

Abstract
Mitochondria are fascinating organelles, which fulfill multiple cellular functions, as diverse as energy production, fatty acid β oxidation, reactive oxygen species (ROS) production and detoxification, and cell death regulation. The coordination of these functions relies on autonomous mitochondrial processes as well as on sustained cross-talk with other organelles and/or the cytosol. Therefore, this implies a tight regulation of mitochondrial functions to ensure cell homeostasis. In many diseases (e.g., cancer, cardiopathies, nonalcoholic fatty liver diseases, and neurodegenerative diseases), mitochondria can receive harmful signals, dysfunction and then, participate to pathogenesis. They can undergo either a decrease of their bioenergetic function or a process called mitochondrial permeability transition (MPT) that can coordinate cell death execution. Many studies present evidence that protection of mitochondria limits disease progression and severity. Here, we will review recent strategies to preserve mitochondrial functions via direct or indirect mechanisms of MPT inhibition. Thus, several mitochondrial proteins may be considered for cytoprotective-targeted therapies.
1. Introduction
Mitochondria are intracellular organelles, whose first discovered function is energy production by oxidative phosphorylation [1]. Depending on the mammalian tissue, mitochondria may have additional functions, such as β oxidation, heat production, reactive oxygen species (ROS) metabolism, and cell death coordination. However, since the emergence of the concept of mitochondrial control of cell death in the 95's (for recent reviews: [2, 3]), it became evident that mitochondria participate to various types of cell death, that are, apoptosis, necrosis, oncosis and mitotic catastrophy via mitochondrial membrane permeabilization (MMP), release of proapoptotic factors contained in the intermembrane space to the cytosol and possibly fission, even if mitochondrial fragmentation is not sufficient per se to induce cell death [4, 5]. Mitochondrial dysfunction has been associated with a series of human diseases such as cancer, cardiopathies, nonalcoholic fatty liver diseases, neurodegenerative diseases, and aging. When due to genetic dysfunction, the diseases have been systematically characterized in animal models [6, 7]. Thus, mitochondrial impairment can be linked either to the metabolic function of these organelles, their role in cell death, or both. In addition, in chronic pathologies, such as cardiac volume overload-induced hypertrophy [8], mitochondrial dysfunction precedes cell loss by apoptosis and necrosis, meaning that both dysfunctions can be separated chronologically during the progression of the disease. This is also observed in the pathogenesis of nonalcoholic steatohepatitis, whatever its initial cause, as extensively reviewed [9]. Hepatic mitochondrial dysfunction would lead to apoptosis or necrosis depending on the energy status of the cell [9].
Metabolic impairment manifests by decreased ATP synthesis capacity, enhanced ROS production due to electron leak from the respiratory chain, change in intracellular pH and frequently, by morphological alterations of mitochondrial network. For instance, heart failure which is defined as the inability of the heart to keep up with the demands and to provide adequate blood flow to other organs such as the brain, liver, and kidneys is accompanied by a decrease in energy production and energy transfer capacity [10]. This leads to a decrease in energy charge of the myocardium that has been described as a prognostic factor in dilated cardiomyopathies [11]. This metabolic impairment also affects the peripheric circulation and was shown to involve decreased mitochondrial biogenesis [10].
MMP corresponds to multiple events that irreversibly lead to cell death [2, 12]. These lethal events are nonexclusive, some of them can occur independently, whereas others are intimately linked. Thus, MMP refers to protein translocation from cytosol to outer membrane (OM), rupture of outer mitochondrial membrane, loss of inner membrane potential (ΔΨm), cristae remodeling and release of intermembrane space proteins such as cytochrome c or apoptosis-inducing factor (AIF). For instance, upon various stress, Bax or tBid, which reside in the cytosol, can translocate to mitochondrial membranes, oligomerize with mitochondrial proteins to form large channels allowing cytochrome c release and activation of the intrinsic apoptosis signaling cascade (for review: [2]).
In many pathophysiological models, but not all, MMP also involves the so-called opening of the permeability transition pore complex (PTPC), which mediates a nonselective permeabilization of the IM and OM to molecules of molecular mass (MM) under 1.5 kDa (see below for more details) [2, 12]. Thus, in chemotherapy-treated tumor cell lines and ischemic neuronal cells, Bax can interact with the adenine nucleotide translocase (ANT) and/or the voltage-dependent anion channel (VDAC) to promote MMP and cell death.
Here, we will review direct and indirect mechanisms or means to protect mitochondrial functions via a closure of PTPC and a prevention of mitochondrial permeability transition (MPT). The discussion of MPT regulatory mechanisms will be based on selected articles focusing on heart diseases and cancer.
2. Mitochondrial Membrane Permeability and PTPC
Mitochondrial membrane permeability is strictly controlled in unicellular and multicellular organisms harboring these organelles. The OM is believed to be freely permeable to ions and metabolites via entry through protein channels (e.g., voltage-dependent anion channel, VDAC), whereas the inner membrane (IM) is considered as impermeable. Thus, the entry and exit of ions or metabolites trough the IM are mediated by integral proteins such as the members of the mitochondrial carrier family [13]. The prototypic protein of this family is ANT or ADP/ATP carrier, which mediates the stoichiometric exchange of ADP and ATP between the matrix and the intermembrane space [14]. Moreover, osmotic movements of water accompany solutes transport from cytosol to matrix, but the molecular basis of these transports is still largely unknown [15]. When excessive stimulation by endogenous signals (excessive ROS, calcium (Ca2+) overload, protease activation, lipid accumulation etc) or activation of harmful signaling pathways (e.g., kinases/phosphatases, proteases, Bax/-Bid-mediated pathways etc.) occur, mitochondria undergo the MPT, a phenomenon that consists in a sudden increase in IM permeability to small molecules. MPT is a phenomenon first studied in isolated beef heart mitochondria in response to Ca2+ overload [16]. Thus, the response of isolated mitochondria to doses of Ca2+ is a nonspecific increase of the permeability of the IM, resulting in entry of water and solutes, loss of ΔΨm, matrix swelling, and simultaneous uncoupling of oxidative phosphorylation (Figure 1). Of note, the doses of Ca2+ depend largely on the tissue origin of mitochondria and the amount of Ca2+ present in the buffers. Ultimately, MPT is accompanied by matrix swelling and OM ruptures as shown by transmission electron microscopy [17–19]. This phenomenon can be blocked by Ca2+ chelation, ATP, Mg2+, and cyclosporin A (CsA) in vitro as well as in vivo [20–22].
Figure 1.
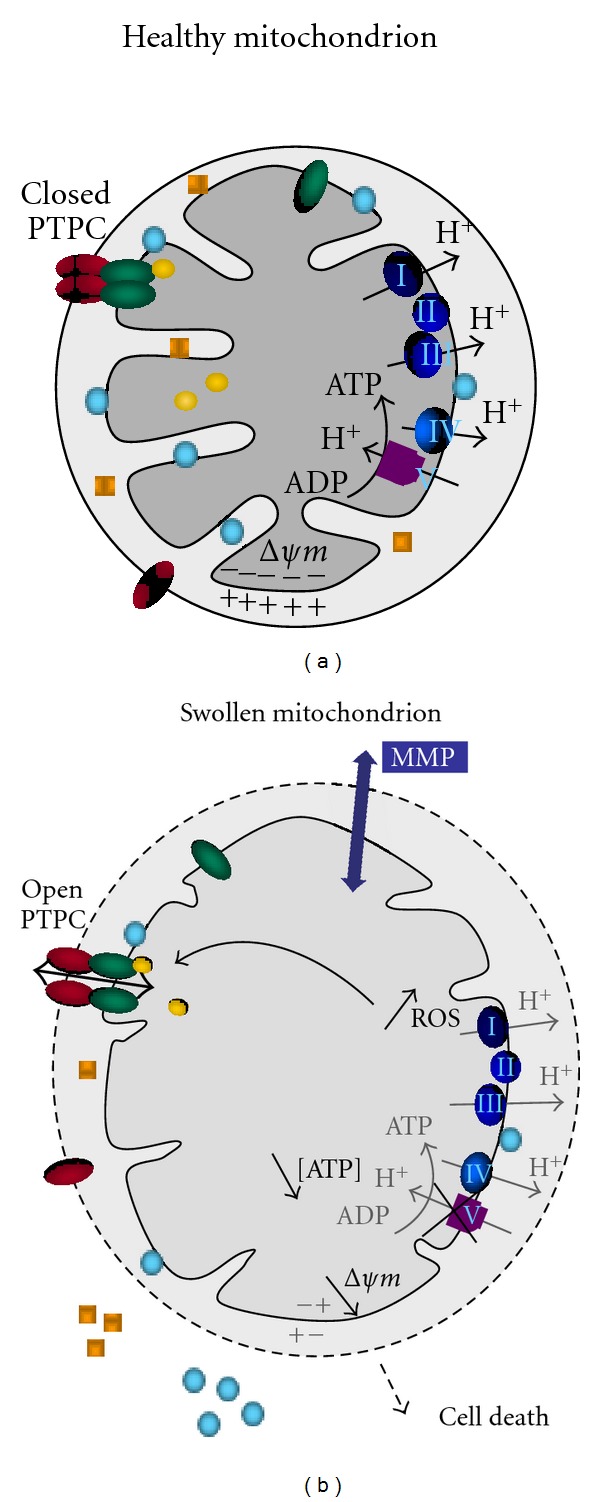
Scheme of mitochondrial alterations following mitochondrial membrane permeabilization (MMP). In this model, in response to the opening of the permeability transition pore (PTPC; green and red ellipses, corresponding to ANT and VDAC resp.), swollen mitochondria exhibit an increase in volume, a more translucide matrix with less cristae and a permeabilized outer membrane. Cytochrome c and apoptosis-inducing factor (AIF) (blue circles and yellow squares), normally confined into the intermembrane space, are released trough ruptures in the outer membrane. The transmembrane inner potential (ΔΨm) is dissipated in response to the arrest of the function of the respiratory complexes (I to V), which contributes to an inhibition of ATP biosynthesis. Altogether, these alterations are lethal, irreversible and lead to cell death.
MPT can be followed experimentally by the loss of absorbance of a suspension of isolated mitochondria by spectrophotometry and by the loss of ΔΨm using suitable fluorescent probes (e.g. tetramethylrhodamine methyl ester (TMRM), rhodamine 123 (Rhod123), 5, 5′, 6, 6′- tetrachloro-1, 1′, 3, 3′-tetraethylbenzimidazol-carbocyanine iodide (JC-1)) [19, 23]. The main interests of the use of isolated organelles are to monitor mitochondrial responses that are directly induced by compounds independently of other cellular compartments and the possibility to automate the measure in the perspective of pharmacological studies [19, 23].
One major pitfall is that, whereas isolation procedures are believed to be nondestructive for liver and cell lines mitochondria [24, 25], mitochondrial responses of isolated mitochondria from skeletal muscle and heart that may rely on the cell architecture are (obviously) lost [26].
Another pitfall is the cross-contamination of the mitochondrial fraction with other cellular compartments and purity of preparations must be checked carefully. MPT can also be measured by imaging with the fluorescent probe calcein in the presence of cobalt in living cell as various as hepatocytes, astrocytes and cardiomyocytes [27–31]. The principle is that calcein (molecular weight, 620 Da) can diffuse into the whole cell, whereas cobalt, a fluorescence quencher, cannot enter into the mitochondrial matrix and diffuses into the rest of the cell. Thus, in physiological conditions mitochondria appear fluorescent and following MPT, the quenching of calcein by cobalt triggers a decrease in fluorescence. For instance, HeLa cells treated by thapsigargin, a SERCA pump inhibitor or A23187, a Ca2+ ionophore [32], undergo MPT as shown by a significant decrease in calcein fluorescence due to IM permeabilization and cobalt quenching [30]. MPT has also been monitored in whole heart by 2-deoxy[3H]glucose entrapment technique [33].
Of note, the full demonstration that the process is mediated by PTPC opening requires its inhibition by pretreatment of cells or isolated mitochondria by CsA, the well-known cyclophilin D (CypD) ligand [20, 34]. Moreover, silencing of CypD by siRNA to prevent the induction of MPT is becoming mandatory in cellulo, since the genetic demonstration that CypD is critical for MPT and cell death [35].
3. Direct Mechanisms of MPT Inhibition
PTPC is defined as a voltage-dependent polyprotein complex, which in certain conditions might form a nonselective channel at contact sites between both mitochondrial membranes [36, 37]. By definition, mitochondria contain all the proteins necessary for MPT induction and then MPT does not necessitate any neosynthesis. Since the initial PTPC identification by electrophysiology, the molecular identity of this pore and its regulators is still controversial [38–40]. ANT, VDAC and CypD, the three former PTPC candidates, have their own functions, irrespective of their association within PTPC or other putative polyprotein complexes such as the ATP synthasome [41] and Bcl-2 family member oligomers [3, 12]. Thus, some of the unknown members of PTPC may have their own role in metabolism (e.g., kinase, peptidyl-prolyl isomerase, deshydrogenase), transport (e.g., mitochondrial carrier, channel) or structure (e.g., dynamic machinery, cytoskeleton, AKAP proteins). This means that lethal MPT needs a stimulation to occur and in pathophysiological conditions, this is mainly achieved by Ca2+ and ROS. Whatever its composition, the PTPC is a widespread phenomenon occurring in many diseases. Although it has been the subject of intense research and therapeutic developments in cancer with the search for PTPC inducers [42], it has emerged only recently as a promising target for cytoprotection in various diseases such as neurodegenerative, cardiovascular and metabolic diseases. PTPC can be modulated directly by a large panel of pharmacological agents, by post-translational modifications and by cooperation with other proteins that may have a major impact on the cell life as discussed below.
3.1. Pharmacological Inhibition of PTPC
Using isolated mitochondria from various sources, an impressive body of literature reports that many molecules or compounds modulate the PTPC in response to Ca2+, ROS, or a disease. Thus, some compounds can activate MPT, whereas a more limited number of them can prevent the opening of PTPC. Some compounds have known mitochondrial targets such as ANT, VDAC, CypD, and translocator protein 18 kDa (Table 1). To summarize, the most investigated inhibitor of PTPC is CsA, which modulates CypD, as discussed below.
Table 1.
List of mitochondrial permeability transition (MPT) inhibitors. CypD, Cyclophilin D; VDAC, voltage-dependent anion channel; ANT, adenine nucleotide translocase; UQ, ubiquinone.
| MPT inhibitor | Target | Model | References |
|---|---|---|---|
| Cyclosporin A | CypD binding with ANT | In vivo, isolated mitochondria, cells | [44, 45] |
| Sanglifehrin A | CypD | In vivo, isolated mitochondria, cells | [46, 47] |
| Bongkrekic acid | ANT | Isolated mitochondria, cells | [48–50] |
| ADP/ATP | ANT | Isolated mitochondria, in vitro | [51, 52] |
| NADH/NAD+ | VDAC | Isolated mitochondria, in vitro | [53–55] |
| DIDS | VDAC | Isolated mitochondria, in vitro, cells | [52–58] |
| glutamate | VDAC | Isolated mitochondria, in vitro | [59–61] |
| Ro 68–3400 | ANT or PiC, not VDAC1 | Isolated mitochondria, in vitro | [61–63] |
| UQ(0) | ANT or PiC | Isolated mitochondria, in vitro | [64, 65] |
| S15176 | unknown, in IM | In vivo, isolated mitochondria | [66, 67] |
| Sildenafil | unknown | In vivo, isolated mitochondria | [68] |
| Debio-025 | CypD | In vivo, isolated mitochondria | [69] |
| TRO19622 | VDAC, translocator protein 18 kDa | In vivo, isolated mitochondria | [70] |
| Carbon monoxide | ANT, unknown | Isolated mitochondria, cells | [71] |
| Antamanide | CypD | Isolated mitochondria, cells | [72] |
One example with future therapeutic applications is cardiac ischaemia/reperfusion injury. During an acute myocardial infarction (AMI), tissue injury occurring after reperfusion represents a significant amount of the whole, irreversible damage. Ischaemia and reperfusion cause a wide array of functional and structural alterations of mitochondria. Some of these responses are directly under the control of the highly conserved transcriptional complex HIF-1 and result from a modulation in expression of genes involved in glycolysis, glucose metabolism, mitochondrial function, cell survival, apoptosis, and resistance to oxidative stress [43].
PTPC opening plays a crucial role in this specific component of myocardial infarction. Strong support for this concept has recently been provided by the reduced infarct size observed in mice lacking CypD [35]. Thus targeting PTPC appears a relevant strategy to reduce ischaemic damages at reperfusion. A large body of evidence has shown that it is possible to reduce infarct size and to protect the heart after an infarct by a postconditioning or pharmacological strategy. Brief episodes of myocardial ischemia-reperfusion employed during reperfusion after a prolonged ischaemic insult may attenuate the total ischaemia-reperfusion injury. Recently, CsA has been shown to dramatically reduce infarct size in many animal species and in human. Recent proof-of-concept clinical trials support the idea that targeting MPT by either coronary intervention postconditioning or CsA can reduce infarct size and improve the recovery of contractile function after reperfusion [21, 73]. Such a strategy was also applied to ischaemia-reperfusion damages in other tissues and cells like vascular endothelial cells [74], hepatocytes [75, 76], and neurons [77–79]. Moreover, some attempts to target CsA to the mitochondrial compartment by conjugation to the lipophilic triphenylphosphonium cation proved to be promising in cytoprotection from glucose and oxygen deprivation in neurons [80], in cardiomyocytes [81], and in other various organs [82].
3.2. Role of Mitochondrial Kinases to Prevent PTPC Opening
Several protective signal pathways involving multiple kinases have been shown to converge on mitochondria and the PTPC [83] to promote cell survival (Table 2). For instance, in cardiomyocytes, pools of kinases such as Akt, protein kinase C-ε (PKCε), extracellular-regulated kinases (ERK), glycogen synthase kinase-3 beta (GSK-3β), and hexokinases (HK) I and II, are localized in or on mitochondria in addition to the cytosol. These mitochondria-associated protein kinases may integrate cytosolic stimuli and in turn, enhance tolerance of myocytes to injury.
Table 2.
List of kinases contributing to a closure of PTP via phosphorylation mechanisms or protein-protein interaction. HK, hexokinase, CK, creatine kinase, PKG, protein kinase G, PKA, protein kinase A, PKC, protein kinase C, ERK, extracellular signal-regulated kinase, GSK3, glucose-regulated kinase 3, PI3K, phosphoinositol3 kinase, and Akt/PKB, protein kinase B.
| Kinase | Effect | Target/pathway | Model | References |
|---|---|---|---|---|
| Akt/PKB, PI3K | Indirect | GSK3 via PI3K or eNos/PKG pathways | Cells | [84, 85] |
| GSK3 | Direct | VDAC, ANT, CypD | Isolated mitochondria, cells, in vivo, in vitro | [8, 37, 86, 87] |
| ERK | Indiret | GSK3 via PI3K pathway | Cells | [85, 88] |
| PKA | Direct | VDAC | Isolated mitochondria | [89] |
| PKC epsilon | Direct | VDAC | Isolated mitochondria, in vivo | [90] |
| PKG | Direct | Unknown | Isolated mitochondria, in vivo | [91, 92] |
| CK | Local regulation of ATP/creatine pools | Energetic metabolism | CK-expressing tissues | [93, 94] |
| HK | Local regulation of glucose/ATP pools | Energetic metabolism | Isolated mitochondria, cells, in vitro | [57, 74, 87, 95] |
Moreover, systematic proteomic approaches revealed that some of these kinases might form hetero-oligomers with putative components of the PTPC. Briefly, GSK-3β and HKs are directly responsible for inhibition of opening of the PTPC and, thus, for myocyte protection from necrosis [96]. As a result, postconditioning, which leads to GSK-3β inhibition, allows the myocardial salvage from reperfusion injury by modulating MPT [86]. In the context of anticancer chemotherapy, β-adrenergic receptors (β-ARs) modulate anthracycline response through crosstalk with multiple signaling pathways. β2-ARs are cardioprotective during exposure to oxidative stress induced by doxorubicin (DOX). DOX cardiotoxicity is mediated in part through a Ca2+-dependent triggering of MPT as clearly shown by a 41% reduction of DOX-induced mortality by CsA [97]. β2-ARs activate prosurvival kinases and attenuate mitochondrial dysfunction caused by oxidative stress. Accordingly, the invalidation of β2-ARs enhances cardiotoxicity via negative regulation of survival kinases and enhancement of intracellular Ca2+, thus predisposing the mitochondria to opening of the PTPC [97].
Moreover, in cancer cell lines, activation of mitochondrial ERK protects cancer cells from death through inhibition of the MPT [88]. ERK inhibition enhanced GSK-3β-dependent phosphorylation of the pore regulator CypD, whereas GSK-3β inhibition protected from PTPC opening.
By different molecular mechanisms, some kinases such as creatine kinase (CK) and HK have also cytoprotective effects and prevent PTPC opening. Depending on the tissue, which supports their expression, these kinases may be cytoprotective via a role in energy transfer to metabolites such as creatine and glucose (Table 2).
3.3. Stabilization of Mitochondrial Membrane Permeability by Bcl-2 Family Members
The Bcl-2 family is composed of more than 25 proteins implicated in the control of life-or-death decision [98]. This protein family has been particularly studied in cancer, which led to the classification of Bcl-2 and Bax as oncogenes and tumor-suppressors, respectively. Some members (e.g., Bax/Bad proteins, BH3-only proteins) favor apoptosis, whereas other members, such as Bcl-2 and Bcl-XL, prevent apoptosis. Moreover, it has been shown that the effects of Bcl-2 family proteins on mitochondria in cancer cells are linked to clinical responses to chemotherapy [99].
The cytoprotective mechanisms of Bcl-2 family members are multiple and include direct mitochondrial effects [3, 12]. Thus, Bcl-2 contributes to the stabilization of the mitochondrial membrane permeability, inhibition of ΔΨm loss and cytochrome c release and, at least in tumor cells, to the stimulation of oxidative phosphorylation [100–103]. Thus, direct protein-protein interactions between Bcl-2 family members, but also with several constitutive mitochondrial proteins such as ANT (IM), VDAC (OM), or FoF1-ATP synthase (IM) have been evidenced [104]. Bcl-2 cooperates directly with ANT, to prevent PTPC opening and to inhibit cell death [101, 105, 106]. In addition, Bcl-2 blocks the Ca2+-induced channel function of ANT and favors ADP/ATP translocase function, which positively impacts the intracellular levels of ATP [107]. Similarly, Bcl-2 and Bcl-XL directly interact with VDAC modulating its channel function [108]. Bcl-XL would bind also to the mitochondrial F0F1-ATP synthase and regulate metabolic efficiency in neurons [104]. Accordingly, recombinant proteins of some members of the Bcl-2 family directly modulate PTPC in isolated mitochondria from various sources, that is, liver and cancer cells [25, 109]. Some specific regions of Bcl-2 (e.g., BH3, BH4 domains) are responsible for these effects and as expected, peptides corresponding to these regions proved to modulate apoptosis in cellulo or in isolated mitochondria, again indicating a direct targeting of mitochondria [25]. Finally, in vivo, the BH4 domain of Bcl-XL exerts antiapoptotic effects and attenuates ischaemia/reperfusion injury through anti-apoptotic mechanism in rat hearts [110, 111].
4. Indirect Mechanisms, Which Lead to an Increased Resistance of PTPC Opening
By definition, several indirect mechanisms may lead to blockade of PTPC opening via modulation of ΔΨm, mitochondrial mass regulation, redox state, fusion/fission processes and calcium retention capacity. This may be due to modification in protein expression, in posttranslational modifications and in their interactome, which consequently affect signaling pathways. Below, we will analyze three indirect mechanisms of PTPC protection that have recently been elucidated.
4.1. Anti-Oxidant Protection
Mitochondria are major sites of ROS production, which may contribute to the development of various diseases including cardiovascular diseases and aging. Several studies have thus described the effects of antioxidant administration in the context of cardiac and liver pathologies in mice [112]. It is widely admitted that natural antioxidants such as resveratrol and curcumin have beneficial effects against ischaemia/reperfusion damages to mitochondria and cells in rat liver or heart [113, 114]. These effects are complex since resveratrol has been proposed to have multiple intracellular targets such as AMPK, SIRT1 and Nrf2, which can influence the transcriptome to increase the anti-oxidant defense (e.g., catalase, GPx, and GCLC), and other genes such eNOS and PGC1α, which favor an increase in mitochondrial mass and bioenergetics and decrease in apoptosis and inflammation (for review: [115]).
Resveratrol treatment exerts beneficial protective effects on survival, endothelium-dependent relaxation, and cardiac contractility and mitochondrial function, suggesting that resveratrol or metabolic activators could be a relevant therapy in hypertension-induced heart failure [116]. Similarly, in the heart, curcumin another polyphenol with antioxidant properties showed cardioprotective effects in catecholamine induced cardiotoxicity through prevention of mitochondrial damage, PTPC opening [117], and ventricular dysfunction [118] and in protecting rat myocardium against ischaemic insult by decreasing oxidative stress [119].
Another promising example of compound is MitoQ10, an ubiquinone derivative, which is a mitochondria-targeted antioxidant [120]. It has proven to be useful for protecting endothelial function and attenuating cardiac hypertrophy in stroke-prone hypertensive rats [121]. Moreover, MitoQ10 potently inhibits cocain-induced cardiac damage via a restoration of oxygen consumption and a stabilization of ROS levels, specifically in interfibrillar mitochondria [122]. However, when used on isolated cardiac mitochondria, MitoQ10 can be enhanced in a dose-dependent-manner MPT in the presence of the prooxidant tert-butyl hydoperoxide (t-BHP) and suboptimal doses of Ca2+ (Figure 2), although it acts as an antioxidant on rat liver mitochondria [123]. This underscores the duality of anti- and prooxidant compounds, whose effects can depend either on the dose, the redox state of the cell, the tissue, and/or the mode of administration. This probably explains, at least in part, the failure of anti-oxidants to protect efficiently the heart function in clinical trials, as recently reviewed in [124].
Figure 2.
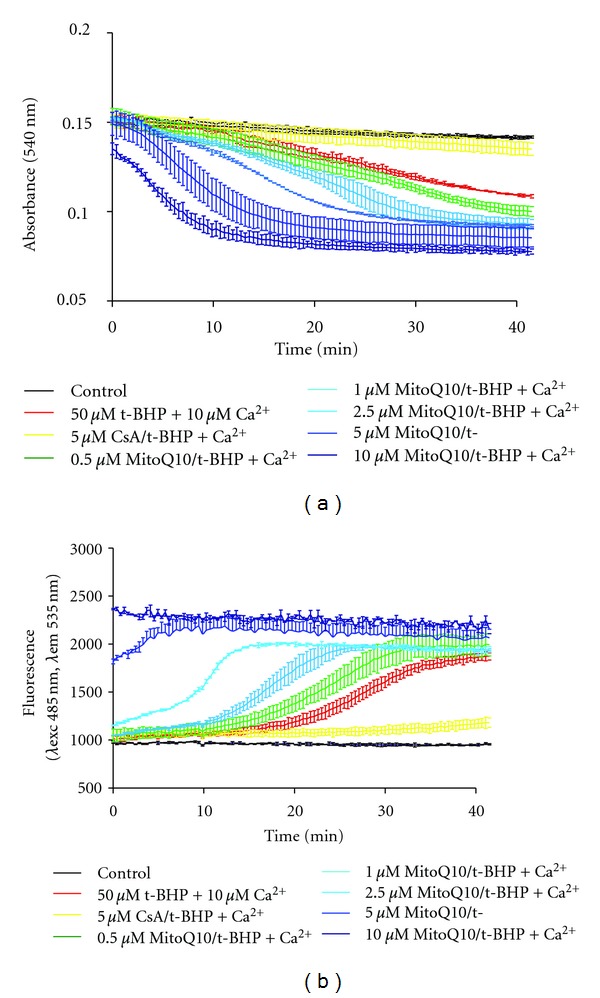
MitoQ10 stimulates MPT in isolated cardiac mitochondria. (a) MitoQ10 increases the mitochondrial swelling induced by an oxidant stress. Mitochondria (25 μg of proteins) have been pretreated by the indicated doses of MitoQ10 and treated by 50 μM t- BHP + 10 μM Ca2+. Absorbance at 540 nm has been registered for 60 min at 37°C. (b) MitoQ10 increases the mitochondrial depolarization induced by an oxidant stress. Mitochondria (25 μg of proteins) have been loaded with 2 μM Rhodamine 123, pretreated by the indicated doses of MitoQ10, and treated by 50 μM t-BHP + 10 μM Ca2+. Fluorescence has been registered for 60 min at 37°C.
4.2. Estrogens Protection
Sex and gender influence the onset and the progression of many human diseases, notably age-related diseases. Thus, estrogens, mainly 17β-estradiol, may have pleiotropic effects depending on the tissue [125]. For instance, certain cardiovascular diseases, such as myocardial hypertrophy and heart failure, differ clearly in their clinical manifestation and prognosis between women and men [126]. As a consequence, hormonal mechanisms underlying sex and gender differences are currently under intense investigation.
Animal and cellular models have been particularly instrumental to better understand estrogen protection at the level of mitochondria [127]. For instance, in cerebral circulation, estrogens mediate an enhancement of vasodilator capacity, suppression of vascular inflammation and increase of mitochondrial efficiency [125, 128, 129]. This effect is, at least in part, due to an increase in mitochondrial biogenesis via gene expression modulation [129] and a decrease in superoxide production [125]. Accordingly, chronic estrogen treatment increases mitochondrial capacity for oxidative phosphorylation while decreasing production of ROS. In breast and lung cancer cells, long-term estradiol treatment activates transcription of NRF-1 and increases mitochondrial biogenesis [130].
Moreover, mitochondrial effects on PTPC might be mediated indirectly by estrogen receptors, α and β, present in nucleus, plasma membrane, endoplasmic reticulum and even mitochondria [131]. In the context of ischaemia-reperfusion injury, it is widely admitted that estrogens protects from myocardial damage via an inhibition of PTPC function. Notably, estradiol may activate the signaling cascade which involves Akt, NO synthase, guanylyl cyclase and protein kinase G, which results in blockade of MPT-induced release of cytochrome c from mitochondria, respiratory inhibition and caspase activation [131]. As a result, estrogens effects are multifactorial, mostly indirect. Even if some estrogen-like molecules can be effective on isolated mitochondria, a precise target of estrogen within PTPC is still unknown [132]. Thus, intriguingly, estrogens may prevent Ca2+-induced cytochrome c release in isolated heart mitochondria, but not mitochondrial swelling [133].
Estrogens also protect from chemotherapy-induced cardiomyopathy in ovariectomized rats. Again, effects on the anti-oxidant cellular defenses have been proposed as one of the target mechanism of estrogen [134].
4.3. Exercise Protection
Exercise training has proven to be beneficial in chronic diseases including heart failure, obesity, diabetes or metabolic syndrome. Because endurance training improves symptoms and quality of life and decreases mortality rate and hospitalization, it is increasingly recognized as a beneficial practice for these patients. Adaptation to endurance training mainly involves energetic remodeling in skeletal and cardiac muscles [135]. The mechanisms involved in the beneficial effects of exercise training are far from being understood. Skeletal muscles adapt to repeated prolonged exercise by marked quantitative and qualitative changes in mitochondria. Endurance training promotes an increase in mitochondrial volume density and mitochondrial proteins by activating mitochondrial biogenesis [136]. Exercise training decreases apoptotic processes, and protects mitochondrial function from oxidative stress and other cardiac insults [137, 138]. Exercise training results in a reduced sensitivity to PTPC opening in heart mitochondria and confers mitochondrial protection. Moreover, even acute exercise protects against cardiac mitochondrial dysfunction, preserving mitochondrial phosphorylation capacity and attenuating DOX-induced decreased tolerance to PTPC opening [139]. Proposed mechanisms to explain the cardioprotective effects of exercise are mediated, at least partially, by redox changes and include the induction of myocardial heat shock proteins, improved cardiac antioxidant capacity and/or elevation of other cardioprotective molecules [137].
5. Conclusion and Open Questions
In the last decade, direct and indirect approaches to protect mitochondrial functions via PTPC modulation have been explored. However, it is still too early to decipher the most efficient strategies in term of cytoprotection. Nevertheless, recent studies and research advances have propelled mitochondria on the scene front of new therapeutic strategies. However, a contradiction emerges between the need to kill tumor cells in cancer therapy and to protect other cells from injuries. Even more worrying is the fact that many anticancer therapies have mitochondrial toxicity that becomes dramatic when highly oxidative nondividing cells like cardiomyocytes are concerned. Indeed, mitochondria are the main target when cardiotoxicity of anticancer drugs is concerned [44, 140]. Thus one challenging issue of cytoprotection directed to mitochondria would be to uncover new molecules or treatments that would selectively target cancer cells without affecting cardiac mitochondria. This should stimulate new studies devoted to increase our basic knowledge of the mechanisms and the tissue specificity of PTPC opening and mitochondrial function. At the same time, this will open the possibility to search for new drugs with tissue-specific effects on mitochondria. Finally, another challenge that basic and clinical research will face in the future is the notion of sex and gender influence that might be decisive for the treatment of many severe diseases.
Acknowledgments
C. Brenner and R. V. Clapier are senior scientists at the Centre National de la Recherche Scientifique. A. Garnier, C. Brenner and R. V. Clapier are supported by LabEx LERMIT. C. Brenner is supported by ANR (ANR-08PCVI 0008-01). C. Martel received a fellowship from Association pour la Recherche sur le Cancer. L. H. Huynh received a fellowship from INSERM.
Abbreviations
- ANT:
Adenine nucleotide translocase
- β-Ars:
β-adrenergic receptors
- Ca2+:
Calcium
- CK:
Creatine kinase
- CsA:
Cyclosporin A
- CypD:
Cyclophilin D
- ΔΨm:
Mitochondrial inner membrane potential
- DOX:
Doxorubicin
- ERK:
Extracellular-signal-regulated kinase
- GSK-3β:
Glycogen synthase kinase-3 beta
- HK:
Hexokinase
- IM:
Inner membrane
- JC-1:
5, 5′, 6, 6′ -tetrachloro-1, 1′, 3, 3′ -tetraethylbenzimidazol-carbocyanine iodine
- MMP:
Mitochondrial membrane permeabilization
- MPT:
Mitochondrial permeability transition
- OM:
Outer membrane
- PTPC:
permeability transition pore complex
- Rhod123:
Rhodamine 123
- ROS:
Reactive oxygen species
- TMRM:
Tetramethylrhodamine methyl ester
- VDAC:
Voltage-dependent anion channel.
References
- 1.Lehninger AL. The transfer of energy in oxidative phosphorylation. Bulletin de la Société de Chimie Biologique. 1964;46:1555–1575. [PubMed] [Google Scholar]
- 2.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological Reviews. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 3.Martinou J-C, Youle R. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Developmental Cell. 2011;21(1):92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majno G, Joris I. Apoptosis, oncosis, and necrosis: an overview of cell death. American Journal of Pathology. 1995;146(1):3–15. [PMC free article] [PubMed] [Google Scholar]
- 5.Papanicolaou KN, Ngoh GA, Dabkowski ER, et al. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. American Journal of Physiology. 2012;302(1):H167–H179. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283(5407):1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 7.Ramachandran A, Jha S, Lefer DJ. Review paper: pathophysiology of myocardial reperfusion injury: the role of genetically engineered mouse models. Veterinary Pathology. 2008;45(5):698–706. doi: 10.1354/vp.45-5-698. [DOI] [PubMed] [Google Scholar]
- 8.Matas J, Tien Sing Young N, Bourcier-Lucas C, et al. Increased expression and intramitochondrial translocation of cyclophilin-D associates with increased vulnerability of the permeability transition pore to stress-induced opening during compensated ventricular hypertrophy. Journal of Molecular and Cellular Cardiology. 2009;46(3):420–430. doi: 10.1016/j.yjmcc.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 9.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6(1):1–38. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Ventura-Clapier R, Garnier A, Veksler V, Joubert F. Bioenergetics of the failing heart. Biochimica et Biophysica Acta. 2011;1813(7):1360–1372. doi: 10.1016/j.bbamcr.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Neubauer S. The failing heart-An engine out of fuel. New England Journal of Medicine. 2007;356(11):1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 12.Indran IR, Tufo G, Pervaiz S, Brenner C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochimica et Biophysica Acta. 2011;1807(6):735–745. doi: 10.1016/j.bbabio.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Archiv European Journal of Physiology. 2004;447(5):689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 14.Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochimica et Biophysica Acta. 2008;1778(10):1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 15.Gena P, Fanelli E, Brenner C, Svelto M, Calamita G. News and views on mitochondrial water transport. Frontiers in Bioscience. 2009;14:4189–4198. doi: 10.2741/3522. [DOI] [PubMed] [Google Scholar]
- 16.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium treated mitochondria. Journal of Biological Chemistry. 1976;251(16):5069–5077. [PubMed] [Google Scholar]
- 17.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-x(L) regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91(5):627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 18.Jacotot E, Ravagnan L, Loeffler M, et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Journal of Experimental Medicine. 2000;191(1):33–45. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belzacq-Casagrande AS, Martel C, Pertuiset C, Borgne-Sanchez A, Jacotot E, Brenner C. Pharmacological screening and enzymatic assays for apoptosis. Frontiers in Bioscience. 2009;14:3550–3562. doi: 10.2741/3470. [DOI] [PubMed] [Google Scholar]
- 20.Halestrap AR, Connern CR, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Molecular and Cellular Biochemistry. 1997;174(1-2):167–172. [PubMed] [Google Scholar]
- 21.Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. New England Journal of Medicine. 2008;359(5):473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 22.Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-D-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75(4):993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- 23.Blattner JR, He L, Lemasters JJ. Screening assays for the mitochondrial permeability transition using a fluorescence multiwell plate reader. Analytical Biochemistry. 2001;295(2):220–226. doi: 10.1006/abio.2001.5219. [DOI] [PubMed] [Google Scholar]
- 24.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured filroblasts. Nature Protocols. 2007;2(2):287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 25.Buron N, Porceddu M, Brabant M, et al. Use of human cancer cell lines mitochondria to explore the mechanisms of BH3 peptides and ABT-737-induced mitochondrial membrane permeabilization. PloS One. 2010;5(3, article e9924) doi: 10.1371/journal.pone.0009924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picard M, Taivassalo T, Gouspillou G, Hepple RT. Mitochondria: isolation, structure and function. Journal of Physiology. 2011;589(18):4413–4421. doi: 10.1113/jphysiol.2011.212712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petronilli V, Miotto G, Canton M, et al. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophysical Journal. 1999;76(2):725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zahrebelski G, Nieminen AL, Al-Ghoul K, Qian T, Herman B, Lemasters JJ. Progression of subcellular changes during chemical hypoxia to cultured rat hepatocytes: a laser scanning confocal microscopic study. Hepatology. 1995;21(5):1361–1372. [PubMed] [Google Scholar]
- 29.Nieminen AL, Saylor AK, Tesfai SA, Herman B, Lemasters JJ. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochemical Journal. 1995;307(1):99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dedkova EN, Blatter LA. Measuring mitochondrial function in intact cardiac myocytes. Journal of Molecular and Cellular Cardiology. 2012;52(1):48–61. doi: 10.1016/j.yjmcc.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rama Rao KV, Jayakumar AR, Norenberg MD. Ammonia neurotoxicity: role of the mitochondrial permeability transition. Metabolic Brain Disease. 2003;18(2):113–127. doi: 10.1023/a:1023858902184. [DOI] [PubMed] [Google Scholar]
- 32.Galluzzi L, Zamzami N, De La Motte Rouge T, Lemaire C, Brenner C, Kroemer G. Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis. 2007;12(5):803–813. doi: 10.1007/s10495-007-0720-1. [DOI] [PubMed] [Google Scholar]
- 33.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochemical Journal. 1995;307(1):93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Current Medicinal Chemistry. 2003;10(16):1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 35.Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 36.Crompton M. Mitochondrial intermembrane junctional complexes and their role in cell death. Journal of Physiology. 2000;529(1):11–21. doi: 10.1111/j.1469-7793.2000.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brdiczka D, Beutner G, Rück A, Dolder M, Wallimann T. The molecular structure of mitochondrial contact sites. Their role in regulation of energy metabolism and permeability transition. BioFactors. 1998;8(3-4):235–242. doi: 10.1002/biof.5520080311. [DOI] [PubMed] [Google Scholar]
- 38.Halestrap AP. What is the mitochondrial permeability transition pore? Journal of Molecular and Cellular Cardiology. 2009;46(6):821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 39.Ricchelli F, Šileikyte J, Bernardi P. Shedding light on the mitochondrial permeability transition. Biochimica et Biophysica Acta. 2011;1807(5):482–490. doi: 10.1016/j.bbabio.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Szabo I, Zoratti M. The mitochondrial megachannel is the permeability transition pore. Journal of Bioenergetics and Biomembranes. 1992;24(1):111–117. doi: 10.1007/BF00769537. [DOI] [PubMed] [Google Scholar]
- 41.Ko YH, Delannoy M, Hullihen J, Chiu W, Pedersen PL. Mitochondrial ATP synthasome: cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. Journal of Biological Chemistry. 2003;278(14):12305–12309. doi: 10.1074/jbc.C200703200. [DOI] [PubMed] [Google Scholar]
- 42.Jacotot E, Deniaud A, Borgne-Sanchez A, et al. Therapeutic peptides: targeting the mitochondrion to modulate apoptosis. Biochimica et Biophysica Acta. 2006;1757(9-10):1312–1323. doi: 10.1016/j.bbabio.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 43.Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death and Differentiation. 2008;15(4):686–690. doi: 10.1038/cdd.2008.13. [DOI] [PubMed] [Google Scholar]
- 44.Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U. New insights into doxorubicin-induced cardiotoxicity: the critical role of cellular energetics. Journal of Molecular and Cellular Cardiology. 2006;41(3):389–405. doi: 10.1016/j.yjmcc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. Journal of Biological Chemistry. 1989;264(14):7826–7830. [PubMed] [Google Scholar]
- 46.Griffiths EJ, Halestrap AP. Protection by cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. Journal of Molecular and Cellular Cardiology. 1993;25(12):1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 47.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. Journal of Biological Chemistry. 2002;277(38):34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 48.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovascular Research. 2003;60(3):617–625. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 49.Henderson PJ, Lardy HA. Bongkrekic acid. An inhibitor of the adenine nucleotide translocase of mitochondria. Journal of Biological Chemistry. 1970;245(6):1319–1326. [PubMed] [Google Scholar]
- 50.Narita M, Shimizu S, Ito T, et al. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(25):14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zamzami N, Hamel CE, Maisse C, et al. Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene. 2000;19(54):6342–6350. doi: 10.1038/sj.onc.1204030. [DOI] [PubMed] [Google Scholar]
- 52.Novgorodov SA, Gudz TI, Milgrom YM, Brierley GP. The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. Journal of Biological Chemistry. 1992;267(23):16274–16282. [PubMed] [Google Scholar]
- 53.Haworth RA, Hunter DR. Control of the mitochondrial permeability transition pore by high- affinity ADP binding at the ADP/ATP translocase in permeabilized mitochondria. Journal of Bioenergetics and Biomembranes. 2000;32(1):91–96. doi: 10.1023/a:1005568630151. [DOI] [PubMed] [Google Scholar]
- 54.Lee AC, Zizi M, Colombini M. β-NADH decreases the permeability of the mitochondrial outer membrane to ADP by a factor of 6. Journal of Biological Chemistry. 1994;269(49):30974–30980. [PubMed] [Google Scholar]
- 55.Deniaud A, Rossi C, Berquand A, et al. Voltage-dependent anion channel transports calcium ions through biomimetic membranes. Langmuir. 2007;23(7):3898–3905. doi: 10.1021/la063105+. [DOI] [PubMed] [Google Scholar]
- 56.Deniaud A, Sharaf El Dein O, Maillier E, et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27(3):285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 57.Thinnes FP, Florke H, Winkelbach H, et al. Channel active mammalian porin, purified from crude membrane fractions of human B lymphocytes or bovine skeletal muscle, reversibly binds the stilbene-disulfonate group of the chloride channel blocker DIDS. Biological Chemistry Hoppe-Seyler. 1994;375(5):315–322. doi: 10.1515/bchm3.1994.375.5.315. [DOI] [PubMed] [Google Scholar]
- 58.Shoshan-Barmatz V, Keinan N, Abu-Hamad S, Tyomkin D, Aram L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochimica et Biophysica Acta. 2010;1797(6-7):1281–1291. doi: 10.1016/j.bbabio.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 59.Rigobello MP, Turcato F, Bindoli A. Inhibition of rat liver mitochondrial permeability transition by respiratory substrates. Archives of Biochemistry and Biophysics. 1995;319(1):225–230. doi: 10.1006/abbi.1995.1286. [DOI] [PubMed] [Google Scholar]
- 60.Gincel D, Silberberg S, Shoshan-Barmatz V. Modulation of the voltagedependent anion channel (VDAC) by glutamate. Journal of Bioenergetics and Biomembranes. 2000;32:571–583. doi: 10.1023/a:1005670527340. [DOI] [PubMed] [Google Scholar]
- 61.Gincel D, Shoshan-Barmatz V. Glutamate interacts with VDAC and modulates opening of the mitochondrial permeability transition pore. Journal of Bioenergetics and Biomembranes. 2004;36(2):179–186. doi: 10.1023/b:jobb.0000023621.72873.9e. [DOI] [PubMed] [Google Scholar]
- 62.Cesura AM, Pinard E, Schubenel R, et al. The voltage-dependent anion channel is the target for a new class of inhibitors of the mitochondrial permeability transition pore. Journal of Biological Chemistry. 2003;278(50):49812–49818. doi: 10.1074/jbc.M304748200. [DOI] [PubMed] [Google Scholar]
- 63.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1−/− mitochondria. Biochimica et Biophysica Acta. 2006;1757(5-6):590–595. doi: 10.1016/j.bbabio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 64.Leung AWC, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. Journal of Biological Chemistry. 2008;283(39):26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fontaine E, Ichas F, Bernardi P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. Journal of Biological Chemistry. 1998;273(40):25734–25740. doi: 10.1074/jbc.273.40.25734. [DOI] [PubMed] [Google Scholar]
- 66.Elimadi A, Sapena R, Settaf A, Le Louet H, Tillement JP, Morin D. Attenuation of liver normothermic ischemia—reperfusion injury by preservation of mitochondrial functions with S-15176, a potent trimetazidine derivative. Biochemical Pharmacology. 2001;62(4):509–516. doi: 10.1016/s0006-2952(01)00676-1. [DOI] [PubMed] [Google Scholar]
- 67.Elimadi A, Jullien V, Tillement JP, Morin D. S-15176 inhibits mitochondrial permeability transition via a mechanism independent of its antioxidant properties. European Journal of Pharmacology. 2003;468(2):93–101. doi: 10.1016/s0014-2999(03)01671-6. [DOI] [PubMed] [Google Scholar]
- 68.Ascah A, Khairallah M, Daussin F, et al. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. American Journal of Physiology. 2011;300(1):H144–H153. doi: 10.1152/ajpheart.00522.2010. [DOI] [PubMed] [Google Scholar]
- 69.Millay DP, Sargent MA, Osinska H, et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nature Medicine. 2008;14(4):442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bordet T, Buisson B, Michaud M, et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. Journal of Pharmacology and Experimental Therapeutics. 2007;322(2):709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- 71.Queiroga CSF, Almeida AS, Martel C, Brenner C, Alves PM, Vieira HLA. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. Journal of Biological Chemistry. 2010;285(22):17077–17088. doi: 10.1074/jbc.M109.065052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Azzolin L, Antolini N, Calderan A, et al. Antamanide, a derivative of amanita phalloides, is a novel inhibitor of the mitochondrial permeability transition pore. PLoS One. 2011;6(1, article e16280) doi: 10.1371/journal.pone.0016280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mewton N, Croisille P, Gahide G, et al. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. Journal of the American College of Cardiology. 2010;55(12):1200–1205. doi: 10.1016/j.jacc.2009.10.052. [DOI] [PubMed] [Google Scholar]
- 74.Okorie M, Bhavsar D, Ridout D, et al. Postconditioning protects against human endothelial ischaemia-reperfusion injury via subtype-specific KATP channel activation and is mimicked by inhibition of the mitochondrial permeability transition pore. European Heart Journal. 2011:1266–1274. doi: 10.1093/eurheartj/ehr041. [DOI] [PubMed] [Google Scholar]
- 75.Kim JS, He L, Qian T, Lemasters JJ. Role of the mitochondrial permeability transition in apoptotic and necrotic death after ischemia/reperfusion injury to hepatocytes. Current Molecular Medicine. 2003;3(6):527–535. doi: 10.2174/1566524033479564. [DOI] [PubMed] [Google Scholar]
- 76.Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. American Journal of Physiology. 1997;273(6):C1783–C1792. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
- 77.Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cyclosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. Journal of Neuroscience. 1998;18(14):5151–5159. doi: 10.1523/JNEUROSCI.18-14-05151.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Khaspekov L, Friberg H, Halestrap A, Viktorov I, Wieloch T. Cyclosporin A and its nonimmunosuppressive analogue N-Me-Val-4-cyclosporin A mitigate glucose/oxygen deprivation-induced damage to rat cultured hippocampal neurons. European Journal of Neuroscience. 1999;11(9):3194–3198. doi: 10.1046/j.1460-9568.1999.00743.x. [DOI] [PubMed] [Google Scholar]
- 79.Matsumoto S, Friberg H, Ferrand-Drake M, Wieloch T. Blockade of the mitochondrial permeability transition pore diminishes infarct size in the rat after transient middle cerebral artery occlusion. Journal of Cerebral Blood Flow and Metabolism. 1999;19(7):736–741. doi: 10.1097/00004647-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 80.Malouitre S, Dube H, Selwood D, Crompton M. Mitochondrial targeting of cyclosporin A enables selective inhibition of cyclophilin-D and enhanced cytoprotection after glucose and oxygen deprivation. Biochemical Journal. 2010;425(1):137–148. doi: 10.1042/BJ20090332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dube H, Selwood D, Malouitre S, Capano M, Simone MI, Crompton M. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochemical Journal. 2012;441(3):901–907. doi: 10.1042/BJ20111301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Porteous CM, Logan A, Evans C, et al. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: implications for mitochondria-specific therapies and probes. Biochimica et Biophysica Acta. 2010;1800(9):1009–1017. doi: 10.1016/j.bbagen.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 83.Miura T, Tanno M, Sato T. Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovascular Research. 2010;88(1):7–15. doi: 10.1093/cvr/cvq206. [DOI] [PubMed] [Google Scholar]
- 84.Feng J, Lucchinetti E, Ahuja P, Pasch T, Perriard JC, Zaugg M. Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3β . Anesthesiology. 2005;103(5):987–995. doi: 10.1097/00000542-200511000-00013. [DOI] [PubMed] [Google Scholar]
- 85.Javadov S, Rajapurohitam V, Kilić A, Zeidan A, Choi A, Karmazyn M. Anti-hypertrophic effect of NHE-1 inhibition involves GSK-3β-dependent attenuation of mitochondrial dysfunction. Journal of Molecular and Cellular Cardiology. 2009;46(6):998–1007. doi: 10.1016/j.yjmcc.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 86.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117(21):2761–2768. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- 87.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Research. 2005;65(22):10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 88.Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(2):726–731. doi: 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pediaditakis P, Kim JS, He L, Zhang X, Graves LM, Lemasters JJ. Inhibition of the mitochondrial permeability transition by protein kinase A in rat liver mitochondria and hepatocytes. Biochemical Journal. 2010;431(3):411–421. doi: 10.1042/BJ20091741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baines CP, Song CX, Zheng YT, et al. Protein kinase Cε interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circulation Research. 2003;92(8):873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takuma K, Phuagphong P, Lee E, Mori K, Baba A, Matsuda T. Anti-apoptotic effect of cGMP in cultured astrocytes: inhibition by cGMP-dependent protein kinase of mitochondrial permeable transition pore. Journal of Biological Chemistry. 2001;276(51):48093–48099. doi: 10.1074/jbc.M108622200. [DOI] [PubMed] [Google Scholar]
- 92.Borutaite V, Morkuniene R, Arandarcikaite O, Jekabsone A, Barauskaite J, Brown GC. Nitric oxide protects the heart from ischemia-induced apoptosis and mitochondrial damage via protein kinase G mediated blockage of permeability transition and cytochrome c release. Journal of Biomedical Science. 2009;16(1, article 70) doi: 10.1186/1423-0127-16-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wyss M, Smeitink J, Wevers RA, Wallimann T. Mitochondrial creatine kinase: a key enzyme of aerobic energy metabolism. Biochimica et Biophysica Acta. 1992;1102(2):119–166. doi: 10.1016/0005-2728(92)90096-k. [DOI] [PubMed] [Google Scholar]
- 94.O’Gorman E, Beutner G, Dolder M, Koretsky AP, Brdiczka D, Wallimann T. The role of creatine kinase in inhibition of mitochondrial permeability transition. FEBS Letters. 1997;414(2):253–257. doi: 10.1016/s0014-5793(97)01045-4. [DOI] [PubMed] [Google Scholar]
- 95.Beutner G, Rück A, Riede B, Brdiczka D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochimica et Biophysica Acta. 1998;1368(1):7–18. doi: 10.1016/s0005-2736(97)00175-2. [DOI] [PubMed] [Google Scholar]
- 96.Miura T, Nishihara M, Miki T. Drug development targeting the glycogen synthase kinase-3β (GSK-3β)-mediated signal transduction pathway: role of GSK-3β in myocardial protection against ischemia/reperfusion injury. Journal of Pharmacological Sciences. 2009;109(2):162–167. doi: 10.1254/jphs.08r27fm. [DOI] [PubMed] [Google Scholar]
- 97.Fajardo G, Zhao M, Berry G, Wong L-J, Mochly-Rosen D, Bernstein D. β2-adrenergic receptors mediate cardioprotection through crosstalk with mitochondrial cell death pathways. Journal of Molecular and Cellular Cardiology. 2011;51(5):781–789. doi: 10.1016/j.yjmcc.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Adams JM, Cory S. Life-or-death decisions by the Bcl-2 protein family. Trends in Biochemical Sciences. 2001;26(1):61–66. doi: 10.1016/s0968-0004(00)01740-0. [DOI] [PubMed] [Google Scholar]
- 99.Chonghaile TN, Sarosiek KA, Vo T-T, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334(6059):1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Llambi F, Moldoveanu T, Tait S, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Molecular Cell. 2011;44(4):517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marzo I, Brenner C, Zamzami N, et al. The permeability transition pore complex: a target for apoptosis regulation by caspases and Bcl-2-related proteins. Journal of Experimental Medicine. 1998;187(8):1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399(6735):483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 103.Chen ZX, Pervaiz S. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell Death and Differentiation. 2007;14(9):1617–1627. doi: 10.1038/sj.cdd.4402165. [DOI] [PubMed] [Google Scholar]
- 104.Alavian KN, Li H, Collis L, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F 1 F O ATP synthase. Nature Cell Biology. 2011;13(10):1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marzo I, Brenner C, Zamzami N, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281(5385):2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 106.Brenner C, Cadiou H, Vieira HLA, et al. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19(3):329–336. doi: 10.1038/sj.onc.1203298. [DOI] [PubMed] [Google Scholar]
- 107.Belzacq AS, Vieira HLA, Verrier F, et al. Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Research. 2003;63(2):541–546. [PubMed] [Google Scholar]
- 108.Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vieira HLA, Boya P, Cohen I, et al. Cell permeable BH3-peptides overcome the cytoprotective effect of Bcl-2 and Bcl-XL. Oncogene. 2002;21(13):1963–1977. doi: 10.1038/sj.onc.1205270. [DOI] [PubMed] [Google Scholar]
- 110.Sugioka R, Shimizu S, Funatsu T, et al. BH4-domain peptide from Bcl-xL exerts anti-apoptotic activity in vivo. Oncogene. 2003;22(52):8432–8440. doi: 10.1038/sj.onc.1207180. [DOI] [PubMed] [Google Scholar]
- 111.Ono M, Sawa Y, Ryugo M, et al. BH4 peptide derivative from Bcl-xL attenuates ischemia/reperfusion injury thorough anti-apoptotic mechanism in rat hearts. European Journal of Cardio-thoracic Surgery. 2005;27(1):117–121. doi: 10.1016/j.ejcts.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 112.Morin D, Assaly R, Paradis S, Berdeaux A. Inhibition of mitochondrial membrane permeability as a putative pharmacological target for cardioprotection. Current Medicinal Chemistry. 2009;16(33):4382–4398. doi: 10.2174/092986709789712871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Plin C, Tillement JP, Berdeaux A, Morin D. Resveratrol protects against cold ischemia-warm reoxygenation-induced damages to mitochondria and cells in rat liver. European Journal of Pharmacology. 2005;528(1–3):162–168. doi: 10.1016/j.ejphar.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 114.Ligeret H, Barthelemy S, Zini R, Tillement JP, Labidalle S, Morin D. Effects of curcumin and curcumin derivatives on mitochondrial permeability transition pore. Free Radical Biology and Medicine. 2004;36(7):919–929. doi: 10.1016/j.freeradbiomed.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 115.Ungvari Z, Sonntag WE, De Cabo R, Baur JA, Csiszar A. Mitochondrial protection by resveratrol. Exercise and Sport Sciences Reviews. 2011;39(3):128–132. doi: 10.1097/JES.0b013e3182141f80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rimbaud S, Ruiz M, Piquereau J, et al. Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One. 2011;6(10, article e26391) doi: 10.1371/journal.pone.0026391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Izem-Meziane M, Djerdjouri B, Rimbaud S, et al. Catecholamine-induced cardiac mitochondrial dysfunction and mPTP opening: protective effect of curcumin. American Journal of Physiology. 2012;302(3):H665–H674. doi: 10.1152/ajpheart.00467.2011. [DOI] [PubMed] [Google Scholar]
- 118.Tanwar V, Sachdeva J, Golechha M, Kumari S, Arya DS. Curcumin protects rat myocardium against isoproterenol-induced ischemic injury: attenuation of ventricular dysfunction through increased expression of hsp27 alongwith strengthening antioxidant defense system. Journal of Cardiovascular Pharmacology. 2010;55(4):377–384. doi: 10.1097/FJC.0b013e3181d3da01. [DOI] [PubMed] [Google Scholar]
- 119.Manikandan P, Sumitra M, Aishwarya S, Manohar BM, Lokanadam B, Puvanakrishnan R. Curcumin modulates free radical quenching in myocardial ischaemia in rats. The International Journal of Biochemistry & Cell Biology. 2004;36(10):1967–1980. doi: 10.1016/j.biocel.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 120.Kelso GF, Porteous CM, Coulter CV, et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. Journal of Biological Chemistry. 2001;276(7):4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 121.Graham D, Huynh NN, Hamilton CA, et al. Mitochondria-targeted antioxidant mitoq10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54(2):322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 122.Vergeade A, Mulder P, Vendeville-Dehaudt C, et al. Mitochondrial impairment contributes to cocaine-induced cardiac dysfunction: prevention by the targeted antioxidant MitoQ. Free Radical Biology and Medicine. 2010;49(5):748–756. doi: 10.1016/j.freeradbiomed.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 123.Plecitá-Hlavatá L, Ježek J, Ježek P. Pro-oxidant mitochondrial matrix-targeted ubiquinone MitoQ10 acts as anti-oxidant at retarded electron transport or proton pumping within Complex I. International Journal of Biochemistry and Cell Biology. 2009;41(8-9):1697–1707. doi: 10.1016/j.biocel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 124.Núñez-Córdoba JM, Martínez-González MA. Antioxidant vitamins and cardiovascular disease. Current Topics in Medicinal Chemistry. 2011;11(14):1861–1869. doi: 10.2174/156802611796235143. [DOI] [PubMed] [Google Scholar]
- 125.Chen JQ, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochimica et Biophysica Acta. 2009;1793(10):1540–1570. doi: 10.1016/j.bbamcr.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Regitz-Zagrosek V, Oertelt-Prigione S, Seeland U, Hetzer R. Sex and gender differences in myocardial hypertrophy and heart failure. Wiener Medizinische Wochenschrift. 2011;161:109–116. doi: 10.1007/s10354-011-0892-8. [DOI] [PubMed] [Google Scholar]
- 127.Maas AHEM, Van Der Schouw YT, Regitz-Zagrosek V, et al. Red alert for womens heart: the urgent need for more research and knowledge on cardiovascular disease in women. European Heart Journal. 2011;32(11):1362–1368. doi: 10.1093/eurheartj/ehr048. [DOI] [PubMed] [Google Scholar]
- 128.Duckles SP, Krause DN. Mechanisms of cerebrovascular protection: oestrogen, inflammation and mitochondria. Acta Physiologica. 2011;203(1):149–154. doi: 10.1111/j.1748-1716.2010.02184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Klinge CM. Estrogenic control of mitochondrial function and biogenesis. Journal of Cellular Biochemistry. 2008;105(6):1342–1351. doi: 10.1002/jcb.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Molecular Endocrinology. 2008;22(3):609–622. doi: 10.1210/me.2007-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Morkuniene R, Arandarcikaite O, Ivanoviene L, Borutaite V. Estradiol-induced protection against ischemia-induced heart mitochondrial damage and caspase activation is mediated by protein kinase G. Biochimica et Biophysica Acta. 2010;1797(6-7):1012–1017. doi: 10.1016/j.bbabio.2010.03.027. [DOI] [PubMed] [Google Scholar]
- 132.Borrás C, Gambini J, López-Grueso R, Pallardó FV, Viña J. Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochimica et Biophysica Acta. 2010;1802(1):205–211. doi: 10.1016/j.bbadis.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 133.Morkuniene R, Jekabsone A, Borutaite V. Estrogens prevent calcium-induced release of cytochrome c from heart mitochondria. FEBS Letters. 2002;521(1-3):53–56. doi: 10.1016/s0014-5793(02)02820-x. [DOI] [PubMed] [Google Scholar]
- 134.Muñoz-Castañeda JR, Montilla P, Muñoz MC, Bujalance I, Muntané J, Túnez I. Effect of 17-β-estradiol administration during adriamycin-induced cardiomyopathy in ovariectomized rat. European Journal of Pharmacology. 2005;523(1–3):86–92. doi: 10.1016/j.ejphar.2005.08.056. [DOI] [PubMed] [Google Scholar]
- 135.Ventura-Clapier R, Mettauer B, Bigard X. Beneficial effects of endurance training on cardiac and skeletal muscle energy metabolism in heart failure. Cardiovascular Research. 2007;73(1):10–18. doi: 10.1016/j.cardiores.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 136.Garnier A, Fortin D, Zoll J, et al. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB Journal. 2005;19(1):43–52. doi: 10.1096/fj.04-2173com. [DOI] [PubMed] [Google Scholar]
- 137.Ascensão A, Ferreira R, Magalhães J. Exercise-induced cardioprotection—biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. International Journal of Cardiology. 2007;117(1):16–30. doi: 10.1016/j.ijcard.2006.04.076. [DOI] [PubMed] [Google Scholar]
- 138.Ciminelli M, Ascah A, Bourduas K, Burelle Y. Short term training attenuates opening of the mitochondrial permeability transition pore without affecting myocardial function following ischemia-reperfusion. Molecular and Cellular Biochemistry. 2006;291(1-2):39–47. doi: 10.1007/s11010-006-9192-9. [DOI] [PubMed] [Google Scholar]
- 139.Marcil M, Bourduas K, Ascah A, Burelle Y. Exercise training induces respiratory substrate-specific decrease in Ca2+-induced permeability transition pore opening in heart mitochondria. American Journal of Physiology. 2006;290(4):H1549–H1557. doi: 10.1152/ajpheart.00913.2005. [DOI] [PubMed] [Google Scholar]
- 140.Ascensão A, Lumini-Oliveira J, Machado NG, et al. Acute exercise protects against calcium-induced cardiac mitochondrial permeability transition pore opening in doxorubicin-treated rats. Clinical Science. 2011;120(1):37–49. doi: 10.1042/CS20100254. [DOI] [PubMed] [Google Scholar]