

Exposure of cells from steroidogenic tissues to trophic peptide hormones results in a rapid increase in the rate of steroid hormone production. Ingrained in the mind of every student of endocrinology is the concept that this stimulation of steroidogenesis hinges on increasing the rate of conversion of cholesterol to pregnenolone. But what are the biochemical events associated with this increased rate of metabolism of cholesterol? The current hypothesis attributes this “acute regulatory response of steroid hormone synthesis” to the mobilization and transport of cholesterol from cytosolic stores to the inner mitochondrial membrane where the enzyme for the first step of steroid hormone formation resides, i.e., cytochrome P450 11A, also called the cholesterol side-chain cleavage enzyme or P450scc. So, the “acute response” is the consequence of a rapid increase in substrate (cholesterol) availability at the active site of P450scc. What then is the nature of the agent(s) that facilitate the transport of cholesterol to the inner mitochondrial membrane? Answering this and related questions has served as a long-standing challenge for many investigators studying the biochemical factors influencing the rapid activation of steroidogenesis, in tissues such as the adrenal, ovary, and testis.
Recently, Clark et al. (1) reported the cloning and expression of a protein that seemed to provide a possible answer—a short-lived 37-kDa protein that they named StAR (for steroidogenic acute regulatory protein). They proposed that StAR, in the form of a mitochondrial precursor protein, plays a critical role in the rapid translocation of cholesterol across the outer and inner mitochondrial membranes. In this way, the rapid import of StAR into the mitochondria would provide, through an undetermined mechanism, cholesterol access to P450scc located on the matrix face of the inner mitochondrial membrane. This interpretation now has been challenged by studies reported in the paper by Arakane et al. (2) in this issue of the Proceedings. Arakane et al. (2) present convincing evidence that the stimulation of cholesterol metabolism by StAR is not necessarily linked to the import of StAR into the mitochondria.
Studies reported over 40 years ago showed that treatment of adrenocortical cells with ACTH (corticotropin) results in a greater than 10-fold increase in the rate of steroid hormone synthesis (3). This stimulation of steroidogenesis was found to occur rapidly, after approximately 3 min exposure of the cells to the trophic hormone ACTH. One of the first clues that gave insight into this phenomenon came from the seminal works by Ferguson (4) and Garren et al. (5). They observed that protein synthesis was necessary for the acute stimulatory response of steroidogenesis, i.e., the increased rate of steroid hormone formation was blocked if adrenal cells were pretreated with inhibitors of translation (puromycin or cycloheximide) before addition of ACTH. These observations led to studies examining cholesterol transport within adrenal cells, which demonstrated that cholesterol became localized in the outer mitochondrial membrane in the presence of protein synthesis inhibitors (6). These studies, and those that followed, led to the hypothesis that trophic hormone action was mediated by a newly synthesized protein that served to enhance the transport of cholesterol from the outer to the inner mitochondrial membrane for metabolism by P450scc. Thus, the availability of the substrate, cholesterol, for hydroxylation by the P450scc was found to be the rate-limiting step of steroidogenesis. This conclusion was supported by the early observations that soluble derivatives of cholesterol [e.g., 22(R)-hydroxycholesterol] could diffuse into mitochondria where they were readily metabolized to pregnenolone in the absence of hormonal stimulation (7).
Armed with this information, the hunt was on. What is this labile protein that is rapidly synthesized following treatment of steroidogenic cells with a trophic hormone and how does it serve to stimulate the rate of delivery of cholesterol across the mitochondrial membranes to P450scc? A number of candidate proteins have been studied, including sterol carrier protein 2, steroidogenesis activator polypeptide (SAP), and the peripheral benzodiazepine receptor (PBR), as summarized in the recent review by Stocco and Clark (8).
Another piece of the puzzle was added by the observation that trophic hormones mediate the acute stimulation of steroidogenesis though a cAMP/protein kinase A-dependent pathway, suggesting that phosphorylation influences the synthesis or activity of the newly synthesized labile protein. An elegant series of studies by Orme-Johnson and colleagues (9, 10) showed that several phosphoproteins (37, 32, and 30 kDa) were generated following trophic hormone stimulus of adrenal and Leydig cells. The synthesis of these phosphoproteins (pp37, pp32, and pp30) was inhibited by cycloheximide. Additional studies showed that pp30 was located in the mitochondria, and was most probably formed from a precursor protein of molecular mass 37 kDa—a protein with a half-life of only 3 to 5 min. A full understanding of the role of protein phosphorylation and its requirement for the acute steroidogenic response following exposure of a cell to a trophic hormone remains an intriguing but poorly understood relationship.
The next piece of the puzzle was provided by Clark et al. (1), who purified the 30-kDa mitochondrial protein followed by the isolation and sequence determination of its tryptic peptides. Screening of a cDNA library of MA-10 Leydig tumor cells led to the identification of a 1456-base pair cDNA. Expression of this cDNA showed that it was for a 37-kDa protein, providing further evidence that the 30-kDa protein found in mitochondria was a processed form of this 37-kDa protein. In addition, transfection of this cDNA into mouse MA-10 Leydig tumor cells resulted in an increase in steroidogenesis. Thus, StAR was born.
A very incisive series of experiments was then carried out by Sugawara et al. (11), who used a heterologous cell model system. COS-1 cells were transfected with the cDNAs for P450scc and adrenodoxin to establish the mitochondrial enzymes necessary to assay the first step of cholesterol metabolism. Sugawara et al. (11) cotransfected these COS-1 cells with a plasmid containing the cDNA for the StAR protein. The rate of pregnenolone formation from cholesterol was significantly increased in those cells expressing StAR. These experiments directly showed the key role played by StAR in stimulating the rate of cholesterol metabolism by P450scc. However, the most convincing evidence that StAR protein was critical for the acute response in steroidogenic cells came from analysis of the disease congenital lipoid adrenal hyperplasia (lipoid CAH). Lipoid CAH is a lethal condition arising from the complete inability of the newborn to synthesize steroid hormones. Isolated mitochondria from the adrenals of such individuals are unable to produce pregnenolone, which led to the early hypothesis that P450scc was mutated or absent. Sequence analysis, however, showed no defect in P450scc. With the sequence for the human StAR cDNA determined, Miller and colleagues (12) demonstrated that in patients with lipoid CAH the StAR gene had been mutated causing loss of function. Their studies gave strong support to the proposition that StAR was the long sought factor regulating the initial step in steroidogenesis, as well as, the consequences if this protein was altered in a manner that destroyed its activity.
In the paper published in this issue of Proceedings, Arakane et al. (2) have modified the cDNA for StAR so portions of the N-terminal and C-terminal regions of the protein are deleted. They then introduced these modified cDNAs into the COS-1 heterologous cell system to determine whether the modified proteins would still stimulate the formation of pregnenolone from cholesterol. Surprisingly, the deletion of up to 62 amino acid residues from the N-terminal domain of StAR (the segment processed when imported into the mitochondria) does not significantly decrease the ability of the StAR protein to stimulate the metabolism of cholesterol to pregnenolone. Further, Arakane et al. (2) showed that the N-62 truncated StAR is not imported into mitochondria but is located on the outer surface of the mitochondrial membrane. These results clearly question the linkage between the transmembrane import of StAR and its role in the movement of cholesterol across the mitochondrial membrane. Arakane et al. (2) also showed that truncating the C-terminal segment of StAR inactivates the protein, so it no longer stimulates the rate of cholesterol metabolism.
As summarized schematically in Fig. 1, a large body of data support the interpretation that the acute steroidogenic response follows the sequence of reactions: (i) A peptide hormone such as ACTH interacts with a cell membrane receptor setting off the cascade of events leading to the formation of cyclic AMP and the activation of protein kinase A (PKA). (ii) PKA stimulates the de novo synthesis of the 37-kDa mitochondrial precursor protein, StAR. At the same time, cholesterol ester hydrolase is activated with a release of free cholesterol from cytosolic stores. (iii) Free cholesterol migrates to the mitochondria where it moves across the mitochondrial membranes for presentation to P450scc. The key question remains unanswered: what is the mechanism by which the 37-kDa labile protein, StAR, facilitates this movement of cholesterol across the mitochondrial membrane? Two alternate processes may be envisioned. Stocco and colleagues (8) proposed that the 37-kDa precursor form of StAR is imported by mitochondria and processed to a 30-kDa mature protein concomitant with the movement of cholesterol across the mitochondrial membrane (Fig. 1, A). In this case, the transport of StAR across the mitochondrial membranes is an integral requirement for the influx of cholesterol. The results of Arakane et al. (2) challenge this interpretation. They propose an alternative explanation based on their recent studies. As shown in Fig. 1, B1, StAR needs only to bind the surface of the outer mitochondrial membrane to facilitate the transport of cholesterol across the mitochondrial membranes. The import of the 37-kDa precursor form of StAR into the mitochondria and its processing is proposed to occur after cholesterol has entered the mitochondria and may serve as a means of shutting down the process of cholesterol movement (Fig. 1, B2). The role of phosphorylation of StAR in either of these proposed mechanisms remains unexplained.
Figure 1.
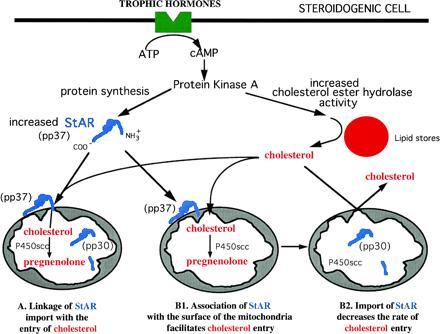
Schematic representation of two proposed mechanisms for the acute regulation of steroidogenesis by StAR protein. Following trophic hormone binding to a cell surface receptor, cAMP increases and activates protein kinase A. Protein kinase A increases the synthesis of StAR and its phosphorylation giving rise to pp37. In proposal A, StAR is imported into the mitochondria and subsequently processed to the mature pp30. During the import process, it is proposed (8) that cholesterol is transported to the inner mitochondrial membranes. Proposal B1 takes into account the data of Arakane et al. (2), which suggests that the interaction of the StAR protein with the mitochondrial surface leads to cholesterol movement. Further, it is proposed (B2) that the importation of StAR and cleavage to pp30 is a mechanism of inactivating cholesterol flux into the mitochondria.
Thus, the centerpiece of the original hypothesis of Stocco and Clark (8), i.e., StAR, as it enters the mitochondria acts to facilitate the movement of cholesterol across the outer and inner membranes of the mitochondria, must now be reconsidered. The results presented by Arakane et al. (2) represent a potentially lethal challenge to this hypothesis. What seemed like a logical and straight forward explanation of the biochemical basis of this “acute response,” now needs rethinking and new experiments.
Many questions remain unanswered. Does StAR react with a unique receptor on the outer surface of the mitochondria so that channels are established for the admission of cholesterol to the mitochondrial matrix? Do stoichiometric experiments provide evidence that StAR can initiate the delivery of sufficient amounts of cholesterol to account for the concentration of pregnenolone formed during the acute steroidogenic response? What is the relationship of the 32-kDa protein to the 37-kDa StAR protein and its 30 kDa processed form? Further, one can question the validity of the COS-1 cell as a faithful assay system; COS-1 cells are nonsteroidogenic cells and may not respond in the same way as steroidogenic cells. And, most important, there remains the unexamined role of phosphorylation of normal or truncated StAR.
Arakane and colleagues (2) have demonstrated that StAR will continue to shine—but a twinkle remains in the eyes of those pursuing the detailed mechanism through which this long sought labile protein participates as an acute regulator of steroidogenesis.
References
- 1.Clark B J, Wells J, King S R, Stocco D M. J Biol Chem. 1994;269:28314–28322. [PubMed] [Google Scholar]
- 2.Arakane F, Sugawara T, Nishino H, Liu Z, Holt J A, Pain D, Stocco D M, Miller W L, Strauss J F., III Proc Natl Acad Sci USA. 1996;93:13731–13736. doi: 10.1073/pnas.93.24.13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stone D, Hechter O. Arch Biochem Biophys. 1954;51:457–469. doi: 10.1016/0003-9861(54)90501-9. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson J J. J Biol Chem. 1963;238:2754–2759. [PubMed] [Google Scholar]
- 5.Garren L D, Ney R L, Davis W W. Proc Natl Acad Sci USA. 1965;53:1443–1450. doi: 10.1073/pnas.53.6.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Privalle C T, Crivello J F, Jefcoate C R. Proc Natl Acad Sci USA. 1983;80:702–706. doi: 10.1073/pnas.80.3.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falke H E, Degenhart H J, Abeln G J, Visser H K. Mol Cell Endocrinol. 1975;3:375–383. doi: 10.1016/0303-7207(75)90037-4. [DOI] [PubMed] [Google Scholar]
- 8.Stocco D M, Clark B J. Endocr Rev. 1996;17:221–244. doi: 10.1210/edrv-17-3-221. [DOI] [PubMed] [Google Scholar]
- 9.Krueger R J, Orme-Johnson N R. J Biol Chem. 1983;258:10159–10167. [PubMed] [Google Scholar]
- 10.Pon L A, Hartigan J A, Orme-Johnson N R. J Biol Chem. 1989;261:13309–13316. [PubMed] [Google Scholar]
- 11.Sugawara T, Holt J A, Driscoll D, Strauss J F, III, Lin D, Miller W L, Patterson D, Clancy K P, Hart I M, Clark B J, Stocco D M. Proc Natl Acad Sci USA. 1995;92:4778–4782. doi: 10.1073/pnas.92.11.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin D, Sugawara T, Strauss J F, III, Clark B J, Stocco D M, Saenger P, Rogol A, Miller W L. Science. 1995;267:1828–1831. doi: 10.1126/science.7892608. [DOI] [PubMed] [Google Scholar]