

Abstract
The glucagon-like peptide-1 receptor (GLP-1R) is a key physiological regulator of insulin secretion and a major therapeutic target for the treatment of type II diabetes. However, regulation of GLP-1R function is complex with multiple endogenous peptides that interact with the receptor, including full-length (1–37) and truncated (7–37) forms of GLP-1 that can exist in an amidated form (GLP-1(1–36)NH2 and GLP-1(7–36)NH2) and the related peptide oxyntomodulin. In addition, the GLP-1R possesses exogenous agonists, including exendin-4, and the allosteric modulator, compound 2 (6,7-dichloro-2-methylsulfonyl-3-tert-butylaminoquinoxaline). The complexity of this ligand-receptor system is further increased by the presence of several single nucleotide polymorphisms (SNPs) that are distributed across the receptor. We have investigated 10 GLP-1R SNPs, which were characterized in three physiologically relevant signaling pathways (cAMP accumulation, extracellular signal-regulated kinase 1/2 phosphorylation, and intracellular Ca2+ mobilization); ligand binding and cell surface receptor expression were also determined. We demonstrate both ligand- and pathway-specific effects for multiple SNPs, with the most dramatic effect observed for the Met149 receptor variant. At the Met149 variant, there was selective loss of peptide-induced responses across all pathways examined, but preservation of response to the small molecule compound 2. In contrast, at the Cys333 variant, peptide responses were preserved but there was attenuated response to compound 2. Strikingly, the loss of peptide function at the Met149 receptor variant could be allosterically rescued by compound 2, providing proof-of-principle evidence that allosteric drugs could be used to treat patients with this loss of function variant.
Introduction
The glucagon-like peptide-1 receptor (GLP-1R) is a key target in the development of treatments for type II diabetes mellitus, with actions including glucose-dependent increases in insulin synthesis and release; decreases in β-cell apoptosis, body mass, and gastric emptying; and a decrease in peripheral resistance to insulin that address fundamental symptoms associated with the condition (DeFronzo, 1992; Drucker, 2006). The GLP-1R is a family B peptide hormone G protein-coupled receptor primarily expressed in pancreatic β-cells that responds to at least four distinct endogenous GLP-1 variants: two full-length GLP-1 peptides [GLP-1(1–36)NH2 and GLP-1(1–37)] and two truncated and more prominent circulating forms [GLP-1(7–36)NH2 and GLP-1(7–37)] (Baggio and Drucker, 2007). In addition, the related endogenous peptide oxyntomodulin and exogenous mimetic peptide exendin-4 act at the GLP-1R to increase the biosynthesis and secretion of insulin, decrease β-cell apoptosis and decrease gastric emptying in a similar manner to the endogenous GLP-1 peptides (Jarrousse et al., 1984; Jarrousse et al., 1985; Göke et al., 1993). Although levels of GLP-1 are reduced in patients with type II diabetes mellitus, the retention of its insulinotropic properties at the GLP-1R make it one of the most promising ligand-receptor systems to target in the development of treatments for type II diabetes (Nauck et al., 1993; Toft-Nielsen et al., 2001). Recent medical developments to target this system include the GLP-1 mimetic liraglutide (Knudsen et al., 2000; Elbrønd et al., 2002) and dipeptidyl peptidase IV inhibitors that prolong the plasma half-life of endogenous GLP-1R peptides (Deacon et al., 1995a,b). The latter compounds fail to achieve the weight loss seen with peptide therapeutics, whereas the peptides have significant potential for reduced patient compliance due to the requirement for subcutaneous administration. To overcome this, small orally active drugs that can augment GLP-1R signaling are continually being pursued. One example, compound 2 (6,7-dichloro-2-methylsulfonyl-3-tert-butylaminoquinoxaline; Novo Nordisk A/S, Bagsværd, Denmark), allosterically enhances peptide binding affinity and subsequently influences insulin secretion (Knudsen et al., 2007; Koole et al., 2010) and provides an exemplar for understanding allosteric modulation of the receptor. However, developing allosteric therapeutics for a pleiotropically coupled receptor with multiple endogenous ligands poses a significant challenge (Koole et al., 2010). In addition, the presence of naturally occurring nonsynonymous single nucleotide polymorphisms (SNPs), add a further element of complexity in the development of these drugs for therapeutic application. The presence of SNPs may be linked to the rate of onset of disease or effectiveness of receptor targeted treatments. Although SNPs have been characterized in great detail for many G protein-coupled receptors, there is limited knowledge on the effects of SNPs at the GLP-1R. Several GLP-1R SNPs have been assessed previously in vitro, although not explored in a wide range of functional outputs (Beinborn et al., 2005; Fortin et al., 2010) and at least one has been reported to have a loose association with type II diabetes mellitus (Tokuyama et al., 2004). A better understanding of the role of these polymorphisms in receptor function is therefore required, not only to gain an insight into the effects they have on receptor function but also to understand their possible association with the onset of disease or effectiveness of drug therapies.
In this study, we found ligand- and pathway-dependent alteration in signaling via select polymorphisms of the GLP-1R. Furthermore, we demonstrate that the major loss of GLP-1R function to peptide agonists at the Met149 polymorphic receptor variant can be restored via allosteric modulation of the receptor, providing potential therapeutic paths to the treatment of diabetic patients carrying this SNP.
Materials and Methods
Materials.
Dulbecco's modified Eagle's medium (DMEM), hygromycin-B, and Fluo-4 acetoxymethyl ester were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Thermo Fisher Scientific (Melbourne, VIC, Australia). The QuikChange site-directed mutagenesis kit was purchased from Stratagene (La Jolla, CA). AlphaScreen reagents, Bolton-Hunter reagent (125I), and 384-well ProxiPlates were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). SureFire extracellular signal-regulated kinases 1 and 2 (ERK1/2) reagents were generously provided by TGR Biosciences (Adelaide, SA, Australia). SigmaFast o-phenylenediamine dihydrochloride tablets and antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Compound 2 was generated according to a method published previously (Teng et al., 2007), to a purity of >95%, and compound integrity was confirmed by NMR. GLP-1 and GLP-1 peptide analogs were purchased from American Peptide Co., Inc. (Sunnyvale, CA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) or BDH Merck (Melbourne, VIC, Australia) and were of an analytical grade.
Receptor Mutagenesis.
Natural variants of the human GLP-1R with supporting nucleotide sequences reported to exist in the population were identified using the SwissProt database (http://www.uniprot.org/uniprot/P43220) and in a previous clinical report (Tokuyama et al., 2004) (Table 1; Fig. 1A). Each of these variants were introduced into a double c-myc-labeled wild-type human GLP-1R in the pEF5/FRT/V5-DEST destination vector (Invitrogen) using oligonucleotides for site-directed mutagenesis from GeneWorks (HindMarsh, SA, Australia) (Supplemental Table 1) and the QuikChange site-directed mutagenesis kit (Stratagene). The integrity of the subsequent receptor clones were confirmed by cycle sequencing as described previously (May et al., 2007). In this study, the wild-type GLP-1R was defined as the form of the receptor comprised of the following residues at the sites of polymorphic variation: Pro7, Arg20, Arg44, Arg131, Thr149, Gly168, Phe260, Ala316, Ser333, and Arg421.
TABLE 1.
Human GLP-1R single nucleotide polymorphisms
Single nucleotide polymorphisms were generated for this study based on those identified in the Swissprot database (www.uniprot.org/uniprot/P43220) in association with those identified to exist in a cohort of normal and diabetic subjects in Tokuyama et al., 2004. Frequency data is reported from the Swissprot database.
| Residue | Amino Acid | Nucleotide Substitution | Frequency of Nucleotide Substitution |
NCBI Identification Number | Reference in Addition to SwissProt | |
|---|---|---|---|---|---|---|
| Homozygous | Heterozygous | |||||
| 7 | Pro | CCG | 0.60 | 0.29 | rs10305420 | Tokuyama et al., 2004 |
| Leu | CTG | 0.11 | ||||
| 20 | Arg | AGG | 0.99 | 0.01 | rs10305421 | |
| Lys | AAG | Unknown | ||||
| 44 | Arg | CGC | 0.99 | 0.01 | rs2295006 | Tokuyama et al., 2004 |
| His | CAC | Unknown | ||||
| 131 | Arg | CGA | 0.92 | 0.08 | rs3765467 | Tokuyama et al., 2004 |
| Gln | CAA | Unknown | ||||
| 149 | Thr | ACG | Unknown | Unknown | 112198 | Tokuyama et al., 2004 |
| Met | ATG | Unknown | ||||
| 168 | Gly | GGC | 0.76 | 0.2 | rs6923761 | |
| Ser | AGC | 0.04 | ||||
| 260 | Phe | TTC or TTT | 0.31a | 0.56 | rs1042044 | Tokuyama et al., 2004 |
| Leu | TTA | 0.13 | ||||
| 316 | Ala | GCC | 0.98 | 0.02 | rs10305492 | |
| Thr | ACC | Unknown | ||||
| 333 | Ser | TCC | 0.99 | 0.01 | rs10305493 | |
| Cys | TGC | Unknown | ||||
| 421 | Arg | CGG | 0.99 | 0.01 | rs10305510 | |
| Gln | CAG | Unknown | ||||
No global information for TTT nucleotide variant.
Fig. 1.

A, schematic diagram of the human GLP-1R; location of residues subject to polymorphic variance is highlighted in gray, B, cell surface expression profiles of the human GLP-1R polymorphisms stably transfected into FlpInCHO cells as determined through antibody detection of the N-terminal c-myc epitope label. Statistical significance of changes in total cell surface expression compared with wild-type human GLP-1R expression (100%) were determined by one-way analysis of variance and Dunnett's post-test and are indicated with an asterisk (*, p < 0.05). All data are mean ± S.E.M. of five to seven independent experiments conducted in triplicate. TM, transmembrane; ICL, intracellular loop.
Transfections and Cell Culture.
Wild-type and polymorphic human GLP-1R were isogenically integrated into FlpIn-Chinese hamster ovary (FlpInCHO) cells (Invitrogen) and selection of receptor-expressing cells accomplished by treatment with 600 μg/ml hygromycin B as described previously (May et al., 2007). Transfected and parental FlpInCHO cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and incubated in a humidified environment at 37°C in 5% CO2.
Radioligand Binding Assay.
FlpInCHO wild-type and polymorphic human GLP-1R cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2. Growth media was replaced with binding buffer [DMEM containing 25 mM HEPES and 0.1% (w/v) BSA] containing 0.5 nM 125I-exendin(9–39) (Ki for the wild-type receptor, 12.5 nM) and increasing concentrations of unlabeled ligand. Cells were then incubated overnight at 4°C, followed by three washes in ice-cold 1× PBS to remove unbound radioligand. Cells were then solubilized in 0.1 M NaOH, and radioactivity determined by γ-counting. For interaction studies, competition of 125I-exendin(9–39) binding by each orthosteric agonist was performed in the presence of 3 μM compound 2, added simultaneously. For all experiments, nonspecific binding was defined by 1 μM exendin(9–39).
cAMP Accumulation Assay.
FlpInCHO wild-type and polymorphic human GLP-1R cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2, and cAMP detection carried out as described previously (Koole et al., 2010). For interaction studies, increasing concentrations of peptide ligand and 3 μM compound 2 were added simultaneously, and cAMP accumulation measured after 30 min of cell stimulation. All values were converted to concentration of cAMP using a cAMP standard curve performed in parallel, and data were subsequently normalized to the response of 100 nM forskolin.
ERK1/2 Phosphorylation Assay.
FlpInCHO wild-type and polymorphic human GLP-1R cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2. Receptor-mediated ERK1/2 phosphorylation was determined by using the AlphaScreen ERK1/2 SureFire protocol as described previously (May et al., 2007). Initial ERK1/2 phosphorylation time-course experiments were performed over 1 h to determine the time at which ERK1/2 phosphorylation was maximal after stimulation by agonists. Subsequent experiments were then performed at the time required to generate a maximal ERK1/2 phosphorylation response (7 min). For interaction studies, increasing concentrations of peptide ligand and 3 μM compound 2 were added simultaneously, and ERK1/2 phosphorylation was measured after 7 min of cell stimulation. Data were normalized to the maximal response elicited by 10% FBS, determined at 7 min (peak FBS response).
Intracellular Ca2+ Mobilization Assay.
FlpInCHO wild-type and polymorphic human GLP-1R cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2, and receptor-mediated intracellular Ca2+ mobilization determined as described previously (Werry et al., 2005). Fluorescence was determined immediately after peptide addition, with an excitation wavelength set to 485 nm and an emission wavelength set to 520 nm, and readings taken every 1.36 s for 120 s. For interaction studies, 3 μM compound 2 was added 30 min before peptide addition because of autofluorescence. Subsequent fluorescence was then determined immediately after peptide addition with the conditions detailed above. Peak magnitude was calculated using five-point smoothing, followed by correction against basal fluorescence. The peak value was used to create concentration-response curves. Data were normalized to the maximal response elicited by 100 μM ATP.
Cell Surface Receptor Expression.
FlpInCHO wild-type and polymorphic human GLP-1R cells, with receptor DNA previously N-terminally labeled with a double c-myc epitope label, were seeded at a density of 25 × 104 cells/well into 24-well culture plates and incubated overnight at 37°C in 5% CO2, washed three times in 1× PBS and fixed with 3.7% paraformaldehyde at 4°C for 15 min. To determine the effects of compound 2 on cell surface receptor expression, adherent cells were treated for 4 or 18 h with 3 μM compound 2 and subsequently fixed as described above. Cell surface receptor detection was performed using a mouse monoclonal (9E10) primary antibody (1:2000) to detect the c-myc tag, and a mouse-raised IgG horseradish peroxidase-linked secondary antibody (1:2000), both diluted in blocking solution [1× PBS containing 2% (w/v) BSA and 0.05% (w/v) Tween 20]. Peroxidase activity was then measured using SigmaFast o-phenylenediamine dihydrochloride tablets (Sigma) according to the manufacturer's instructions, and fluorescence detected at an emission wavelength of 492 nm. Data were normalized to the basal fluorescence detected in FlpInCHO parental cells.
Data Analysis.
All data were analyzed in Prism 5.02 (GraphPad Software Inc., San Diego, CA). Concentration response signaling data were analyzed using a three-parameter logistic equation as described previously (May et al., 2007):
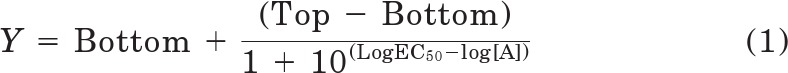 |
where Bottom represents the Y value in the absence of ligand(s), Top represents the maximal stimulation in the presence of ligand/s, [A] is the molar concentration of ligand, and EC50 represents the molar concentration of ligand required to generate a response halfway between Top and Bottom. Likewise, this equation was used in the analysis of inhibition binding data, replacing EC50 with IC50. In this case, Bottom defines the specific binding of the radioligand that is equivalent to nonspecific ligand binding, whereas Top defines radioligand binding in the absence of a competing ligand, and the IC50 value represents the molar concentration of ligand required to generate a response halfway between Top and Bottom.
Statistics.
Changes in peptide affinity, efficacy, potency, and cell surface expression of polymorphic variants compared with wild-type control were statistically analyzed with one-way analysis of variance and Dunnett's post test, and significance was accepted at p < 0.05. Differential modulation of oxyntomodulin potency by the allosteric ligand compound 2 at each of the polymorphic variants was assessed in comparison with an oxyntomodulin control using a paired t test, and statistical significance was accepted at p < 0.05.
Results
Cell Surface Expression of Human GLP-1R Polymorphic Variants.
Each of the human GLP-1R polymorphisms was isogenically integrated into FlpInCHO host cells (Invitrogen) by recombination, allowing comparison of relative cell surface expression without complication from variation in gene transcription. In this study we observed an interesting trend in the cell surface expression profiles of the GLP-1R polymorphisms as assessed by antibody labeling of the N-terminally incorporated c-myc tag (Fig. 1B). Although the polymorphisms occurring in the N-terminal domain of the receptor seemed to have cell surface expression similar to that of the wild-type human GLP-1R, most other polymorphic variants distributed across the receptor had reduced cell surface expression, with greatest effect seen for the Thr316 variant.
The Human GLP-1R Met149 Polymorphic Variant Displays a Reduction in Orthosteric Agonist Affinity.
To establish the binding profiles at each of the reported human GLP-1R polymorphisms, we performed equilibrium binding studies with the orthosteric GLP-1R agonists or the allosteric agonist compound 2, in competition with the radiolabeled orthosteric antagonist 125I-exendin(9–39). There was no significant influence of the polymorphisms on orthosteric agonist potency for inhibition of the radiolabel, except for the Met149 receptor variant, which had reductions in the affinities of GLP-1(7–36)NH2, exendin-4, oxyntomodulin (Fig. 2; Table 2), and GLP-1(7–37) (data not shown)1. It is noteworthy that the extent of apparent affinity reduction differed between agonists, with a 251-fold shift for GLP-1(7–36)NH2 and oxyntomodulin (Table 2) but only a 32-fold shift for exendin-4 (Table 2). Minimal effects were observed on GLP-1(1–36)NH2, compound 2 (Fig. 2), and GLP-1(1–37) (data not shown)2 binding within the concentration range measured, suggesting the affinity of these agonists is not greatly affected in any of the human GLP-1R variants compared with wild-type.
Fig. 2.

Characterization of the binding of GLP-1(1–36)NH2 (A), GLP-1(7–36)NH2 (B), exendin-4 (C), oxyntomodulin (D), and compound 2 (E) in competition with the radiolabeled antagonist 125I-exendin(9–39) in whole FlpInCHO cells stably expressing each of the human GLP-1R polymorphisms or the wild-type GLP-1R. Data are normalized to the maximum 125I-exendin(9–39) binding of each individual data set, with nonspecific binding measured in the presence of 1 μM exendin(9–39). Data are analyzed with a three-parameter logistic equation as defined in eq. 1. All values are mean ± S.E.M. of three to four independent experiments conducted in duplicate.
TABLE 2.
Effects of naturally occurring human GLP-1R polymorphisms on agonist binding
Data were analyzed using a three-parameter logistic equation as defined in eq. 1. pIC50 values represent the negative logarithm of the concentration of agonist that inhibits binding of half the total concentration of radiolabeled antagonist 125I-exendin(9–39). Data are normalized to maximum 125I-exendin(9–39) binding of each individual data set, with nonspecific binding measured in the presence of 1 μM exendin(9–39). All values are mean ± S.E.M. of three to four independent experiments, conducted in duplicate. Data were analyzed with one-way analysis of variance and Dunnett's post test.
| pIC50 |
|||
|---|---|---|---|
| GLP-1(7–36)NH2 | Exendin-4 | Oxyntomodulin | |
| Wild type | 8.9 ± 0.1 | 9.4 ± 0.1 | 7.9 ± 0.1 |
| Leu7 | 8.9 ± 0.2 | 9.5 ± 0.1 | 8.2 ± 0.1 |
| Lys20 | 8.8 ± 0.1 | 9.6 ± 0.1 | 7.9 ± 0.2 |
| His44 | 8.9 ± 0.1 | 9.5 ± 0.1 | 7.9 ± 0.1 |
| Gln131 | 8.8 ± 0.1 | 9.3 ± 0.1 | 7.9 ± 0.1 |
| Met149 | 6.5 ± 0.2* | 7.9 ± 0.2* | 5.5 ± 0.2* |
| Ser168 | 9.3 ± 0.1 | 9.5 ± 0.1 | 8.2 ± 0.1 |
| Leu260 | 9.2 ± 0.2 | 9.4 ± 0.2 | 7.9 ± 0.1 |
| Thr316 | 9.5 ± 0.2 | 9.7 ± 0.2 | 8.4 ± 0.2 |
| Cys333 | 9.0 ± 0.1 | 9.6 ± 0.1 | 8.0 ± 0.1 |
| Gln421 | 9.1 ± 0.2 | 9.6 ± 0.2 | 8.1 ± 0.2 |
Statistically significant at p < 0.05.
The Human GLP-1R Met149 Polymorphic Variant Displays Reduced Orthosteric but Not Allosteric Agonist Potency in cAMP Accumulation.
The cAMP accumulation profile of all orthosterically binding peptides at each of the GLP-1R polymorphic variants was similar to that of wild-type human GLP-1R, with the exception of the Met149 polymorphism, where each peptide exhibited reduced potency, with observed decreases in potency of 158-fold for GLP-1(7–36)NH2, 200-fold for exendin-4, and >500-fold for oxyntomodulin (Fig. 3; Table 3). Full concentration-response relationships for the lower potency agonists GLP-1(1–36)NH2, GLP-1(1–37), oxyntomodulin, and compound 2 could not be determined; however, the effect appeared more profound for oxyntomodulin relative to the truncated GLP-1 peptides and exendin-4 (Fig. 3). In contrast, the potency and efficacy of the allosteric agonist compound 2 at the Met149 polymorphic variant, was similar to that observed with the Thr149 variant, with pEC50 values of 5.5 ± 0.1 and 5.5 ± 0.1, respectively (n = 4) (Fig. 3; Table 3). However, at the Cys333 polymorphism, where orthosteric peptide profiles were equivalent to those of wild-type, compound 2 displayed a reduced cAMP response (Fig. 3; Table 3).
Fig. 3.

Characterization of cAMP accumulation in the presence of GLP-1(1–36)NH2 (A), GLP-1(7–36)NH2 (B), exendin-4 (C), oxyntomodulin (D), and compound 2 (E) in FlpInCHO cells stably expressing each of the human GLP-1R polymorphisms or the wild-type GLP-1R. Data are normalized to the response elicited by 100 nM forskolin and analyzed with a three-parameter logistic equation as defined in eq. 1. All values are mean ± S.E.M. of 4 to 12 experiments conducted in duplicate.
TABLE 3.
Effects of naturally occurring human GLP-1R polymorphisms on agonist signaling via cAMP
Data were analyzed using a three-parameter logistic equation as defined in eq. 1. pEC50 values represent the negative logarithm of the concentration of agonist that produces half the maximal response. Emax represents the maximal response normalized to the response elicited by that of 100 nM forskolin. All values are mean ± S.E.M. of four to ten independent experiments, conducted in duplicate. Data were analyzed with one-way analysis of variance and Dunnett's post test.
| cAMP Accumulation |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pEC50 |
Emax |
|||||||||
| GLP-1 (1–36)NH2 | GLP-1 (7–36)NH2 | Exendin-4 | Oxyntomodulin | Compound 2a | GLP-1 (1–36)NH2 | GLP-1 (7–36)NH2 | Exendin-4 | Oxyntomodulin | Compound 2b | |
| Wild type | 6.8 ± 0.2 | 10.2 ± 0.2 | 10.7 ± 0.1 | 8.7 ± 0.1 | 5.5 ± 0.1 | 270 ± 36 | 343 ± 21 | 299 ± 9 | 368 ± 9 | 254 ± 44 |
| Leu7 | 7.1 ± 0.2 | 10.5 ± 0.2 | 10.4 ± 0.2 | 8.5 ± 0.1 | 5.6 ± 0.1 | 217 ± 18 | 294 ± 18 | 277 ± 14 | 283 ± 13* | 242 ± 50 |
| Lys20 | 7.3 ± 0.2 | 10.3 ± 0.2 | 10.6 ± 0.2 | 8.5 ± 0.2 | 5.6 ± 0.1 | 244 ± 26 | 339 ± 19 | 249 ± 12 | 279 ± 18* | 200 ± 26 |
| His44 | 7.1 ± 0.2 | 10.4 ± 0.2 | 10.4 ± 0.2 | 8.3 ± 0.1 | 5.7 ± 0.1 | 244 ± 27 | 321 ± 18 | 302 ± 16 | 322 ± 17 | 291 ± 46 |
| Gln131 | 7.4 ± 0.3 | 10.4 ± 0.2 | 10.6 ± 0.2 | 8.3 ± 0.2 | 5.6 ± 0.1 | 209 ± 31 | 322 ± 21 | 253 ± 15 | 271 ± 14* | 225 ± 35 |
| Met149 | N.D. | 8.0 ± 0.3* | 8.4 ± 0.1* | N.D. | 5.5 ± 0.1 | N.D. | N.D. | 223 ± 13 | N.D. | 217 ± 24 |
| Ser168 | 6.9 ± 0.2 | 10.5 ± 0.2 | 10.4 ± 0.2 | 8.3 ± 0.2 | 5.8 ± 0.1 | 314 ± 33 | 320 ± 17 | 242 ± 11 | 273 ± 14* | 283 ± 13 |
| Leu260 | 6.9 ± 0.2 | 10.4 ± 0.3 | 10.5 ± 0.2 | 8.4 ± 0.1 | 5.3 ± 0.1 | 297 ± 28 | 291 ± 21 | 290 ± 13 | 302 ± 12 | 184 ± 30 |
| Thr316 | 7.2 ± 0.2 | 10.5 ± 0.3 | 10.5 ± 0.4 | 8.7 ± 0.3 | 5.3 ± 0.1 | 304 ± 33 | 228 ± 16* | 301 ± 30 | 277 ± 22* | 182 ± 18 |
| Cys333 | 6.9 ± 0.2 | 10.2 ± 0.3 | 10.2 ± 0.2 | 8.2 ± 0.2 | N.D. | 259 ± 24 | 331 ± 26 | 257 ± 12 | 288 ± 16 | 46 ± 8* |
| Gln421 | 6.7 ± 0.2 | 10.1 ± 0.2 | 10.3 ± 0.2 | 8.1 ± 0.1 | 5.6 ± 0.1 | 242 ± 23 | 275 ± 15 | 267 ± 13 | 332 ± 15 | 236 ± 45 |
N.D., data unable to be experimentally defined or with incomplete curves.
For estimation of pEC50 values, Emax was set to ≤300% of the 100 nM forskolin response.
Compound 2 response at 10−5 M.
Statistically significant at p < 0.05.
Human GLP-1R Polymorphisms Have Variable Intracellular Ca2+ Mobilization Responses.
In accord with previous studies (Koole et al., 2010), no Ca2+ response was seen for full-length GLP-1 peptides or compound 2 at the wild-type receptor, and this was true also for each of the polymorphic receptor variants (Fig. 4). In the case of oxyntomodulin, only a weak response was observed, and this was submaximal at 1 μM, even at the wild-type receptor. The weak response made interpretation of the effect of polymorphic variation difficult; however, a reduced response at 1 μM was seen for the Met149 and Thr316 variants (Fig. 4; Table 4).
Fig. 4.

Characterization of intracellular Ca2+ mobilization in the presence of GLP-1(1–36)NH2 (A), GLP-1(7–36)NH2 (B), exendin-4 (C), and oxyntomodulin (D) in FlpInCHO cells stably expressing each of the human GLP-1R polymorphisms or the wild-type GLP-1R. Data are normalized to the maximal response elicited by 100 μM ATP and analyzed with a three-parameter logistic equation as defined in eq. 1. All values are mean ± S.E.M. of five to nine experiments conducted in duplicate.
TABLE 4.
Effects of naturally occurring human GLP-1R polymorphisms on agonist signaling via intracellular Ca2+ mobilization
Data were analyzed using a three-parameter logistic equation as defined in eq. 1. pEC50 values represent the negative logarithm of the concentration of agonist that produces half the maximal response. Emax represents the maximal response normalized to the response elicited by that of 10−4 M ATP. All values are mean ± S.E.M. of 5 to 10 independent experiments, conducted in duplicate. Data were analyzed with one-way analysis of variance and Dunnett's post test.
| Intracellular Ca2+ Mobilization |
||||||
|---|---|---|---|---|---|---|
| pEC50 |
Emax |
|||||
| GLP-1(7–36)NH2 | Exendin-4 | Oxyntomodulin | GLP-1(7–36)NH2 | Exendin-4 | Oxyntomodulina | |
| Wild type | 7.8 ± 0.2 | 8.0 ± 0.3 | N.D. | 25.8 ± 1.5 | 23.1 ± 2.5 | 25.9 ± 5.5 |
| Leu7 | 8.0 ± 0.2 | 7.9 ± 0.2 | N.D. | 25.2 ± 1.4 | 20.4 ± 1.4 | 21.0 ± 2.5 |
| Lys20 | 7.9 ± 0.2 | 8.1 ± 0.1 | N.D. | 21.3 ± 1.4 | 15.0 ± 0.7* | 15.7 ± 1.6 |
| His44 | 8.1 ± 0.2 | 8.1 ± 0.2 | N.D. | 24.5 ± 1.4 | 18.9 ± 1.3 | 18.8 ± 2.1 |
| Gln131 | 8.2 ± 0.2 | 7.9 ± 0.3 | N.D. | 17.3 ± 1.4* | 12.8 ± 1.1* | 13.7 ± 1.5 |
| Met149 | N.D. | N.D. | N.D. | N.D. | N.D. | 2.6 ± 0.4* |
| Ser168 | 8.1 ± 0.2 | 8.0 ± 0.2 | N.D. | 18.3 ± 1.0* | 13.9 ± 0.8* | 14.4 ± 0.9 |
| Leu260 | 8.2 ± 0.1 | 8.2 ± 0.1 | N.D. | 27.8 ± 1.2 | 21.3 ± 1.0 | 19.0 ± 1.9 |
| Thr316 | 7.8 ± 0.2 | 7.7 ± 0.5 | N.D. | 12.4 ± 1.0* | 7.3 ± 0.9* | 11.9 ± 1.9* |
| Cys333 | 7.8 ± 0.3 | 7.8 ± 0.2 | N.D. | 23.6 ± 2.4 | 14.6 ± 1.1* | 15.0 ± 2.4 |
| Gln421 | 7.9 ± 0.3 | 7.9 ± 0.3 | N.D. | 15.6 ± 1.7* | 13.6 ± 1.3* | 16.8 ± 7.8 |
N.D., data unable to be experimentally defined or with incomplete curves.
Statistically significant at p < 0.05.
Response at 10−6 M oxyntomodulin.
No significant alteration in the potency of GLP-1(7–36)NH2, GLP-1(7–37), or exendin-4 peptides was observed; however, marked reduction in peptide efficacy was found for selected receptor variants (Fig. 4; Table 4). For each of the higher affinity peptides, there was reduced efficacy at the Gln131, Met149, Ser168, Thr316, and Gln421 variants, the greatest reduction in efficacy being seen at the Met149 variant, which exhibited no measurable agonist-induced response (Fig. 4; Table 4). In addition, for exendin-4 only, there were reductions in efficacy at the Cys333 and Lys20 variants; for Lys20, this occurred despite wild-type levels of cell surface receptor expression. In systems with low receptor reserve, changes in receptor density can affect observed efficacy and potency. In many instances, there was evidence of reduced cell surface receptor expression, as assessed by antibody binding to the c-myc epitope (Fig. 1B), that probably contributed to the observed decrease in efficacy, because there is little reserve for coupling to this pathway. Intriguingly, the Leu260 variant had essentially wild-type response to the peptides, despite significantly reduced cell surface receptor expression, suggesting that the Leu260 substitution may actually favor coupling via Gαq.
Effect of Receptor Polymorphisms on ERK1/2 Phosphorylation.
Agonist-induced ERK1/2 phosphorylation was determined at 7 min for each of the human GLP-1R polymorphic variants. Most receptor variants exhibited ERK1/2 responses similar to those of peptide agonists or compound 2 at the wild-type GLP-1R with the exception of the Met149 variant (Fig. 5, Table 5). There was a decrease in the potency and/or efficacy of all orthosteric peptides with the Met149 polymorphic variant, but this was not as pronounced as in the other pathways, suggesting alteration to signal bias of the receptor. The allosteric ligand compound 2 retained its weak agonism at this variant, sharing an ERK1/2 phosphorylation profile similar to that of compound 2 at the wild-type human GLP-1R (Fig. 5; Table 5).
Fig. 5.

Characterization of ERK1/2 phosphorylation in the presence of GLP-1(1–36)NH2 (A), GLP-1(7–36)NH2 (B), exendin-4 (C), oxyntomodulin (D), and compound 2 (E), in FlpInCHO cells stably expressing each of the human GLP-1R polymorphisms or the wild-type GLP-1R. Data are normalized to the maximal response elicited by 10% FBS and analyzed with a three-parameter logistic equation as defined in eq. 1. All values are mean ± S.E.M. of four to five independent experiments conducted in duplicate.
TABLE 5.
Effects of naturally occurring human GLP-1R polymorphisms on agonist signaling via ERK1/2 phosphorylation
Data were analyzed using a three-parameter logistic equation as defined in eq. 1. pEC50 values represent the negative logarithm of the concentration of agonist that produces half the maximal response. Emax represents the maximal response normalized to the response elicited by that of 10% FBS. All values are mean ± S.E.M. of four to five independent experiments, conducted in duplicate. Data were analyzed with one-way analysis of variance and Dunnett's post test.
| ERK1/2 Phosphorylation |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pEC50 |
Emax |
|||||||||
| GLP-1 (1–36)NH2 | GLP-1 (7–36)NH2 | Exendin-4 | Oxyntomodulin | Compound 2 | GLP-1 (1–36)NH2 | GLP-1 (7–36)NH2 | Exendin-4 | Oxyntomodulin | Compound 2 | |
| Wild type | 7.4 ± 0.4 | 8.5 ± 0.3 | 8.7 ± 0.2 | 7.6 ± 0.1 | 6.1 ± 0.3 | 2.3 ± 0.4 | 5.4 ± 0.4 | 6.6 ± 0.4 | 6.4 ± 0.3 | 1.7 ± 0.3 |
| Leu7 | 7.1 ± 0.2 | 8.8 ± 0.2 | 9.2 ± 0.2 | 7.7 ± 0.1 | 6.1 ± 0.2 | 3.3 ± 0.4 | 7.1 ± 0.4 | 7.2 ± 0.4 | 8.1 ± 0.3 | 2.6 ± 0.3 |
| Lys20 | 6.9 ± 0.3 | 8.7 ± 0.1 | 9.3 ± 0.2 | 7.4 ± 0.1 | 5.9 ± 0.2 | 2.8 ± 0.5 | 5.7 ± 0.2 | 6.9 ± 0.4 | 8.4 ± 0.4 | 1.9 ± 0.2 |
| His44 | 7.2 ± 0.2 | 8.6 ± 0.2 | 9.3 ± 0.2 | 7.6 ± 0.2 | 6.3 ± 0.3 | 2.6 ± 0.3 | 6.2 ± 0.3 | 6.7 ± 0.4 | 7.8 ± 0.6 | 1.8 ± 0.2 |
| Gln131 | 7.3 ± 0.2 | 8.9 ± 0.2 | 9.5 ± 0.2 | 7.8 ± 0.2 | 6.2 ± 0.3 | 2.8 ± 0.2 | 6.4 ± 0.3 | 6.9 ± 0.4 | 8.2 ± 0.5 | 1.9 ± 0.3 |
| Met149 | N.D. | 7.8 ± 0.3 | 8.0 ± 0.2 | 7.1 ± 0.2 | 5.9 ± 0.3 | 0.6 ± 0.1 | 4.1 ± 0.5 | 3.4 ± 0.3* | 3.7 ± 0.3* | 2.0 ± 0.4 |
| Ser168 | 7.3 ± 0.2 | 8.8 ± 0.2 | 9.3 ± 0.3 | 7.8 ± 0.1 | 5.9 ± 0.2 | 2.0 ± 0.2 | 5.4 ± 0.3 | 6.7 ± 0.5 | 6.1 ± 0.3 | 2.3 ± 0.3 |
| Leu260 | 7.3 ± 0.4 | 8.4 ± 0.4 | 8.9 ± 0.4 | 7.5 ± 0.3 | 6.0 ± 0.3 | 2.2 ± 0.3 | 5.1 ± 0.6 | 5.4 ± 0.7 | 5.9 ± 0.7 | 1.9 ± 0.3 |
| Thr316 | 7.3 ± 0.4 | 8.8 ± 0.3 | 8.8 ± 0.3 | 7.8 ± 0.3 | 6.0 ± 0.3 | 2.0 ± 0.3 | 4.2 ± 0.4 | 4.9 ± 0.5 | 5.1 ± 0.6 | 1.8 ± 0.3 |
| Cys333 | 7.7 ± 0.2 | 8.7 ± 0.2 | 9.1 ± 0.3 | 7.7 ± 0.2 | 6.0 ± 0.2 | 1.7 ± 0.2 | 5.9 ± 0.4 | 6.1 ± 0.5 | 7.0 ± 0.5 | 1.4 ± 0.2 |
| Gln421 | 7.2 ± 0.4 | 8.9 ± 0.3 | 9.1 ± 0.2 | 7.7 ± 0.4 | 6.1 ± 0.3 | 2.3 ± 0.4 | 5.0 ± 0.5 | 5.7 ± 0.4 | 6.3 ± 0.9 | 2.9 ± 0.4 |
N.D., data unable to be experimentally defined or with incomplete curves.
Statistically significant at p < 0.05.
Effect of Receptor Polymorphisms on the Allosteric Modulation of the cAMP Response to Oxyntomodulin by Compound 2.
We have demonstrated previously that compound 2 positively modulates binding affinity and, as a result cAMP potency, of the endogenous peptide agonists oxyntomodulin and, to a lesser extent, GLP-1(7–36)NH2 and GLP-1(7–37) (Koole et al., 2010). Oxyntomodulin was the most robustly enhanced peptide and was used to explore whether allosteric regulation of the peptide response was retained at the polymorphic receptor variants. Compound 2 significantly enhanced the oxyntomodulin response at the wild-type receptor, Lys20, and His44 receptor variants (Table 6). At the Cys333 variant, which had attenuated cAMP agonism to compound 2, the allosteric modulator failed to enhance the oxyntomodulin response. It is noteworthy that despite retaining cAMP agonism upon activation with compound 2, the oxyntomodulin response at the Leu7 and Thr316 variants was not significantly modified by compound 2, and although there was a trend toward enhancement with the Gln131, Ser168, Leu260, and Gln421 variants, this was attenuated relative to the wild-type receptor.
TABLE 6.
Differential modulation of oxyntomodulin at naturally occurring human GLP-1R polymorphisms in cAMP accumulation
Data were analyzed using a three-parameter logistic equation as defined in eq. 1. pEC50 values represent the negative logarithm of the concentration of agonist that produces half the maximal response. All data are normalized to the response elicited by that of 10−7M forskolin, and are mean ± S.E.M. of three to five independent experiments, conducted in duplicate.
| pEC50 |
||
|---|---|---|
| Oxyntomodulin | + 3 μM Compound 2 | |
| Wild type | 8.7 ± 0.1 | 10.2 ± 0.7* |
| Leu7 | 8.7 ± 0.1 | 9.1 ± 0.7 |
| Lys20 | 8.6 ± 0.2 | 9.5 ± 0.5* |
| His44 | 8.4 ± 0.1 | 10.2 ± 1.2* |
| Gln131 | 8.7 ± 0.1 | 9.2 ± 0.4 |
| Met149 | N.D. | 7.7 ± 1.5* |
| Ser168 | 8.9 ± 0.1 | 9.4 ± 0.4 |
| Leu260 | 8.6 ± 0.2 | 9.2 ± 0.5 |
| Thr316 | 8.6 ± 0.1 | 8.7 ± 0.6 |
| Cys333 | 8.2 ± 0.1 | 8.3 ± 0.2 |
| Gln421 | 8.4 ± 0.1 | 9.1 ± 0.6 |
N.D., data unable to be experimentally defined or with incomplete curves.
Statistically significant, P < 0.05 compared with oxyntomodulin control, paired t test.
Allosteric Rescue of GLP-1R Function at the Met149 Polymorphic Variant.
Compound 2 exhibits differential modulation of individual peptide agonists at the “wild-type” GLP-1R, a behavior termed “probe dependence” (Keov et al., 2011), with greatest modulation of the oxyntomodulin response relative to that of the more potent peptides, GLP-1(7–36)NH2, GLP-1(7–37), and exendin-4 and indeed has minimal effect on exendin-4-mediated cAMP responses (Koole et al., 2010). Remarkably, compound 2 rescued the binding and cAMP signal of most peptide agonists at the Met149 receptor variant (excluding full-length GLP-1 peptides) (Figs. 6 and 7; Tables 7 and 8), with binding affinity and potency of truncated GLP-1 and exendin-4 recovered to within 8-fold of the wild-type receptor (Figs. 6 and 7; Tables 7 and 8). Because agonist functional potency is a composite of efficacy and affinity, this parameter can be influenced by receptor expression and receptor reserve. Consequently, we assessed the role of compound 2 on cell surface expression of the Met149 variant of the receptor. Compound 2 had no significant effect on the level of cell surface expression of Met149 over the time scale of the assay (data not shown). We also performed interaction studies between compound 2 and GLP-1(7–36)NH2, oxyntomodulin, or exendin-4 for ERK1/2 phosphorylation and intracellular Ca2+ mobilization, but no modulation of these responses was observed at this receptor variant (data not shown). This indicates that compound 2 retains the pathway-specific modulation of response that we had previously observed at the wild-type human GLP-1R (Koole et al., 2010). No modulation of GLP-1(1–36)NH2 at the Met149 variant was observed (not shown).
Fig. 6.

Characterization of the binding of GLP-1(7–36)NH2 (A and B), exendin-4 (C and D), and oxyntomodulin (E and F) in whole FlpInCHO cells stably expressing the human GLP-1R wild-type (A, C, and E) or human GLP-1R Met149 variant (B, D, and F) in the presence (♦) or absence (●) of 3 μM compound 2 and in competition with the radiolabeled antagonist 125I-exendin(9–39). Data are normalized to the maximum 125I-exendin(9–39) binding of each individual data set, with nonspecific binding measured in the presence of 1 μM exendin(9–39). Data are analyzed with a three-parameter logistic equation as defined in eq. 1. All values are mean ± S.E.M. of three to four independent experiments conducted in duplicate.
Fig. 7.

Characterization of cAMP accumulation generated by GLP-1(7–36)NH2 (A and B), exendin-4 (C and D), and oxyntomodulin (E and F) in FlpInCHO cells stably expressing the human GLP-1R wild-type (A, C, and E) or human GLP-1R Met149 variant (B, D, and F) in the presence (♦) or absence (●) of 3 μM compound 2. Data are normalized to the response elicited by 100 nM forskolin and analyzed with a three-parameter logistic equation as defined in eq. 1. All values are mean ± S.E.M. of three to five independent experiments conducted in duplicate.
TABLE 7.
Differential modulation of agonist binding at the human GLP-1R Met149 receptor variant by compound 2
Data were analyzed using a three-parameter logistic equation as defined in eq. 1. pIC50 values represent the negative logarithm of the concentration of agonist that inhibits binding of half the total concentration of radiolabeled antagonist, 125I-exendin(9–39). All data are normalized to the maximum 125I-exendin(9–39) binding in each individual data set, with non-specific binding measured in the presence of 1 μM exendin(9–39). All values are mean ± S.E.M. of three to four independent experiments, conducted in duplicate.
| pIC50 |
||||
|---|---|---|---|---|
| Wild Type |
Met149 |
|||
| Peptide | + 3 μM Compound 2 | Peptide | + 3 μM Compound 2 | |
| GLP-1(7–36)NH2 | 8.4 ± 0.1 | 8.9 ± 0.1* | 6.4 ± 0.2 | 8.4 ± 0.2* |
| Exendin-4 | 8.9 ± 0.1 | 9.4 ± 0.1 | 7.6 ± 0.1 | 9.0 ± 0.2* |
| Oxyntomodulin | 7.4 ± 0.1 | 8.5 ± 0.2* | 6.0 ± 1.0 | 7.2 ± 0.3 |
Statistically significant, P < 0.05 compared with peptide control, paired t test.
TABLE 8.
Differential modulation of agonist peptides at the human GLP-1R Met149 receptor variant by compound 2 in cAMP accumulation
Data were analyzed using a three-parameter logistic equation as defined in eq. 1. pEC50 values represent the negative logarithm of the concentration of agonist that produces half the maximal response. All data are normalized to the response elicited by 10−7 M forskolin and are mean ± S.E.M. of three to five independent experiments, conducted in duplicate.
| pEC50 |
||||
|---|---|---|---|---|
| Wild Type |
Met149 |
|||
| Peptide | + 3 μM Compound 2 | Peptide | + 3 μM Compound 2 | |
| GLP-1(7–36)NH2 | 10.2 ± 0.3 | 10.8 ± 0.6 | 7.0 ± 0.2 | 9.7 ± 0.8* |
| Exendin-4 | 10.4 ± 0.2 | 10.8 ± 0.6 | 8.5 ± 0.2 | 9.6 ± 0.7† |
| Oxyntomodulin | 8.9 ± 0.2 | 9.8 ± 1.0† | N.D. | N.D. |
N.D., data unable to be experimentally defined or with incomplete curves.
Statistically significant, P < 0.05 compared with peptide control, paired t test.
P = 0.05 compared with peptide control, paired t test.
Discussion
There is a paucity of information examining either the prevalence or influence of GLP-1R polymorphisms in, and on, susceptibility to diseases such as type II diabetes and obesity. Indeed, only a single study identified the Met149 polymorphism in a patient with type II diabetes (Tokuyama et al., 2004), and study of other polymorphisms in vitro has been very limited (Beinborn et al., 2005; Fortin et al., 2010). Greater understanding of the potential pharmacological impact of GLP-1R polymorphisms is thus required to drive clinical research in this area. In this study, we have identified major pharmacological differences in the signaling profile or allosteric modulation of multiple human GLP-1R polymorphic variants and, importantly, that the loss of function associated with the most detrimental substitution, Met149, can be allosterically rescued with a small molecule modulator.
The GLP-1R is pleiotropically coupled to signaling pathways, and the physiological response to peptides represents a convergence of all pathways activated. In this study, we chose three well characterized second messenger pathways, each linked to physiological outcomes from the receptor, to assess the functional impact of the GLP-1R polymorphisms. Both cAMP production and intracellular Ca2+ mobilization are critical to the incretin response (Baggio and Drucker, 2007), whereas ERK1/2 signaling is involved in pancreatic β-cell growth and survival (Klinger et al., 2008; Quoyer et al., 2010). Furthermore, the GLP-1R can respond to multiple endogenous peptides, as well as mimetics such as exendin-4 that are used clinically (Göke et al., 1993), requiring detailed assessment to infer the impact of polymorphic receptor variance clinically. Our analysis has revealed major new findings on the pharmacology of GLP-1R polymorphisms that have implications both physiologically and for development of therapeutic interventions for treatment of patients carrying these receptor variants.
All polymorphic receptors were well expressed at the cell surface although total expression was reduced with individual variants, relative to “wild-type” receptor. The greatest effect was observed with the Thr316 substitution, located at the second extracellular loop/transmembrane 5 interface. The majority of the polymorphic variants had minimal impact on receptor interaction with agonists and consequent signaling. However, with respect to intracellular Ca2+ mobilization, the loss in Ca2+ signaling was largely paralleled by a decrease in cell surface receptor expression, particularly for the Thr316 variant, which exhibited the lowest level of cell surface expression and a weak Ca2+ response to each of the agonists. For this variant, there was also a parallel loss in efficacy of the low-potency peptide agonist oxyntomodulin. In the current study, receptor constructs were isogenically integrated into the host cell genome and thus the loss of cell surface receptor expression is probably linked to changes in protein stability or trafficking. Although the prevalence of the Thr316 polymorphism is relatively rare (Table 1), homozygote expression in people could potentially lead to impaired GLP-1 responses.
The greatest impact on Ca2+ signaling occurred with the Met149 variant where responses to all peptides were effectively abolished. There was also greater impact of polymorphic variation on exendin-4 Ca2+ signaling relative to the GLP-1(7–36)NH2 peptide. In particular, there was significant attenuation of exendin-4 signaling with the Lys20 and Cys333 variants that had no effect on GLP-1(7–36)NH2 response. Although the mechanistic basis for this is unclear, it nonetheless provides additional evidence for a differential mode of receptor activation by exendin-4, relative to the GLP-1 peptides, that is not affinity driven. It is interesting to note that one of these variants, Cys333, also displayed selectively reduced cAMP signaling by compound 2.
Assessment of ligand binding revealed minimal effect of GLP-1R polymorphic variants on potency, with the exception of the Met149 variant. It is noteworthy that the loss of peptide potency at this variant was limited to GLP-1(7–36)NH2/GLP-1(7–37), exendin-4, and oxyntomodulin, with no observed effect on the antagonist exendin(9–39) [Beinborn et al., 2005; pIC50 values of 8.0 and 8.1, n = 1 for the wild-type and Met149 variant, respectively (data not shown)], full-length GLP-1 peptides, or compound 2 binding. These data are consistent with the critical involvement of amino acid 149 in activation transition of the receptor by peptide agonists, with the higher relative affinity of the truncated GLP-1 peptides, exendin-4, and oxyntomodulin linked to their ability to interact/induce an activated state of the receptor.
Consistent with previous data, the most detrimental of all polymorphisms that we studied was the Met149 polymorphic variant (Beinborn et al., 2005). This variant has previously been associated with a case of type II diabetes (Tokuyama et al., 2004) and a loss of binding affinity and cAMP potency in the presence of GLP-1(7–36)NH2 and exendin-4 only (Beinborn et al., 2005). By performing similar binding and cAMP accumulation assays in the presence of all peptide agonists of the GLP-1R, we observed that the effect of the Met149 polymorphic variant is ligand-dependent, with a greater effect on oxyntomodulin (251-fold decrease in binding affinity, >500-fold decrease in cAMP potency) relative to GLP-1(7–36)NH2 (decreases of 251- and 158-fold in binding affinity and cAMP potency, respectively) or exendin-4 (decreases of 32- and 200-fold in binding affinity and cAMP potency, respectively). We also found that effects on signaling at this polymorphic variant are pathway-dependent, in that the loss of function was more pronounced in cAMP accumulation and Ca2+ mobilization than ERK1/2 phosphorylation. Intriguingly, both the binding and signaling responses to compound 2 were unaltered, providing supporting evidence for a distinct molecular mechanism for receptor activation from that of orthosteric-acting peptide agonists. In parallel, and perhaps most importantly, compound 2 retained its ability to modulate peptide-mediated binding and cAMP responses at the Met149 polymorphic variant, indicating that there is potential for developing therapeutics to treat patients possessing polymorphic receptor variants that are refractory to peptide-mimetic therapy. The data from the Met149 polymorphic variant is also informative, suggesting that the principal allosteric effect of compound 2 is to lower the energy barrier for activation transition, in particular with respect to the receptor conformation(s) linked to Gαs coupling. At the “wild-type” receptor, GLP-1(7–36)NH2, GLP-1(7–37), and exendin-4 are all highly potent and efficacious peptide ligands, and addition of compound 2 has a limited effect on receptor binding and signaling output because the receptor is already in a highly active state. The weaker agonist, oxyntomodulin, which has a lower response in the system, however, was positively modulated to a greater extent by compound 2. At the Met149 variant, our results suggest that there is a higher barrier to establish an active state transition as engaged by the peptide agonists. The addition of compound 2, which modifies the receptor in a manner that is insensitive to the Met149 substitution, lowers the barrier to allow the recovery of the response, so that an active conformation can more readily be achieved. As also previously noted by Beinborn et al. (2005), the Met149 polymorphic variant does not alter antagonist [exendin(9–39)] binding to the receptor. This is consistent with the involvement of residue 149 in an activation transition rather than directly disrupting peptide binding interactions.
The recovery of GLP-1(7–36)NH2 and exendin-4 potency at the Met149 polymorphic variant in cAMP accumulation to within 8-fold of the GLP-1R wild-type range provides an excellent example of the potential to rescue detrimental receptor polymorphisms with the application of allosteric ligands. Nonetheless, broad assessment of the impact of different GLP-1R polymorphisms on the allosteric potentiation of oxyntomodulin cAMP response indicated that multiple receptor variants had attenuated responses. Not surprisingly, the C333 variant had minimal allosteric response because this substitution also led to attenuation of cAMP signaling by compound 2, suggesting that it interferes with the ability of compound 2 to promote a Gαs-interacting conformation of the receptor; previous work has demonstrated a correlation between allosteric potentiation and the ability of compounds to promote an active state of the receptor (Leach et al., 2010; Keov et al., 2011). Intriguingly, the Leu7, Gln131, Ser168, Leu260, Thr316, and Gln421 also exhibited attenuated allosteric enhancement of oxyntomodulin cAMP signaling in the presence of compound 2. The mechanistic basis for the loss of effect is unclear, but it highlights the potential complexity of the allosteric interaction between peptides and small molecule ligands. Although this effect is likely to be chemotype-dependent, it nonetheless highlights the need for careful consideration in clinical trial design where potential allosteric drugs are being assessed. In the case of the GLP-1R, at least one of the variants with loss of oxyntomodulin modulation, Leu7, is reported in the SwissProt database to occur in 40% of assessed populations in either homozygous or heterozygous form.
In conclusion, we have demonstrated important pharmacological effects arising from polymorphisms of the GLP-1R. For the Met149 variant, this is likely to be clinically relevant, albeit for a small percentage of patients. Importantly, we have demonstrated that loss of function arising from polymorphisms such as the Met149 substitution can be rescued by allosteric modulation of the receptor, with small molecule compounds providing scope for therapeutic intervention for patients whose disease is linked to such polymorphic variation.
Supplementary Material
Acknowledgments
We thank Prof. Roger Summers for helpful discussions on this work and Drs. Ron Osmond and Michael Crouch of TGR BioSciences (Hindmarsh, SA, Australia), who generously provided the SureFire ERK1/2 reagents.
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was funded in part by the National Health and Medical Research Council (NHMRC) of Australia [Grants 519461, 1002180]; and by an NHMRC Australian Principal Research Fellowship (to P.M.S.) and a Senior Research Fellowship (to A.C.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.072884.
1 The response to GLP1(7–36)NH2 and GLP-1(7–37) are effectively equivalent; therefore, only a limited analysis was performed for the GLP-1(7–37) peptide with data limited to one or two experiments only.
2 The response to GLP1(1–36)NH2 and GLP-1(1–37) are effectively equivalent, therefore, only a limited analysis was performed for the GLP-1(1–37) peptide with data limited to one or two experiments only.
- GLP-1R
- glucagon-like peptide-1 receptor
- SNP
- single nucleotide polymorphism
- DMEM
- Dulbecco's modified Eagle's medium
- ERK
- extracellular signal-regulated kinase
- FBS
- Fetal bovine serum
- CHO
- Chinese hamster ovary
- BSA
- bovine serum albumin
- compound 2
- 6,7-dichloro-2-methylsulfonyl-3-tert-butylaminoquinoxaline.
Authorship Contributions
Participated in research design: Koole, Wootten, Simms, Christopoulos, and Sexton.
Conducted experiments: Koole.
Contributed new reagents or analytical tools: Valant.
Performed data analysis: Koole.
Wrote or contributed to writing of the manuscript: Koole, Wootten, Simms, Miller, Christopoulos, Sexton.
References
- Baggio LL, Drucker DJ. (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132:2131–2157 [DOI] [PubMed] [Google Scholar]
- Beinborn M, Worrall CI, McBride EW, Kopin AS. (2005) A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul Pept 130:1–6 [DOI] [PubMed] [Google Scholar]
- Deacon CF, Johnsen AH, Holst JJ. (1995a) Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab 80:952–957 [DOI] [PubMed] [Google Scholar]
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. (1995b) Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 44:1126–1131 [DOI] [PubMed] [Google Scholar]
- DeFronzo RA. (1992) Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia 35:389–397 [DOI] [PubMed] [Google Scholar]
- Drucker DJ. (2006) The biology of incretin hormones. Cell Metab 3:153–165 [DOI] [PubMed] [Google Scholar]
- Elbrønd B, Jakobsen G, Larsen S, Agersø H, Jensen LB, Rolan P, Sturis J, Hatorp V, Zdravkovic M. (2002) Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diabetes Care 25:1398–1404 [DOI] [PubMed] [Google Scholar]
- Fortin JP, Schroeder JC, Zhu Y, Beinborn M, Kopin AS. (2010) Pharmacological characterization of human incretin receptor missense variants. J Pharmacol Exp Ther 332:274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göke R, Fehmann HC, Linn T, Schmidt H, Krause M, Eng J, Göke B. (1993) Exendin-4 is a high potency agonist and truncated exendin-(9–39)-amide an antagonist at the glucagon-like peptide 1-(7–36)-amide receptor of insulin-secreting beta-cells. J Biol Chem 268:19650–19655 [PubMed] [Google Scholar]
- Jarrousse C, Audousset-Puech MP, Dubrasquet M, Niel H, Martinez J, Bataille D. (1985) Oxyntomodulin (glucagon-37) and its C-terminal octapeptide inhibit gastric acid secretion. FEBS Lett 188:81–84 [DOI] [PubMed] [Google Scholar]
- Jarrousse C, Bataille D, Jeanrenaud B. (1984) A pure enteroglucagon, oxyntomodulin (glucagon 37), stimulates insulin release in perfused rat pancreas. Endocrinology 115:102–105 [DOI] [PubMed] [Google Scholar]
- Keov P, Sexton PM, Christopoulos A. (2011) Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 60:24–35 [DOI] [PubMed] [Google Scholar]
- Klinger S, Poussin C, Debril MB, Dolci W, Halban PA, Thorens B. (2008) Increasing GLP-1-induced beta-cell proliferation by silencing the negative regulators of signaling cAMP response element modulator-alpha and DUSP14. Diabetes 57:584–593 [DOI] [PubMed] [Google Scholar]
- Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, Kodra JT, Holst JJ, Jeppesen CB, Johnson MD, de Jong JC, et al. (2007) Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc Natl Acad Sci USA 104:937–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thøgersen H, Wilken M, Agersø H. (2000) Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem 43:1664–1669 [DOI] [PubMed] [Google Scholar]
- Koole C, Wootten D, Simms J, Valant C, Sridhar R, Woodman OL, Miller LJ, Summers RJ, Christopoulos A, Sexton PM. (2010) Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol Pharmacol 78:456–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. (2010) Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology 35:855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Avlani VA, Langmead CJ, Herdon HJ, Wood MD, Sexton PM, Christopoulos A. (2007) Structure-function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol Pharmacol 72:463–476 [DOI] [PubMed] [Google Scholar]
- Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. (1993) Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 36:741–744 [DOI] [PubMed] [Google Scholar]
- Quoyer J, Longuet C, Broca C, Linck N, Costes S, Varin E, Bockaert J, Bertrand G, Dalle S. (2010) GLP-1 mediates anti-apoptotic effect by phosphorylating Bad through a beta-arrestin 1-mediated ERK1/2 activation in pancreatic beta-cells. J Biol Chem 285:1989–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M, Johnson MD, Thomas C, Kiel D, Lakis JN, Kercher T, Aytes S, Kostrowicki J, Bhumralkar D, Truesdale L, et al. (2007) Small molecule ago-allosteric modulators of the human glucagon-like peptide-1 (hGLP-1) receptor. Bioorg Med Chem Lett 17:5472–5478 [DOI] [PubMed] [Google Scholar]
- Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, Holst JJ. (2001) Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 86:3717–3723 [DOI] [PubMed] [Google Scholar]
- Tokuyama Y, Matsui K, Egashira T, Nozaki O, Ishizuka T, Kanatsuka A. (2004) Five missense mutations in glucagon-like peptide 1 receptor gene in Japanese population. Diabetes Res Clin Pract 66:63–69 [DOI] [PubMed] [Google Scholar]
- Werry TD, Gregory KJ, Sexton PM, Christopoulos A. (2005) Characterization of serotonin 5-HT2C receptor signaling to extracellular signal-regulated kinases 1 and 2. J Neurochem 93:1603–1615 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.