release medium.
Fig. 13.

Water absorption of samples. Each data represents mean±SD of six determinations (n = 6)
CONCLUSIONS
Patches based on SIS thermoplastic elastomer copolymer were prepared. MS, CS, and DPH were selected as model drugs. The FITR spectra, DSC, and XRD analysis indicate the inert nature of the matrix and a good compatibility between drugs and matrix. The different drug absorption in matrix accounts for the variations in release behaviors of the three drugs. Furthermore, morphological and rheological tests indicated that the diffusion mechanism solely controls MS and CS release rates. High [SI] diblock content in SIS and high plasticizer content in formulation provides a quicker release rate for MS and CS. But for DPH, higher plasticizer amounts in matrix lead to a less water absorption, which cause a reduction in release rate due to the water-soluble property of DPH. Hence DPH release profile is controlled by two inter-restraint mechanisms: a matrix diffusion-controlled mechanism and a water-content-dependent mechanism.
ACKNOWLEDGMENTS
This research was supported by the National Science and Technology Key Program for the 11th five-year Plan of China (2008BAI53B075).
REFERENCES
- 1.Hock ST, William RP. Pressure-sensitive adhesives for transdermal drug delivery systems. Pharm Sci Technol Today. 1999;2:60–69. doi: 10.1016/S1461-5347(99)00119-4. [DOI] [PubMed] [Google Scholar]
- 2.Webster I. Recent developments in pressure-sensitive adhesives for medical applications. Int J Adhesion &Adhesives. 1997;17:69–73. doi: 10.1016/S0143-7496(96)00024-3. [DOI] [Google Scholar]
- 3.Galán C, Gómez Fatou JM, Delgado JA. A hot-melt pressure-sensitive adhesive based on styrene-butadiene-styrene rubber. The effect of adhesive composition on the properties. J Appl Polym Sci. 1996;62:1263–1275. doi: 10.1002/(SICI)1097-4628(19961121)62:8<1263::AID-APP17>3.0.CO;2-8. [DOI] [Google Scholar]
- 4.Nazhat SN, Parker S, Patel MP. Isoprene-styrene copolymer elastomer and tetrahydrofurfuryl methacrylate mixtures for soft prosthetic applications. Biomaterials. 2001;22:2411–2416. doi: 10.1016/S0142-9612(00)00428-2. [DOI] [PubMed] [Google Scholar]
- 5.O’Connor AE, Willenbacher N. The effect of molecular weight and temperature on tack properties of model polyisobutylenes. Int J Adhes Adhes. 2004;24:335–346. doi: 10.1016/j.ijadhadh.2003.11.005. [DOI] [Google Scholar]
- 6.Kim DJ, Kim HJ, Yoon G. Effect of substrate and tackifier on peeling strength of SIS (styrene-isoprene-styrene)-based HMPSAs. Int J Adhes Adhes. 2005;25:288–295. doi: 10.1016/j.ijadhadh.2004.10.001. [DOI] [Google Scholar]
- 7.Laurer JH, Khan SA, Spontak RJ. Morphology and rheology of SIS and SEPS triblock copolymers in the presence of a midblock-selective solvent. Langmuir. 1999;15:7947–7955. doi: 10.1021/la981441n. [DOI] [Google Scholar]
- 8.Gibert FX, Marin G, Derail C, Allal A, Lechat A. Rheological properties of hot melt pressure-sensitive adhesives based on styrene–isoprene copolymers. Part I: a rheological model for [SIS-SI] formulations. J. Adhesion. 2003;79:825–852. doi: 10.1080/00218460309552. [DOI] [Google Scholar]
- 9.Lee WL, Kim HD, Kim EY. Morphological reorientation by extensional flow deformation of a triblock copolymer styrene–isoprene–styrene. Curr Appl Phys. 2006;6:718–722. doi: 10.1016/j.cap.2005.04.026. [DOI] [Google Scholar]
- 10.Efrain RP, Michael CW. Thermodynamics of plasticized triblock copolymers. Part II: model verification by light transmittance and rheology. Polym Eng & Sci. 1977;17:573–581. doi: 10.1002/pen.760170814. [DOI] [Google Scholar]
- 11.Zhang JY, Park YJ, Kim HY. A new approach on the thermal stability of SDS copolymer for HMPSA, Part I: oxidation kinetics for the whole process. Polym Degrad Stab. 2008;93:1008–1023. doi: 10.1016/j.polymdegradstab.2007.12.016. [DOI] [Google Scholar]
- 12.Hayashi T, Yamazaki T, Yamaguchi Y, Sugibayashi K, Morimoto Y. Release kinetics of indomethacin from pressure sensitive adhesive Matrices. J Control Release. 1997;43:213–221. doi: 10.1016/S0168-3659(96)01486-1. [DOI] [Google Scholar]
- 13.Cheng Y, Huang Y, Alexander K, Dollimore D. A thermal analysis study of methyl salicylate. Thermochim Acta. 2001;367–368:23–28. doi: 10.1016/S0040-6031(00)00689-4. [DOI] [Google Scholar]
- 14.Fang JY, Wu PC, Huang YB, Tsai YH. In vitro permeation study of capsaicin and its synthetic derivatives from ointment bases using various skin types. Int J Pharm. 1995;126:119–128. doi: 10.1016/0378-5173(95)04105-2. [DOI] [Google Scholar]
- 15.Akram ME, Moftah AM. Spectrophotometric determination of diphenhydramine hydrochloride in pharmaceutical preparations and biological fluids via ion-pair formation. Arabian J Chem. 2010;3:265–270. doi: 10.1016/j.arabjc.2010.06.014. [DOI] [Google Scholar]
- 16.Yat HK, Dodou K. Rheological studies on pressure-sensitive silicone adhesives and drug-in-adhesive layers as a means to characterize adhesive performance. Int J Pharm. 2007;333:24–33. doi: 10.1016/j.ijpharm.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 17.Kokubo T, Sugibayashi K, Morimoto Y. Mathematical models describing the drug release kinetics from pressure sensitive adhesive matrix. J Control Release. 1992;20:3–12. doi: 10.1016/0168-3659(92)90133-C. [DOI] [Google Scholar]
- 18.Mclean RS, Sauer BB. Tapping-mode AFM studies using phase detection for resolution of nanophases in segmented polyurethanes and other block copolymers. Macromolecules. 1997;30:8314–8317. doi: 10.1021/ma970350e. [DOI] [Google Scholar]
- 19.Magonov SN, Cleveland J, Elings V, Denley D, Whangbo M-H. Tapping-mode atomic force microscopy study of the near-surface composition of a styrene–butadiene–styrene triblock copolymer film. Surface Sci. 1997;389:201–211. doi: 10.1016/S0039-6028(97)00412-3. [DOI] [Google Scholar]
- 20.Motomastu M, Mizutani W, Tokumoto H. Microphase domains of poly (styrene-block-ethylene/butylene-block-styrene) triblock copolymers studies by atomic force microscopy. Polymer. 1996;38:1779–1785. [Google Scholar]
- 21.Varghese HT, Panicker CY, Philip D, Mannekutla JR, Inamdar SR. IR, Raman and SERS studies of methyl salicylate. Spectrochimica Acta Part A. 2007;66:959–963. doi: 10.1016/j.saa.2006.04.034. [DOI] [PubMed] [Google Scholar]
- 22.Songsurang K, Pakdeebumrung J, Praphairaksit N, Muangsin N. Sustained release of amoxicillin from ethyl cellulose-coated amoxicillin/chitosan–cyclodextrin-based tablets. AAPS Pharm Sci Tech. 2010;12:35–45. doi: 10.1208/s12249-010-9555-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang JC, Chen SH. Preparation and characterization of microcapsules containing capsaicin. App Polym Sci. 2010;116:2234–2241. doi: 10.1002/app.31443. [DOI] [Google Scholar]
- 24.Nandgude TD, Bhise KS. Characterization of hydrochloride and tannate salts of diphenhydramine. Indian J Pharm Sci. 2008;70:482–486. doi: 10.4103/0250-474X.44598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang JH, Liu ZP, Du H, Zeng Y, Dong AJ. A novel hydrophilic adhesive matrix with self-enhancement for drug percutaneous permeation through rat skin. Pharm Res. 2009;26:1398–1406. doi: 10.1007/s11095-009-9850-1. [DOI] [PubMed] [Google Scholar]
- 26.Kokubo T, Sugibayshi K, Morimoto Y. Diffusion of drug in acrylic-type pressure sensitive adhesive matrices. I. Influence of physical property of matrices on the drug diffusion. J Control Release. 1991;17:69–78. doi: 10.1016/0168-3659(91)90132-W. [DOI] [Google Scholar]
- 27.Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60:110–111. [PubMed] [Google Scholar]
- 28.Siepmann J, Siepmann F. Mathematical modeling of drug delivery. Int J Pharm. 2008;364:328–343. doi: 10.1016/j.ijpharm.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Morimoto Y, Kokubo T, Sugibayashi K. Diffusion of drugs in acrylic-type pressure-sensitive adhesive matrix II.Influence of interaction. J Control Release. 1992;18:113–122. doi: 10.1016/0168-3659(92)90180-Y. [DOI] [Google Scholar]
- 30.Sawant PD, Luu D, Ye R, Buchta R. Drug release from hydroethanolic gels. Effect of drug’s lipophilicity (log P), polymer–drug interactions and solvent lipophilicity. Int J Pharm. 2010;396:45–52. doi: 10.1016/j.ijpharm.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC) Adv Drug Deliver Rev. 2001;48:139–157. doi: 10.1016/S0169-409X(01)00112-0. [DOI] [PubMed] [Google Scholar]
- 32.Shucai L, Jӓrvelӓ PK, Jӓrvelӓ PA. A comparison between apparent viscosity and dynamic complex viscosity for polypropylene/maleated polypropylene blends. Polym Eng Sci. 1997;37:18–23. doi: 10.1002/pen.11641. [DOI] [Google Scholar]
- 33.Liu DZ, Sheu MT, Chen CH, Yang YR, Ho HO. Release characteristics of lidocaine from local implant of polyanionic and polycationic hydrogels. J Control Release. 2007;118:333–339. doi: 10.1016/j.jconrel.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Gal A, Nussinovitch A. Plasticizers in the manufacture of novel skin-bioadhesive patches. Int J Pharm. 2009;370:103–109. doi: 10.1016/j.ijpharm.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 35.Glaessl B, Siepmann F. Deeper insight into the drug release mechanisms in Eudragit RL-based delivery systems. Int J Pharm. 2010;389:139–146. doi: 10.1016/j.ijpharm.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 36.Schulz M, Fussnegger B, Bodmeier R. Drug release and adhesive properties of crospovidone-containing matrix patches based on polyisobutene and acrylic adhesives. Eur J Pharm Sci. 2010;41:675–684. doi: 10.1016/j.ejps.2010.09.011. [DOI] [PubMed] [Google Scholar]