

Abstract
A novel mutagenesis technique using error-prone DNA polymerase δ (polδ), the disparity mutagenesis model of evolution, has been successfully employed to generate novel microorganism strains with desired traits. However, little else is known about the spectra of mutagenic effects caused by disparity mutagenesis. We evaluated and compared the performance of the polδ MKII mutator, which expresses the proofreading-deficient and low-fidelity polδ, in Saccharomyces cerevisiae haploid strain with that of the commonly used chemical mutagen ethyl methanesulfonate (EMS). This mutator strain possesses exogenous mutant polδ supplied from a plasmid, tthereby leaving the genomic one intact. We measured the mutation rate achieved by each mutagen and performed high-throughput next generation sequencing to analyze the genome-wide mutation spectra produced by the 2 mutagenesis methods. The mutation frequency of the mutator was approximately 7 times higher than that of EMS. Our analysis confirmed the strong G/C to A/T transition bias of EMS, whereas we found that the mutator mainly produces transversions, giving rise to more diverse amino acid substitution patterns. Our present study demonstrated that the polδ MKII mutator is a useful and efficient method for rapid strain improvement based on in vivo mutagenesis.
1. Introduction
Random mutagenesis is a powerful tool for generating enzymes, proteins, metabolic pathways, or even entire genomes with desired or improved properties [1]. Due to the technical simplicity and applicability to almost any organism, chemical or radiation mutagenesis is frequently used for the generation of genetic variability in a microorganism. However, these methods tend to be inefficient because they can cause substantial cell damage when performed in vivo [2].
A novel mutagenesis technique using error-prone DNA polymerase δ (polδ), based on the disparity mutagenesis model of evolution [3] has been successfully employed to generate novel microorganism strains with desired traits [4–11]. In the disparity model, mutations occur preferentially on the lagging strand, due to the more complex, discontinuous DNA replication that takes place there. Computer simulation shows that the disparity model accumulates more mutations than the parity model, in which mutations occur stochastically and evenly in both strands [3]. In addition, the disparity model produces greater diversity because some offspring will have mutant DNA while some offspring will have nonmutated, wild-type DNA.
Several studies have shown that the disparity mutagenesis method often achieved more satisfactory results (i.e., higher mutation rate and quick attainment of the desired phenotype) than conventional methods such as the chemical mutagen, ethyl methanesulfonate (EMS) [5, 10], which is known to produce mainly G/C to A/T transitions [12]. However, little else is known about the spectra of mutagenic effects caused by disparity mutagenesis.
polδ is involved in the synthesis of the lagging strand of DNA [13]. Several mutants, including the proofreading-deficient pol3-01 strain and several low-fidelity mutants, have been shown to elevate the mutation rate [14–18]. To generate the strains with the greatest mutagenicity, Neo-Morgan Laboratory (Kanagawa, Japan) has developed the plasmid YCplac33/polδMKII, expressing the polδ mutant allele with 2 mutations: one mutation to inactivate the proofreading activity (D321A and E323A) [15] and another mutation to decrease the fidelity of replication (L612M) [14, 17, 18].
With the recent advent of next-generation sequencing technologies, an accurate characterization of the mutant genome, relative to the parental reference strain, is now achievable. In fact, Flibotte et al. have analyzed the mutation spectra induced by various mutagens, such as EMS, ENU, and UV/TMP, in the whole genome of Caenorhabditis elegans [12]. Another group has also used these sequencing technologies to analyze the genetic variations between a parental and EMS-mutagenized strain of yeast [19].
In this study, we evaluate the performance of the polδMKII mutator, which expresses the proofreading-deficient and low-fidelity polδ in S. cerevisiae haploid strain, compared with the commonly used chemical mutagen EMS. This mutator strain possesses exogenous mutant polδ supplied from a plasmid, thereby leaving the genomic one intact. We measured the mutation rate of this mutator strain and found that the mutation frequency of polδMKII was approximately 7 times higher than that of EMS. We also performed high-throughput next generation sequencing with Illumina GAII to analyze the genome-wide mutation spectra produced by the 2 different mutagenesis methods and found that the mutator strain exhibited more pleiotropy and gave rise to more diverse amino acid substitution patterns. Our present study has demonstrated that a proofreading-deficient and low-fidelity polδMKII mutator is a useful and efficient method for rapid strain improvement based on in vivo mutagenesis. This mutator is also useful for studying the acceleration of evolution.
2. Materials and Methods
2.1. Plasmid
Plasmid YCplac33/polδMKII was constructed as follows: a 4.8 kb DNA fragment containing the S. cerevisiae BY2961 pol3 gene, plus the UTR 1 kb upstream and 0.5 kb downstream, (Matα ura3-52, his3-Δ300, trp1-Δ901, leu2-3, 112 lys2-801, ade2-2) was inserted into the SalI-EcoRI site of YCplac33, and 3 amino acid substitutions, D321A, E323A, and L612M, were introduced into the pol3 gene using site-directed mutagenesis [20]. YCplac33 is low-copy number plasmid and is stably maintained in S. cerevisiae [20].
2.2. Mutator Mutagenesis
YCplac33/polδMKII vector (and YCplac33 empty vector as nonmutator control) was introduced into S. cerevisiae BY2961 strain cells using the LiCl method, and the transformants (mutator strains) were selected on synthetic complete (SC)-agar plates without uracil. Five mutator strains were picked and independently cultivated in 1 mL SC medium at 30°C for 24 h (about 30 generations) in order to introduce mutations into their chromosomes. To determine the mutation frequencies of the 5 mutator strains, aliquots were spread on SC-agar plates containing L-canavanine sulfate salt (0.06 mg/mL) (Sigma, St. Louis, MO, USA) to identify CAN1 mutants, and incubated until resistant colonies were formed. The mutation frequencies were calculated as the number of drug-resistant colonies divided by the number of colonies on SC-agar plate without drug. Forward mutation rates at CAN1 were determined by fluctuation analysis using these 5 independent cultures [21]. In order to fix mutations, another aliquot of the mutator culture was spread on SC-agar plates containing 5-fluoroorotic acid monohydrate (Wako) to obtain demutatorized cells curing from YCplac33/polδMKII vector. The genomic DNA was prepared from the demutatorized cells using the procedure described in the following section.
2.3. EMS Mutagenesis
S. cerevisiae BY2961 strain cells were suspended in 0.1 M phosphate-buffered saline (PBS) (pH 7.0) containing 1.5, 2.0, 2.5, or 3.0% ethyl methanesulfonate (EMS) and were incubated at 30°C for 1 h to introduce chromosomal mutations. The cells were washed 3 times with 5% sodium thiosulfate, suspended in sterilized water, and spread on SC-agar plates containing L-canavanine sulfate salt (0.06 mg/mL) (Sigma) to identify CAN1 mutants. The mutation frequencies were calculated as described above. Another aliquot of the EMS-treated cell suspension was spread on a YPD-agar plate to isolate single clones. The genomic DNA was prepared from 5 single clones derived from the cells treated with 1.5% EMS using the procedure described in the following section.
2.4. Library Preparation for Illumina Sequencing
The genomic DNA from S. cerevisiae was extracted using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA). Each sequenced sample was prepared according to the Illumina protocols. Briefly, 3 μg of genomic DNA was fragmented to an average length of 200 bp by using the Covaris S2 system (Covaris, Woburn, MA, USA). The fragmented DNA was repaired, a single “A” nucleotide was ligated to the 3′ end, Illumina Index PE adapters (Illumina, San Diego, CA, USA) were ligated to the fragments, and the sample was size selected for a 300 bp product using E-Gel SizeSelect 2% (Invitrogen, Grand Island, NY, USA). The size-selected product was amplified by 18 cycles of PCR with the primers InPE1.0, InPE2.0, and the Index primer containing 6-nt barcodes (Illumina). The final product was validated using the Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA).
2.5. Sequencing and Data Analysis
The 11 barcoded libraries (the parental strain BY2961, 5 colonies from the mutator strain, and 5 colonies from the EMS-treated strain) were used for cluster generation in several multiplexed flow cell lanes in the Illumina Genome Analyzer II system. Ninety-one cycles of multiplexed paired-end sequencing was performed, running phi X 174 genomic DNA as a control in a separate lane of the flow cell. After the sequencing reactions were complete, Illumina analysis pipeline (CASAVA 1.6.0) was used to carry out image analysis, base calling, and quality score calibration. Reads were sorted by barcode and exported in the FASTQ format. The quality of each sequencing library was assessed by evaluating the quality score chart and the nucleotide distribution plot using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/).
Once the raw sequence data were curated, the reads of each sample were aligned to the S288c reference genome (http://www.yeastgenome.org/) using the BWA software (Ver. 0.5.1) with default parameters [22]. To avoid false positives and mutations from repetitive regions, we removed repetitive reads from the alignment files. We then used the SAMtools software (Ver. 0.1.9) [23] to produce the lists of mutations. To identify mutations that were produced by mutagenesis, we applied the following filtering criteria to the lists of mutations:
the coverage at the mismatch positions should be at least 10;
the variant is not present in the sequenced parental strain;
indels meet a SNP quality threshold of 50 and substitutions meet a SNP quality threshold of 20 (SAMtools assigns SNP quality, which is the Phred-scaled probability that the consensus is identical to the reference);
samples meet a mapping quality of 30 (SAMtools assigns Mapping quality, which is the Phred-scaled probability that the read alignment is wrong);
the percentage of reads showing the variant allele exceeds 90%.
A variant must pass this filter to be considered a mutation. Alignments of all mutations were inspected by Integrative Genomics Viewer (IGV) [24]. The lists of mutations were then annotated using COVA (comparison of variants and functional annotation) (http://sourceforge.net/projects/cova). COVA was specifically designed to annotate the large number of identified mutants using the Genbank annotation files.
3. Results
3.1. Determination of Mutation Frequencies
In this study, we evaluated the performance of the polδMKII mutator, compared with that of the commonly used chemical mutagen, EMS. To assess EMS efficiency, S. cerevisiae BY2961 cells were treated with different concentrations of EMS. The lethality and mutation frequencies of the canavanine resistant colonies are shown in Table 1. At an EMS concentration of 1.5%, the mutation frequency was approximately 18-fold higher than that in the control (untreated) strain. Above 2.0% EMS, the survival rate decreased with no increase in mutation frequency. Based on this result, we decided to use cells treated with 1.5% EMS for whole-genome sequencing.
Table 1.
Relationship between mutation frequency and survival after EMS treatment.
| EMS concentration (%) |
Mutation frequency of canavanine resistant (×10−7) | Fold elevation* | Survival (%) |
|---|---|---|---|
| 0.0 | 2 | 1 | 100 |
| 1.5 | 35 | 18 | 51 |
| 2.0 | 36 | 19 | 30 |
| 2.5 | 33 | 17 | 21 |
| 3.0 | 37 | 19 | 12 |
* Fold elevation is relative to untreated cells.
To assess the effectiveness of the mutator, we transformed the haploid BY2961 strain with a yeast expression plasmid, YCplac33/polδMKII, expressing the polδ mutant allele containing both the mutation to inactivate the proofreading activity (D321A and E323A) and the mutation to decrease the fidelity of replication (L612M). The mutator strain harboring the YCplac33/polδMKII plasmid will be referred to from here on as “mutator.” We determined the mutation frequency by resistance to canavanine. As summarized in Table 2, the mutation frequency of the mutator was approximately 132-fold higher than in the cells containing the empty vector. The forward mutation rate at the CAN1 (arginine permease) locus was calculated to be 7.9 × 10−6/cell division. These results show that the plasmid-generated mutated polδ protein effectively competes with the endogenous wild-type polδ protein that is produced from the chromosome, and the semidominant negative expression of mutated polδ was effective in introducing mutations. These results also demonstrate that the mutation frequency of the mutator was approximately 7 times higher than that of EMS.
Table 2.
Frequency of drug-resistant mutants in the mutator strains.
| Plasmids | Mutation frequency of canavanine resistant (×10−7) | Fold elevation* |
|---|---|---|
| YCplac33 | 3.70 | 1 |
| YCplac33/polδMKII | 486.7 ± 145.0# | 132 |
*Fold elevation is relative to empty vector.
#Mean ± standard deviation of 3 SC plates.
3.2. Whole-Genome Sequencing
To analyze the genome-wide mutation spectra of the 2 different mutagenesis methods, we implemented a parallel sequencing approach with the Illumina Solexa technology (GAII instrument). We sequenced the parental haploid strain BY2961, each of the 5 clones from the mutator strains, and each of the 5 clones from the EMS-treated strains under nonselective conditions. Sequencing reads were aligned to the S288c reference genome using the BWA software [22]. To avoid false positives due to mutations from repetitive regions, reads mapped to multiple locations were discarded, and only uniquely mapped reads were used for subsequent analysis.
In the current study, the average genomic coverage ranged from 32× to 87× (Table 3). On average, 94.18% of the S288c reference genome was covered with at least 1 uniquely mapped read at each base. Subsequently, we analyzed the data for 2 kinds of mutational events: single nucleotide variants (SNVs) and small insertions and deletions (Indels). Illumina sequencing found 6,766 genetic differences between our parental strain BY2961 and the S288c. Mutations induced by these mutagens were identified by subtracting the parental mutations. Sequence-processing details can be found in Section 2.
Table 3.
Sequencing and mapping statistics.
| Sample name | Number of mapped unique reads | % mapped reads | % genome covered* by unique reads | Average coverage by unique reads |
|---|---|---|---|---|
| BY2961 | 11,155,487 | 96.13 | 94.97 | 87.9× |
| EMS1 | 5,406,681 | 96.94 | 94.81 | 42.2× |
| EMS2 | 6,240,554 | 97.26 | 94.85 | 48.7× |
| EMS3 | 5,275,583 | 98.12 | 94.81 | 41.2× |
| EMS4 | 4,502,271 | 97.17 | 94.80 | 35.2× |
| EMS5 | 4,113,345 | 96.27 | 94.83 | 32.1× |
| Mutator 1 | 9,612,541 | 93.93 | 94.90 | 75.8× |
| Mutator 2 | 5,111,531 | 92.39 | 94.79 | 39.9× |
| Mutator 3 | 5,649,822 | 96.11 | 94.95 | 44.1× |
| Mutator 4 | 4,226,405 | 98.79 | 94.85 | 33.0× |
| Mutator 5 | 9,855,938 | 97.36 | 95.10 | 77.6× |
*Coverage is defined as the percentage of bases in the genome that have at least 1 uniquely mapped read at that position.
3.3. The Mutation Spectra of Mutator and EMS
We compared the average number of mutations between mutator strains and EMS-mutagenized strains (Figure 1). Mutator produced fewer SNVs than EMS (7.2 versus 55.8 per strain, resp., P < 0.05). Mutator and EMS produced few deletions (1.6 versus 2.8 per strain, resp.), as well as few insertions (0.2 versus 0.6 per strain, resp.). An average of 1.14 × 107 nucleotide sites fulfilled our criteria of read depth (≥10), with an average base-substitutional mutation rate estimate of EMS: 4.87 (SE = 1.34) × 10−6 per site, Mutator: 2.09 (0.55) × 10−8 per site per cell division (about 30 generations). The rate we calculated for the mutator is 100-fold higher than the previously reported spontaneous mutation rate, 3.3 (0.8) × 10−10, based on 454 analyses of 4 mutation-accumulation (MA)-lines [26]. The 2 mutagens generate mutations that are distributed similarly across the various gene features although the mutator did produce more SNVs within exons than did EMS (Figure 2).
Figure 1.
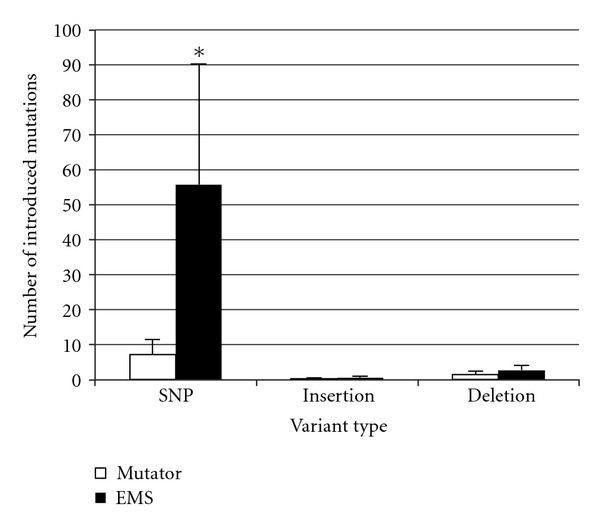
Average number of introduced mutations. By subtracting parental mutations from each mutagenized strain, we determined the number of mutations that were introduced by each mutagen. Bars represent mean ± standard error for 5 clones. *P < 0.05 versus mutator in a two-sample t-test.
Figure 2.
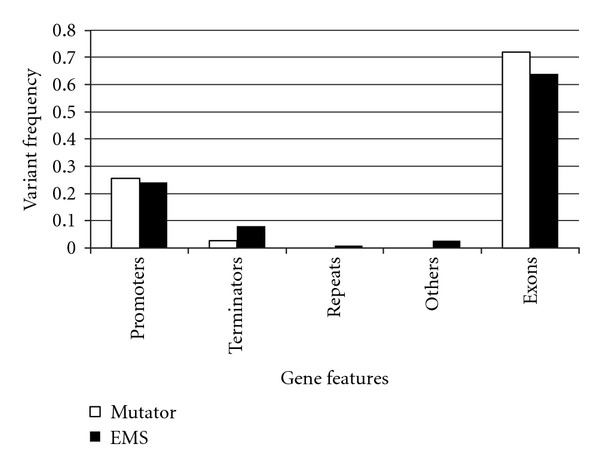
Relative frequency of SNVs affecting various gene features. The mutator and EMS generated mutations that were distributed similarly across the various gene features. The data for individual strains were combined according to the mutagen used. Promoters indicate the region 1 kb upstream of each gene. Terminators indicate the region 200 bp downstream of each gene.
The mutation spectra are shown in Figure 3(a). In the genome-wide profile, we found that the mutator primarily induced transversions (72%) while EMS primarily induced transitions (97%), well in accord with the known mutagenic specificity of EMS [12]. Similarly, the mutator primarily induced transversions (69%) in the nonsynonymous substitutions in exons (Figure 3(b)), similar to what has been seen in pol3-01 study using URA3 reporter gene [16]. EMS treatment was also in agreement with the genome-wide spectra, induced transitions with a prevalence of 98%.
Figure 3.
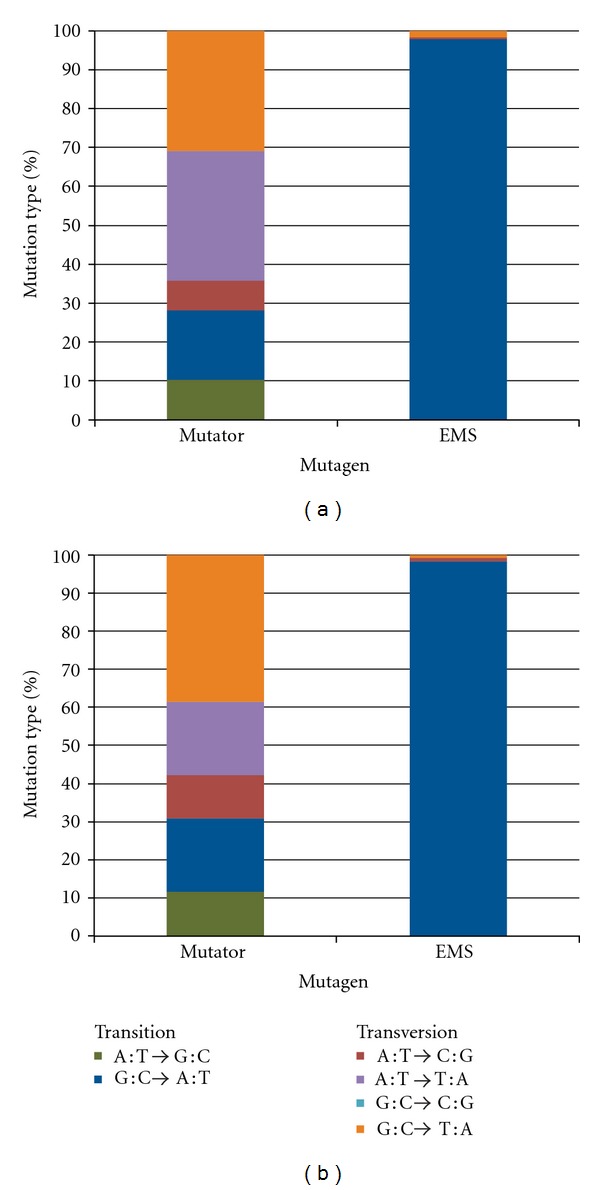
Relative frequency of transitions and transversions induced by polδ and EMS. The mutations spectra show the frequency of transitions and transversions generated by the mutator and EMS. The data for the individual strains were combined according to the mutagen used. The color key identifying the type of mutation is provided in the inset. Complementary mutations, such as A→C and T→G, are pooled. (a) Genome-wide profile; (b) Non-synonymous substitutions only.
3.4. Amino Acid Substitution Patterns
The mutation spectra of a given mutagenesis method influences the repertoire of changed amino acids at the protein level, and we were able to evaluate the amino acid substitution patterns generated by our 2 protocols (Table 4). Initially, we classified mutations into those that preserved the corresponding amino acid, changed the amino acid, or generated a stop codon. A clear difference was seen between mutator and EMS. Of the total mutations, the mutator changed the amino acid in approximately 85%, whereas EMS changed the amino acid in approximately 61%. The mutator also generated more stop codons than EMS (7% versus 2%, resp.). While mutator generated more changes to the first or second nucleotide of the codon, EMS generated changes in all 3 positions in approximately equal proportions.
Table 4.
Mutations at protein level.
| Mutator | EMS | |||
|---|---|---|---|---|
| n | % | n | % | |
| Total mutations | 28 | 100 | 201 | 100 |
| Preserved amino acids | 2 | 7.1 | 74 | 36.8 |
| Amino acid changes | 24 | 85.7 | 123 | 61.2 |
| Stop | 2 | 7.1 | 4 | 2.0 |
| Changes in codon letter | 28 | 100 | 201 | 100 |
| 1st | 11 | 39.3 | 64 | 31.8 |
| 2nd | 13 | 46.4 | 65 | 32.3 |
| 3rd | 4 | 14.3 | 72 | 35.8 |
| Impact of amino acid change | 24 | 100 | 123 | 100 |
| Conservativea | 4 | 16.7 | 57 | 46.3 |
| Nonconservative | 20 | 83.3 | 66 | 53.7 |
| Stop and Gly/Pro codons | 4 | 15.4 | 4 | 3.1 |
| Stop | 2 | 50.0 | 4 | 100.0 |
| Gly/Pro | 2 | 50.0 | 0 | 0.0 |
aConservative and nonconservative amino acid substitutions were defined according to the BLOSUM62 matrix [25].
Amino acid changes were classified into conservative and nonconservative substitutions, where a conservative substitution changed the encoded amino acid to a similar amino acid according to the criteria of the BLOSUM62 matrix [25]. Of the amino acid changes, mutator produced more nonconservative substitutions than EMS (83% and 53%). For the comparison of random mutagenesis methods, Wong et al. [27] proposed a useful structure indicator that takes into account Gly and Pro substitutions as well as stop codons. In our study, the mutator produced an equivalent number of Gly/Pro and stop codon substitutions, whereas EMS generated only stop codon substitutions.
4. Discussion
In this study, we evaluated the performance of a novel mutagenesis technique using error-prone proofreading-deficient and low-fidelity DNA polymerase δ by determining the mutation rate of the strain harboring the enzyme. We also analyzed the spectra of mutations across the entire S. cerevisiae genome and then assessed the diversity of mutation types at the amino acid level.
Proofreading-deficient polδ mutants, such as pol3-01 strain, and several low-fidelity polδ mutants, such as L612M, have been shown to present a mutator phenotype and to elevate the mutation rate [14–18]. We generated a BY2961 strain expressing a polδMKII mutator, polδ mutant allele containing a combination of mutations to inactivate the proofreading activity (D321A and E323A) and to decrease the fidelity of replication (L612M). This mutant allele acts as a strong mutator, as evidenced by the high frequency of spontaneous mutations (131-fold over control, compared to 18-fold for EMS strains). Vencatesan et al. reported the forward CAN1 mutation rates of polδ mutants as 1.5 × 10−6 in L612M, and 5.6 × 10−6 in pol3-01 [18]. These mutant strains were constructed by integrating the pol3-01 or pol3-L612M allele into the chromosomal POL3 gene by targeted integration, thereby disrupting the endogenous POL3 gene. In contrast, our mutator plasmid expressing the polδ mutant allele produced a mutation rate of 7.9 × 10−6, which shows a high mutation rate as well as chromosomal integration. The use of the polδMKII mutator plasmid allows the continued expression of the endogenous wild-type POL3 and provides for an efficient restoration of the wild-type mutation rate by curing the yeast strains of the mutator plasmid. Once the desired trait(s) has been selected, curing the cells from the mutator plasmid can stabilize the newly obtained phenotype.
In general, all random mutagenesis methods developed to date are biased toward transition mutations, although efforts have been made to overcome this [28]. While transition bias was observed in EMS, we actually observed transversion bias with the mutator (Figure 3(a)). Because of this, the mutator yielded a broader spectrum of nucleotide changes across the entire genome. The mutator was also biased toward transversions in the nonsynonymous substitutions (Figure 3(b)). For EMS, the spectrum of mutation events we observed is similar to what has been reported by others [12].
At the protein level, the amino acid substitution pattern differed between the mutator and EMS (Table 4). Mutations generated by the mutator resulted in amino acid substitutions more often than did mutations generated by EMS (85% versus 61%, resp.). Most of the substitutions made by the mutator were nonconservative, whereas only half of the substitutions made by EMS were nonconservative. In addition, the mutator generated more structure-disturbing amino acid changes (Gly/Pro). The transversion bias of non-synonymous substitutions by the mutator generates more diverse amino acid substitution patterns than does the transition bias of EMS.
Although the average base-substitution mutation rate of EMS was approximately 100 times higher than that of the mutator, the mutation frequency of the mutator was approximately 7 times higher than that of EMS. This gap between a higher apparent mutation frequency and fewer mutations may be explained by the higher proportion of amino acid changes and the diversity of amino acid substitutions by the mutator. This suggests one plausible explanation for the effectiveness of the disparity mutagenesis.
The disparity mutagenesis technique has been successfully applied to not only eukaryotic microorganisms such as S. cerevisiae [5, 7–9], S. pombe [9], and Ashbya gossypii [10], but also to prokaryotic microorganisms such as Escherichia coli [4] and Bradyrhizobium japonicum [6]. We believe that this novel mutagenesis technique has the potential to be applied to a wide variety of microorganisms.
Our present study has demonstrated that a proofreading-deficient and low-fidelity polδMKII mutator is a useful and efficient method for rapid strain improvement based on in vivo mutagenesis. It has been suggested that organisms may accelerate evolution by decreasing the fidelity of the proofreading activity of polδ in nature [29]; therefore, this mutator may also be useful for studying the acceleration of evolution.
Data Access
The raw reads used in this study are available on the DDBJ Sequence Read Archive (DRA) under accession DRA000522.
Acknowledgments
Authors thank Dr. M. Furusawa for critical reading of the manuscript and for useful suggestions. This study was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (S0801025).
Abbreviations
- EMS:
Ethyl methanesulfonate
- SNV:
Single nucleotide variant
- Indel:
Insertions and deletions
- SC:
Synthetic complete.
References
- 1.Labrou NE. Random mutagenesis methods for in vitro directed enzyme evolution. Current Protein and Peptide Science. 2010;11(1):91–100. doi: 10.2174/138920310790274617. [DOI] [PubMed] [Google Scholar]
- 2.Selifonova O, Valle F, Schellenberger V. Rapid evolution of novel traits in microorganisms. Applied and Environmental Microbiology. 2001;67(8):3645–3649. doi: 10.1128/AEM.67.8.3645-3649.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Furusawa M, Doi H. Promotion of evolution: disparity in the frequency of strand-specific misreading between the lagging and leading DNA strands enhances disproportionate accumulation of mutations. Journal of Theoretical Biology. 1992;157(1):127–133. doi: 10.1016/s0022-5193(05)80761-1. [DOI] [PubMed] [Google Scholar]
- 4.Tanabe K, Kondo T, Onodera Y, Furusawa M. A conspicuous adaptability to antibiotics in the Escherichia coli mutator strain, dnaQ49. FEMS Microbiology Letters. 1999;176(1):191–196. doi: 10.1111/j.1574-6968.1999.tb13661.x. [DOI] [PubMed] [Google Scholar]
- 5.Shimoda C, Itadani A, Sugino A, Furusawa M. Isolation of thermotolerant mutants by using proofreading-deficient DNA polymerase δ as an effective mutator in Saccharomyces cerevisiae . Genes and Genetic Systems. 2006;81(6):391–397. doi: 10.1266/ggs.81.391. [DOI] [PubMed] [Google Scholar]
- 6.Itakura M, Tabata K, Eda S, et al. Generation of Bradyrhizobium japonicum mutants with increased N2O reductase activity by selection after introduction of a mutated dnaQ gene. Applied and Environmental Microbiology. 2008;74(23):7258–7264. doi: 10.1128/AEM.01850-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abe H, Fujita Y, Chiba Y, Jigami Y, Nakayama KI. Upregulation of genes involved in gluconeogenesis and the glyoxylate cycle suppressed the drug sensitivity of an N-glycan-deficient Saccharomyces cerevisiae mutant. Bioscience, Biotechnology and Biochemistry. 2009;73(6):1398–1403. doi: 10.1271/bbb.90069. [DOI] [PubMed] [Google Scholar]
- 8.Abe H, Fujita Y, Takaoka Y, et al. Ethanol-tolerant Saccharomyces cerevisiae strains isolated under selective conditions by over-expression of a proofreading-deficient DNA polymerase δ . Journal of Bioscience and Bioengineering. 2009;108(3):199–204. doi: 10.1016/j.jbiosc.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 9.Abe H, Takaoka Y, Chiba Y, et al. Development of valuable yeast strains using a novel mutagenesis technique for the effective production of therapeutic glycoproteins. Glycobiology. 2009;19(4):428–436. doi: 10.1093/glycob/cwn157. [DOI] [PubMed] [Google Scholar]
- 10.Park EY, Ito Y, Nariyama M, et al. The improvement of riboflavin production in Ashbya gossypii via disparity mutagenesis and DNA microarray analysis. Applied Microbiology and Biotechnology. 2011;91(5):1315–1326. doi: 10.1007/s00253-011-3325-0. [DOI] [PubMed] [Google Scholar]
- 11.Matsuzawa T, Fujita Y, Tanaka N, Tohda H, Itadani A, Takegawa K. New insights into galactose metabolism by Schizosaccharomyces pombe: isolation and characterization of a galactose-assimilating mutant. Journal of Bioscience and Bioengineering. 2011;111(2):158–166. doi: 10.1016/j.jbiosc.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Flibotte S, Edgley ML, Chaudhry I, et al. Whole-genome profiling of mutagenesis in Caenorhabditis elegans . Genetics. 2010;185(2):431–441. doi: 10.1534/genetics.110.116616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukui T, Yamauchi K, Muroya T, et al. Distinct roles of DNA polymerases delta and epsilon at the replication fork in Xenopus egg extracts. Genes to Cells. 2004;9(3):179–191. doi: 10.1111/j.1356-9597.2004.00716.x. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Murphy KM, Kanevets U, Reha-Krantz LJ. Sensitivity to phosphonoacetic acid: a new phenotype to probe DNA polymerase δ in Saccharomyces cerevisiae . Genetics. 2005;170(2):569–580. doi: 10.1534/genetics.104.040295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrison A, Johnson AL, Johnston LH, Sugino A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae . The EMBO Journal. 1993;12(4):1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrison A, Sugino A. The 3′→5′exonucleases of both DNA polymerases δ and ε participate in correcting errors of DNA replication in Saccharomyces cerevisiae . Molecular and General Genetics. 1994;242(3):289–296. doi: 10.1007/BF00280418. [DOI] [PubMed] [Google Scholar]
- 17.McElhinny SAN, Stith CM, Burgers PMJ, Kunkel TA. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase. The Journal of Biological Chemistry. 2007;282(4):2324–2332. doi: 10.1074/jbc.M609591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkatesan RN, Hsu JJ, Lawrence NA, Preston BD, Loeb LA. Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase δ . The Journal of Biological Chemistry. 2006;281(7):4486–4494. doi: 10.1074/jbc.M510245200. [DOI] [PubMed] [Google Scholar]
- 19.Timmermann B, Jarolim S, Russmayer H, et al. A new dominant peroxiredoxin allele identified by whole-genome re-sequencing of random mutagenized yeast causes oxidant-resistance and premature aging. Aging. 2010;2(8):475–486. doi: 10.18632/aging.100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74(2):527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 21.Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. Journal of Genetics. 1949;49(3):264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nature Biotechnology. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lynch M, Sung W, Morris K, et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(27):9272–9277. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong TS, Roccatano D, Zacharias M, Schwaneberg U. A statistical analysis of random mutagenesis methods used for directed protein evolution. Journal of Molecular Biology. 2006;355(4):858–871. doi: 10.1016/j.jmb.2005.10.082. [DOI] [PubMed] [Google Scholar]
- 28.Rasila TS, Pajunen MI, Savilahti H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Analytical Biochemistry. 2009;388(1):71–80. doi: 10.1016/j.ab.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Furusawa M. Implications of double-stranded DNA structure for development, cancer and evolution. Open Journal of Genetics. 2011;01(03):78–87. [Google Scholar]