

Abstract
Down syndrome (DS) is the most common genetic cause of mental disability. Based on the homology of Hsa21 and the murine chromosomes Mmu16, Mmu17 and Mmu10, several mouse models of DS have been developed. The most commonly used model, the Ts65Dn mouse, has been widely used to investigate the neural mechanisms underlying the mental disabilities seen in DS individuals. A wide array of neuromorphological alterations appears to compromise cognitive performance in trisomic mice. Enhanced inhibition due to alterations in GABAA-mediated transmission and disturbances in the glutamatergic, noradrenergic and cholinergic systems, among others, has also been demonstrated. DS cognitive dysfunction caused by neurodevelopmental alterations is worsened in later life stages by neurodegenerative processes. A number of pharmacological therapies have been shown to partially restore morphological anomalies concomitantly with cognition in these mice. In conclusion, the use of mouse models is enormously effective in the study of the neurobiological substrates of mental disabilities in DS and in the testing of therapies that rescue these alterations. These studies provide the basis for developing clinical trials in DS individuals and sustain the hope that some of these drugs will be useful in rescuing mental disabilities in DS individuals.
1. Introduction
Trisomy 21, or Down syndrome (DS), is the most common genetic cause of intellectual disability. It affects 1 in 850–1000 infants [1] and is characterized by a number of phenotypes, including cardiovascular, skeletal, and motor alterations. However, the most prominent feature of DS is an intellectual disability that affects 100% of the individuals with this condition. DS individuals typically display an average Intelligence Quotient (IQ) of 50 (ranging from 30 to 70) [2] and show an array of altered cognitive and behavioral phenotypes, including the incomplete and delayed acquisition of motor [3], linguistic [3, 4] and visual-spatial abilities [3], impairments in learning and memory [3–6], and neurobehavioral disorders [4] and have a higher risk of developing Alzheimer-like dementia by the age of 40 [7, 8]. Great interindividual variability, however, is present in both the nature and the intensity of all of these conditions.
In recent years, the question of how trisomy of Hsa21 leads to this set of phenotypes has been a matter of debate. Two hypotheses have been proposed to account for this phenomenon: the “amplified developmental instability” hypothesis [9] and the “gene-dosage effect” hypothesis [10–12]. The first hypothesis proposes that trisomy of Hsa21 causes a general alteration in developmental homeostasis that leads to the DS phenotypes; the “gene-dosage effect” proposal maintains that these alterations result from the overexpression of a subset of genes and their encoded proteins.
The analysis of DS cases resulting from partial trisomies of Hsa21 and the development of a number of mouse models of this condition have provided insight on the causative role of dosage-sensitive genes on DS phenotypes. These studies have yielded evidence that support both theories; while the role of single dosage-sensitive genes on different phenotypes has been confirmed, research has also demonstrated that many of these DS features are due to the complex effects of multiple Hsa21 genes (see [13]) and their interactions with genes of other chromosomes.
To identify the biological mechanisms underlying different pathologies and to evaluate the efficacy of novel therapies, thousands of animal models of human disorders have been developed. For an animal model of a human disorder to be considered valid, it has to satisfy three criteria: construct, face, and predictive validity [14, 15]. Construct validity relates to the similarity between the etiology of the human and the animal disorder (e.g., in the case of mouse models of DS, the triplication of Hsa21 genes). Face validity refers to how well the model mimics the molecular, cellular, physiological, and behavioral phenotypes observed in humans. Predictive validity requires that the new knowledge obtained in the animal model makes accurate predictions of what will be found in the human condition. This validity is particularly important for unraveling the neurobiological causes of the cognitive deficits found in DS that cannot be assessed in humans for ethical or practical reasons and for developing and testing new therapies.
In the following sections, we will summarize (1) the similarities between the genetic overdose of various mouse models of DS and human trisomy 21; (2) the concordance between the behavioral, neuromorphological, and neurochemical phenotypes of DS mouse models and the human condition; (3) the knowledge obtained in these animals about the neurobiology of DS that have yielded the development and analysis of several therapeutic strategies that could potentially be used to attenuate cognitive impairments in DS individuals.
2. Mouse Models of Down Syndrome
The long arm of Hsa21 contains approximately 552 genes, 166 of which are orthologous to genes localized in syntenic regions of three mouse chromosomes: Mmu16 (110 orthologous genes), Mmu17 (19 orthologous genes), and Mmu10 (37 orthologous genes) [16]. Based on these homologies, several mouse models that are trisomic for different sets of Hsa21 genes have been developed (Figure 1). The first attempt to create a mouse model of DS was to develop a mouse, named Ts16, which was trisomic for the entire Mmu16 [17]. However, this model does not resemble the DS aneuploidy because Mmu16 presents syntenies with regions of Hsa3, Hsa8, Hsa16, and Hsa21; thus, it has triplicated many genes that are not in trisomy in DS and, consequently, does not exhibit good construct validity. Furthermore, Ts16 embryos die in utero, making it impossible to test phenotypes in young and adult mice, thus restricting the face and predictive validities of this model.
Figure 1.
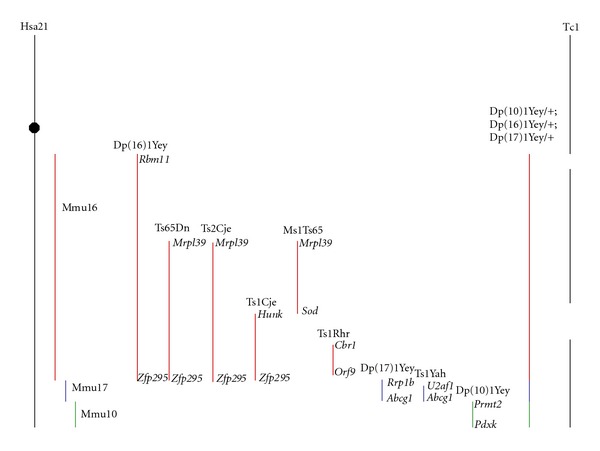
Schematic representation of Hsa21 and syntenic regions of Mmu16, Mmu17, and Mmu10 and the different mouse models trisomic for different sets of genes orthologous to those of Hsa21. The flanking genes found at the boundaries of the triplicated region in each model are written in italics. Modified from [3, 137].
The next approach adopted was the generation of mouse models with partial trisomies of sets of Mmu16 genes orthologous to those found in Hsa21. In 1993, Davisson et al. [18] created the Ts65Dn mouse, which is now the most commonly used and best characterized model of DS. This mouse bears a partial trisomy of a segment of Mmu16, extending from the Mrp139 to the Znf295 genes, and contains approximately 92 genes orthologous to Hsa21 genes [16]. Additionally, Ts65Dn mice also carry a trisomy of ~10 Mb of Mmu17 containing 60 genes nonhomologous to Hsa21 [19]. Thus, this model does not have perfect construct validity because many of the orthologous genes found in Hsa21 are not triplicated in this mouse and because a set of genes not triplicated in DS are in trisomy in this model. However, as detailed below, the Ts65Dn mouse is currently the model that displays the best face validity. Additionally, in some cases, DS results from a partial trisomy of different regions of Hsa21, and there is strong evidence that some regions of this chromosome contribute more to the DS phenotype [12, 20]. Moreover, according to the “gene-dosage effect” hypothesis, different DS phenotypes are determined by the increased dosage of only a subset of genes. A comparison of the phenotypes in Ts65Dn mice with those of other partial trisomic models (see below) suggests that the set of genes triplicated in this model contribute to several DS phenotypes, including cognitive and neuroanatomical impairments (Tables 1 and 2).
Table 1.
Behavioural and cognitive alterations in DS and in the different DS mouse models.
| Trisomy | Hsa21 | Segment of Mmu16 | Segment of Mmu17 | Segment of Mmu10 | Segment of Mmu16, Mmu17, and Mmu10 | Hsa21 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DS | Dp(16)1Yey/+ | Ts65Dn | Ts2Cje | Ts1Cje | Ms1Ts65 | Ts1Rhr | Dep(17)1Yey/+ | Ts1Yah | Dp(10)1Yey/+ | Dp(10)1Yey/+; Dp(16)1Yey/+; Dep(17)1Yey/+ Yu et al., 2010 | Tc1 | |
| Motor skills | Delayed acquisition | Delayed acquisition | ||||||||||
|
| ||||||||||||
| Motor coordination | Impaired | Impaired | Impaired | |||||||||
|
| ||||||||||||
| Activity and attention | Reduced attention | Hyperactivity and reduced attention | Normal activity | Normal activity | Normal activity | Increased spontaneous activity | ||||||
|
| ||||||||||||
| Context discrimination | Impaired | Impaired | Unchanged | Unchanged | Impaired | |||||||
|
| ||||||||||||
| Spatial learning and memory | Impaired | Impaired | Impaired | Impaired | Impaired | Unchanged | Enhanced | Unchanged | Impaired | Impaired | ||
|
| ||||||||||||
| Working and reference memory | Impaired | Impaired | Impaired | |||||||||
|
| ||||||||||||
| Novel object recognition | Impaired | Impaired | Impaired | |||||||||
|
| ||||||||||||
| Operant conditioning | Impaired | |||||||||||
Table 2.
Neuromorphological and electrophysiological alterations DS and DS mouse models.
| Trisomy | Hsa21 | Segment of Mmu16 | Segment of Mmu17 | Segment of Mmu10 | Segment of Mmu16, Mmu17, and Mmu10 | Hsa21 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DS | Dp(16)1Yey/+ | Ts65Dn | Ts2Cje | Ts1Cje | Ms1Ts65 | Ts1Rhr | Dep(17)1Yey/+ | Ts1Yah | Dp(10)1Yey/+ | Dp(10)1Yey/+; Dp(16)1Yey/+; Dep(17)1Yey/+ Yu et al., 2010 | Tc1 | |
| Brain volume | Reduced | Reduced | Reduced during the embryonic period | Reduced during the embryonic period | Reduced | Reduced at 4 months of age | ||||||
|
| ||||||||||||
| Neuronal density | Reduced | Reduced | Reduced | Not affected | ||||||||
|
| ||||||||||||
| Cerebellar volume | Reduced | Reduced | Reduced | Not affected | Reduced | Reduced | ||||||
|
| ||||||||||||
| Cerebellar neuronal density | Reduced | Reduced | Reduced | Reduced | ||||||||
|
| ||||||||||||
| Neurogenesis | (i) Impaired neural precursor proliferation | (i) Impaired neural precursor proliferation | (i) Impaired neural precursor proliferation | (i) Impaired neural precursor proliferation | ||||||||
| (ii) Slowing of the cell cycle | (ii) Slowing of the cell cycle | (ii) Impaired neurodifferentiation | ||||||||||
| (iii) Impaired neurodifferentiation | (iii) Impaired neurodifferentiation | (iii) Impaired cerebellar neurogenesis | ||||||||||
| (iv) Impaired cerebellar neurogenesis | (iv) Impaired cerebellar neurogenesis | |||||||||||
|
| ||||||||||||
| Dendrites and dendritic spines | (i) Impaired morphology | (i) Impaired morphology | (i) Impaired morphology | (i) Impaired morphology | (i) Impaired morphology | |||||||
| (ii) Reduced density | (ii) Reduced density | (ii) Reduced density | (ii) Reduced density | (ii) Reduced density | ||||||||
|
| ||||||||||||
| Synaptic density | (i) Reduced | (i) Size of presynaptic boutons and average length of synaptic clefts are increased | ||||||||||
| (ii) Size of presynaptic boutons and average length of synaptic clefts are increased | ||||||||||||
|
| ||||||||||||
| Inhibition | (i) Increased number of inhibitory synapses | (i) Redistribution of inhibitory synapses | ||||||||||
| (ii) Decreased number of excitatory synapses | ||||||||||||
| (iii) Redistribution of inhibitory synapses | ||||||||||||
| (iv) Increased number of GABAergic interneurons | ||||||||||||
|
| ||||||||||||
| Electrophysiology | (i) EEG abnormalities | (i) Impaired hippocampal LTP | (i) Impaired hippocampal LTP | (i) Impaired hippocampal LTP | (i) Impaired hippocampal LTP | (i) Enhanced hippocampal LTP | (i) Unchanged LTP | (i) Impaired hippocampal LTP | ||||
| (ii) EEG coherence differences | ||||||||||||
| (iii) Alterations in event-related potentials | ||||||||||||
The Ts2Cje model carries the same segment of Mmu16 triplicated in the Ts65Dn mouse but is translocated to chromosome 12 [21]. Although this model also shows some of the DS-relevant phenotypes found in the Ts65Dn mouse, it has not been fully characterized.
Several other segmental trisomic models of different segments of Mmu16, 17, and 10 have been created. In the late 90s, Sago and coworkers generated two mouse models with the triplication of two different regions of Mmu16: the Ts1Cje mouse, which presents a trisomy of 81 genes localized in the region of Mmu16 that extend from Sod1 to Znf295 [22]; the Ms1Ts65 mouse, which has a partial trisomy of 33 genes mapped in the region of Hsa21 that extend from App to Sod1 [23]. In addition, to evaluate the influence of the so-called Down syndrome critical region (DSCR), Olson et al. [24] developed the Ts1Rhr mouse, a model that is trisomic for the Cbr1-Orf9 region of Mmu16, which contains 33 genes. Finally, Li et al. [25] generated a mouse trisomic for the complete Hsa21 syntenic region on Mmu16 (between Lipi and Zfp295) containing 110 orthologous genes, the Dp(16)1Yey/+ mouse [16].
To model the trisomy of Hsa21 orthologous genes located in Mmu17, two mouse models have been created: the Ts1Yah mouse, trisomic for 12 genes in the Mmu17 region, syntenic to the subtelomeric region of Hsa21 [26] and the Dep(17)1Yey/+ mouse which is trisomic for the entire Hsa21 syntenic region on Mmu17 that contains 19 orthologous genes [16, 27, 28]. Additionally, Vacík et al. [29] created the Ts43H model, a mouse that is trisomic for 30 Mb of Mmu17 containing over 300 genes but only ~20 of them are orthologs of Hsa21 genes. Therefore, this is not a valid DS mouse model.
The last segmental trisomic mouse generated is a mouse that models the trisomy of Hsa21 orthologous genes located in Mmu10. The Dp(10)1Yey mouse is trisomic for the region of Mmu10 syntenic to the distal part of Hsa21 containing 37 orthologous genes [16, 27].
After the tree partial trisomic models for all the Hsa21 syntenic regions on Mmu10 (Dp(10)1Yey/+), Mmu16 (Dp(16)1Yey/+) and Mmu17 (Dep(17)1Yey/+) were established, Yu et al. [27] cross-breeded them to generate a mouse that is trisomic for the entire Hsa21 syntenic regions on Mmu10, Mmu16, and Mmu17 chromosomes: the Dp(10)1Yey/+Dp(16)1Yey/+; Dep(17)1Yey/+ mouse. This is a promising new model with excellent construct and face validities, as it shows several DS phenotypes [27].
The Tc1 model is a mouse in which the entire human Hsa21 has been triplicated [30]. This mouse shows different DS-relevant phenotypes [30–32], although its characterization is not as complete as those performed on the different segmental trisomic models. However, the Tc1 mouse presents variable levels of mosaicism of the extra chromosome in different tissues, confounding the analysis of phenotypic consequences. In addition, although the starting material was an intact Hsa21, delections occurred and this mouse has triplicated ~83% of the genes on Hsa21.
Finally, to study the role of particular genes in the DS phenotypes, a number of mouse models with the triplication of single genes and trisomic mice in which the expression of only one of the genes triplicated in DS have been normalized have been created (see [13]).
3. Cognitive and Behavioral Deficits in Mouse Models of DS
This section describes the similarities between the cognitive and behavioral disturbances found in various mouse models of DS compared to the human condition (Table 1).
Motor dysfunction is a hallmark of DS. Hypotonia, hyporeflexia, reduced muscular strength, disturbances in striate muscle control, and delays in the acquisition of fine and gross motor skills are found in DS individuals from early childhood [33–35].
Ts65Dn mice are not impaired in sensorimotor abilities such as forelimb strength, postural skills, equilibrium, and climbing [36, 37]. However, these mice show poorer balance and motor coordination [38]. Ts65Dn mice are hyperactive in the dark [36, 39, 40] and in other settings that provoke caution and lack of movement in normal animals, such as in open-field and plus-maze tests [36, 41–43]. This hyperactivity has been proposed to be due to a failure to inhibit activity or as a deficit in the ability to attend to relevant stimuli [44–46]. Attention deficits in Ts65Dn mice have been confirmed by Driscoll et al. [47]. Unlike Ts65Dn, the other models that are partially trisomic for different segments of Mmu16 are not hyperactive. Ts1Cje and Ms1Ts65 mice do not exhibit altered spontaneous activity [23], and Ts1Rhr mice display normal performances in the open-field test [48]. However, Tc1 mice present higher spontaneous locomotor activity, reduced ability to habituate to new environments, and several deficits of motor coordination and balance in the rotarod and static-rod tests [32].
Similar to DS [6], Ts65Dn mice are impaired in performing hippocampal-dependent tasks, such as spontaneous alterations in the T-maze, contextual fear conditioning, novel object recognition [49, 50], and spatial memory in the radial arm maze [51–54] and the Morris water maze tests [23, 40, 42, 45, 55]. Ts65Dn mice also show deficits in learning an operant conditioning paradigm [56].
Ts1Cje and Ms1Ts65 mice display poor performance in hippocampal-dependent tasks such as the T-maze [52] and the Morris water maze [22, 23]. Ts1Rhr mice are impaired in spontaneous alterations in the T-maze task [48] and show altered long-term memory in the novel object recognition test [48] but not in the Morris water maze [57]. Dp(16)1Yey/+ mice showed impaired performance in the Morris water maze and in the contextual fear conditioning test [27].
Regarding the two mouse models trisomic for segments of Mmu17, Ts1Yah mice are impaired in the novel object recognition and Y-maze test, but their performance in the Morris water maze is enhanced [26]; however, Dp(17)1Yey+ do not show alterations in performance in the later task or in the contextual fear conditioning test [28].
The Dp(10)1Yey/+Dp(16)1Yey/+; Dep(17)1Yey/+ mouse is impaired in the Morris water maze and in the contextual fear conditioning test [27]. Finally, Tc1 mice show altered performance in the novel object recognition test but not in the T-maze [30].
In summary, most of the above-mentioned mouse models show characteristic DS cognitive and behavioral phenotypes, although they differ in the degree of impairment.
4. Neuromorphological Alterations in DS and DS Mouse Models
Several mechanisms have been proposed to be the neurobiological correlates of intellectual disability in DS, including altered neurogenesis, hypocellularity, altered synaptic development, increased inhibition, and neurodegeneration. Table 2 summarizes the main neuromorphological and electrophysiological alterations found in DS individuals and in the different mouse models of DS, and Table 3 describes the outcome of several studies that tested the ability of different therapeutical approaches to rescue different phenotypic alterations in the Ts65Dn mouse and in DS individuals.
Table 3.
Therapeutical approaches tested in DS and in the Ts65Dn mouse.
| Therapies | DS | Ts65Dn |
|---|---|---|
| Targeting neurogenesis | ||
| (i) Fluoxetine (ii) Lithium (iii) SAG 1.1 (iv) Environmental enrichment |
(i) Not tested (ii) Not tested (iii) Not tested (iv) Improves cognition |
(i) Restores BDNF levels, neurogenesis, dendritic maturation and branching and cognition (ii) Restores neurogenesis (iii) Restores neurogenesis and cognition (iv) Restores neurogenesis, improves cognition, no effect on dendritic arborization in TS mice |
|
| ||
| Targeting inhibition | ||
| (i) Picrotoxin (ii) Bilobalide (iii) PTZ (iv) α5IA |
(i) Not tested (ii) Not tested (iii) Not tested (iv) Not tested |
(i) Rescues LTP and cognition (ii) Rescues LTP and cognition (iii) Rescues LTP and cognition (iv) Rescues cognition |
|
| ||
| Targeting NMDA receptor functioning | ||
| (i) Memantine | (i) No effect | (i) Improves cognition, reduces APP levels |
|
| ||
| Targeting NA functioning | ||
| (i) L-DOPS (ii) Xamoterol |
(i) Not tested (ii) Not tested |
(i) Rescues cognition (ii) Rescues cognition |
|
| ||
| Targeting neurotrophins | ||
| (i) NGF | (i) Not tested | (i) Rescues BFCNs altered size and number |
| (ii) Peptide 6 (CNFT) | (ii) Not tested | (ii) Improves learning and memory, enhanced neurogenesis |
| (iii) EGCG | (iii) Not tested | (iii) Rescued BDNF levels, brain size, and LTP in the Dyr1A Tg mouse |
| (iv) Neurotrophin | (iv) Not tested | (iv) Prevents decline in BDNF expression, improves cognition |
|
| ||
| Targeting inflammatory activity | ||
| (i) Minocycline | (i) Not tested | (i) Inhibits microglia activation, prevents neuron loss, improves working, and reference memory |
|
| ||
| Neuropeptides | ||
| (i) NAP and SAL | (i) Not tested | (i) Rescues acquisition of neurodevelopmental milestones, increases ADNP levels and rescued ADNP levels |
|
| ||
| Targeting oxidative stress: antioxidants | ||
| (i) SGS111 (ii) Vitamin E (iii) Combined antioxidant supplementation (iv) Folinic acid/ folinic acid + antioxidants |
(i) Not tested (ii) No effect (iii) No effect (iv) Beneficial effect on developmental age/no effect |
(i) No effect on cognition (ii) Reduced oxidative stress, improved cognitive performance, reduced cholinergic neuron pathology, and increased cell density in the DG |
|
| ||
| Estrogens | (i) Not tested | (i) In females enhanced cognition, increased the size and number of cholinergic neurons and NGF levels |
|
| ||
| Targeting AD neuropathology | ||
| (i) DAPT (ii) Donepezil |
(i) Not tested (ii) No effect/small effect |
(i) Reduced beta-amyloid levels, rescued cognition (ii) No effect |
4.1. Reduced Volume and Hypocellularity
In DS individuals, the volume of the brain is reduced, beginning at early developmental stages [57–62]; in adults, the reduction in size reaches approximately 20% [63, 64], and, during aging, neurodegeneration further deteriorates this scenario [65].
The brain volume of Ts65Dn, Ts1Cje, and Ts2Cje mice is also reduced during the embryonic period, but not after birth or during adulthood [39, 52, 66–68]. Ms1Rhr mice also show reduced brain volume [66], as does the Ts1Rhr mouse, at 4 months of age [69] but not at later stages [48]. Thus, most DS mouse models do not show changes in total brain volume during adulthood.
A number of studies demonstrated that brain areas are dissimilarly affected. Reduced volumes of the hippocampus, entorhinal, frontal, prefrontal, and temporal cortices, amygdala, cerebellum, brain stem nuclei, and mammillary bodies of the hypothalamus have been reported in children and adults with DS [63, 64, 70–78].
Consistently, size and anatomical alterations have been found in some brain regions of trisomic mice. The hippocampus and the cerebellum appear to be the most affected structures. Spatial learning is known to depend on the functional integrity of the hippocampus, a structure that plays a key role in information encoding and retrieving in the CNS [79, 80]. In Ts65Dn mice, the hippocampal granule cell layer and hilus show reduced volume [54, 81–83]. The hippocampal area of Ts1Cje mouse is not reduced [52], and the Ts1Rhr mice show greater volume of the posterior hippocampus [48].
In DS individuals, cell density is not compromised in early gestation [60, 62, 84], but neuron number is reduced in late gestation (after weeks 19–23). Indeed, the number of neurons in the hippocampus, parahippocampal gyrus, cerebellum and neocortex of fetuses [60, 61, 85], and newborn DS individuals [60, 62, 84] is reduced. Hypocellularity persists in different areas of the brain [86] and cerebellum [37] of children and adults with DS.
Ts65Dn mice show reduced cell density during prenatal (E 18.5) and early postnatal stages in the neocortex (P8) [67]. At 1 month of age, Ts65Dn mice display a normal number of neurons in the hippocampal CA1–CA3 areas [81]. However, CA1 neuron density is lower in older (17-18 month) Ts65Dn mice [87, 88]. The dentate gyrus (DG) of Ts65Dn mice has fewer granule cells at all examined ages [54, 81, 87, 89, 90]. However, in 18-month-old Ts1Cje mice, the thickness of the granule cell layer and molecular layer of the DG is not affected [52].
The cerebellum has been traditionally implicated in motor coordination, but there is increasing evidence for the role of this structure in higher cognitive processes, including attention, cognitive flexibility, and memory [91]. Consistent with what is found in DS, the volume of the cerebellum is significantly reduced in Ts65Dn, Ts1Cje, and Ts1Rhr mice [37, 48, 66, 92]. However, Ms1Ts65 mice do not show changes in cerebellar volume [92].
As expected from the reduced cerebellar volume of DS and trisomic mice, the cerebellum of Ts65Dn mice has a lower number of granule and Purkinje cells from early postnatal stages to adulthood [37, 93, 94]. Ts1Cje, Ms1Rs65, and Tc1 mice also show a decrease in cerebellar granule cell density [30, 92].
One of the anatomical substrates of learning and attention is the septohippocampal cholinergic system [95, 96]. In the aging DS brain, there is a loss of cholinergic neurons [97, 98]. Several studies have demonstrated an age-dependent decline in cholinergic markers in Ts65Dn mice. Starting at 6 months of age, Ts65Dn mice basal forebrain cholinergic neurons show a loss of the cholinergic phenotype. These neurons show a decrease in choline acetyltransferase (ChAT) and p75NGFR, a neurotrophin receptor localized in basal forebrain cholinergic neurons, immunoreactivity. Furthermore, there is a decrease in the size of these cholinergic neurons cell bodies [82, 99–102].
In conclusion, persistent hypocellularity is one cause of intellectual disability in the trisomic condition. The following sections will describe the evidence showing that these neuroanatomical alterations could be caused by impaired cell proliferation, increased apoptosis and/or neurodegeneration.
4.2. Neurogenesis
4.2.1. Neurogenesis in Trisomic States
Neurogenesis is severely compromised in DS from early developmental stages. Impaired neuronal precursor proliferation, slowing of the cell cycle, and altered differentiation are thought to account for altered neurogenesis.
In DS fetuses, a reduced number of dividing cells is found in the dentate gyrus (DG) and lateral ventricle [60, 89]. Reduced proliferation of neural precursor cells is also found in mouse models of DS. In Ts65Dn mice, reduced neural precursor proliferation is found in the neocortical ventricular zone (VZ) during embryonic stages [67]. However, in these mice, a larger progenitor population of inhibitory neurons has been found in the embryonic medial ganglionic eminence [103].
Reductions in neural progenitor cells and neuroblasts, leading to altered neurogenesis, in the embryonic neocortex and subventricular zone (SVZ) of Ts1Cje and Ts2Cje mice have also been reported [68, 104, 105].
It has been proposed that the extra copy of Hsa21 in DS delays the mitotic cell cycle of neuronal precursors, thereby affecting cell proliferation. Accordingly, a slowing of the cell cycle in different trisomic conditions has been demonstrated. In Ts65Dn mice, the cell cycle is extended during embryonic stages in CA3 [67] and in early postnatal life in the DG [89]. The expression of two regulators of the G2/M and G1/S transitions, Ccnb1 and Skp2, is decreased in newbornTs65Dn cerebellar granule cell precursors [82]. Hewitt et al. [104] also observed dysregulated expression of genes involved in cell cycle control in Ts1Cje mice.
Impaired differentiation also appears to contribute to the smaller number of neurons in DS brains [106, 107]. Ts1Cje neural progenitors have a reduced capacity to differentiate into neurons [104, 105].
Adult hippocampal neurogenesis has been demonstrated in many species, including rodents [108–111]. During the entire life span, cell proliferation takes place in the SVZ and in the subgranular zone (SGZ) of the DG, where a pool of multipotent progenitor cells is located. In the SGZ, newborn neurons migrate into the granular cell layer (GCL) and establish functional connections in the dentate molecular region, where they receive excitatory synaptic input from perforant path afferents [111]. Increasing evidence indicates that adult hippocampal neurogenesis is implicated in the establishment of long-term potentiation (LTP) and has a role in hippocampal-dependent learning and memory [112–114]. Interestingly, we have showed a negative correlation between performance in the Morris water maze and the number of proliferating cells in the DG of Ts65Dn mice and euploid littermates (Figure 2).
Figure 2.
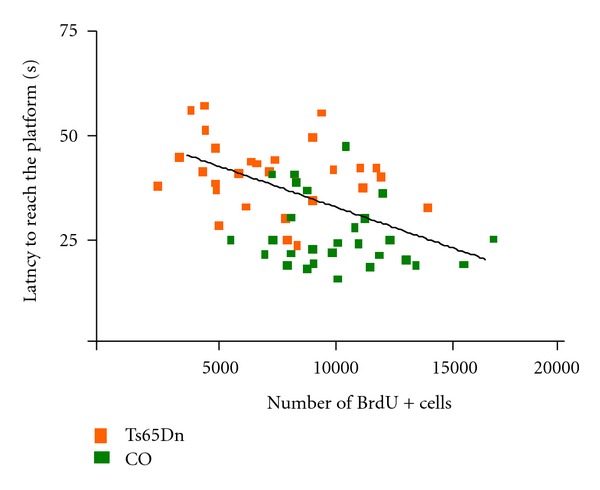
Correlation between performance in the Morris water maze (mean latency to reach the platform) and the number of BrdU+ cells in the DG of Ts65Dn and euploid littermates (Rueda et al., unpublished results; Pearson's R: −0.4647; P < 0.001).
In Ts65Dn mice, cell proliferation in the SVZ is reduced from birth to adulthood [54, 114, 115]. In the DG, proliferation impairments have also been reported in newborn [81, 89], young [54, 116], middle-aged [117], and aged [83] Ts65Dn mice. Adult (3-month-old) Ts1Cje and Ts2Cje mice also present severe neurogenesis reduction in the SGZ [118].
Cerebellar neurogenesis is also affected by trisomy. DS fetuses show reduced neurogenesis in the external granular layer (EGL) of the cerebellum and in the VZ [61]. Newborn (P0, P2, and P6) Ts65Dn mice also show reduced proliferation of cerebellar granule cell precursors in the EGL [93, 119]; their cell cycle is also dramatically slowed, and the G1 and G2 phases are the most affected [119]. One-month-old Ts65Dn mice show reduced proliferation of the granule neurons of the internal granular layer [94]. In Ts1Cje mice, proliferation of cerebellar granule cells is reduced at birth but normal at postnatal days 3 and 7 [120]. Differentiation is also altered in the cerebellum of Ts65Dn mice because a smaller percentage of cells acquire a neuronal phenotype [119]. Reductions in cerebellar neurogenesis in Ts65Dn mice seem to be due to the decreased response of granule cell precursors to the mitogenic factor Sonic hedgehog (Shh) [93].
It can be concluded that neurogenesis impairment, due to reductions in neural precursors, cell cycle timing and differentiation, is a hallmark of trisomic conditions from prenatal to adult stages. This altered proliferation is likely to be one of the mechanisms responsible for the widespread hypocellularity leading to altered synaptogenesis, connectivity, synaptic plasticity, and cognitive disabilities.
4.2.2. Trisomic Genes and Neurogenesis Impairment
A number of trisomic genes in DS have been proposed to play a role in the proliferation impairment found in this condition. One of the genes overexpressed in the DS brain is DYRK1A (dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A), an orthologous gene to the Drosophila gene minibrain [121]. DYRK1A codes for a serine-threonine protein kinase [122], which has important transcription factors as substrates and, consequently, appears to be implicated in multiple biological pathways. DYRK1A is essential for normal postembryonic neurogenesis [123, 124]. This gene plays a role in neuronal progenitor proliferation, neurogenesis, and neurodifferentiation, and regulates neuronal development, brain volume and cellular density in different brain areas [124–127]. The DYRK1A protein also modulates CREB (cAMP response element-binding protein) activity, which participates in synaptic plasticity signal transduction pathways [126]. Overexpression of DYRK1A inhibits proliferation, induces premature differentiation of neural progenitor cells in the developing mouse cerebral cortex, and impairs G1/G0-S phase transition in rat hippocampal progenitor cells [128, 129]. Recent studies have suggested that the DYRK1A gene could be a potential therapeutic target in DS because the inhibition of DYRK1A expression rescued several DS relevant phenotypes. Ortiz-Abalia et al. [130] demonstrated that the normalization of the Dyrk1A expression in the striatum of TgDyrk1A mice, through the injection of an adeno-associated virus type 2-mediated Dyrk1A RNA inhibitor (AAVshDyrk1A), rescued motor alterations in these animals.
The Olig1 and Olig2 genes are also overexpressed in DS individuals. These genes encode transcription factors that have been implicated in neurogenesis and oligodendrogenesis [131–133]. Chakrabarti et al. [103] have demonstrated the implication of these genes in the regulation of the number of inhibitory neurons during embryonic development. The normalization of Olig1 and Olig2 expression in Ts65Dn mice rescued the abnormal interneuron production and the balance between excitatory and inhibitory transmission [103].
The APP gene is triplicated in DS and in most DS mouse models, and it is thought to play a role in different DS phenotypes, such as the development of AD pathology. It has been proposed that the APP gene may also be involved in the altered neurogenesis characteristic of the trisomic condition. Trazzi et al. [115] have recently related increased levels of the APP fragment AICD to the overexpression of the negative regulator of the Shh pathway, Ptch1, in Ts65Dn mice neural precursors and to their proliferation impairment. APP overexpression may also alter the differentiation of newly born cells by acting upon the Notch pathway, which is implicated in the acquisition of a glial phenotype [134]. Notch is upregulated in the cortex of DS and AD patients and in DS fibroblasts [135]; therefore, it could shift the balance towards a glial phenotype rather than a neuronal phenotype in newly born cells.
4.2.3. Therapies Targeting Neurogenesis
The implication of adult neurogenesis defects in DS-related cognitive impairments suggests that therapies targeted to rescue neurogenesis may be of value in treating intellectual disability in DS individuals.
The selective serotonin reuptake inhibitor fluoxetine is an antidepressant that has been shown to increase neurogenesis in the mouse DG and SVZ [113, 136]. Chronic treatment with fluoxetine restored neurogenesis in adult Ts65Dn mice [116]. Bianchi et al. [54] have recently shown that Ts65Dn mice treated with fluoxetine during the first two weeks of postnatal life showed rescued proliferation in the DG and SVZ, differentiation, and survival. Furthermore, this treatment restored brain derived neurotrophic factor (BDNF) expression, total granule cell number, and cognitive performance in a contextual fear conditioning task.
Another drug that markedly increases neurogenesis in the DG of adult normal mice is lithium, a drug prescribed for the treatment of bipolar depression [113]. Lithium treatment for 1 month rescued neurogenesis in the SVZ of 12-month-old Ts65Dn mice [114].
As mentioned above, the Shh pathway plays a key role in granule precursor cell (GPC) proliferation. Drugs targeting this pathway rescue neurogenesis alterations. Treatment of Ts65Dn mice with an activator of the Shh pathway, SAG 1.1, increased mitosis, restored cerebellar granule cell precursor populations [93], and rescued the cell proliferation of neural progenitors from the SVZ and DG [115]. Furthermore, a single injection of SAG 1.1 to newborn Ts65Dn mice restored cognition in these mice when they became adults [137].
Active care programs are one of the most successful therapeutic interventions used in DS individuals. In rodents, environmental enrichment has been associated with morphological, physiological, and cognitive improvements. These changes include increases in cortical weight and thickness, hippocampal neurogenesis, dendritic branching, length, number of dendritic spines and size of dendritic spines [138–140], facilitation of long-term potentiation [141, 142], and more efficient learning during different tasks [143–145].
Exposure of Ts65Dn mice to environmental enrichment for 7 weeks modulated spatial memory in a sex-dependent manner [55]. Environmental enrichment improved the performance of Ts65Dn females in the Morris water maze but lowered the performance of Ts65Dn male mice. In a subsequent study [42], it was shown that the deterioration found after environmental enrichment in Ts65Dn males was likely due to the stress induced by increased intermale aggression when the animals were housed in large groups. Enriching TS males in large groups (8–10) produced a large deterioration of performance in the Morris water maze and an increase in corticosterone plasma levels, effects that were not found when TS mice were housed in standard laboratory conditions or enriched in groups of 2-3.
Chakrabarti et al. [146] have recently shown that environmentally enriching groups of 2-3 Ts65Dn mice per cage increased cell proliferation and neurogenesis in the DG and SVZ of both male and female mice. It was proposed that this cellular response could underlie the cognitive improvements seen after special care programs in DS individuals.
Voluntary exercise is beneficial for cognition in both normal rodents and mouse models of altered cognition [110, 140, 147–149]. It has been suggested that these beneficial effects could be mediated, at least in part, by enhanced hippocampal neurogenesis [148, 150]. We have demonstrated that voluntary physical exercise improved the performance of TS mice in the Morris water maze but did not restore the neuromorphological phenotype (neurogenesis and hypocellularity in the DG), which suggests that the cognitive improvements produced by exercise were not mediated by neurogenesis-dependent mechanisms [83].
4.3. Apoptosis
Apoptosis or programmed cell death is physiologically involved in nervous system development and aging. It has been proposed that the hypocellularity found in DS brains could also be due to increased cell death. However, thus far, studies on the apoptotic processes in the trisomic condition have led to contradictory results. Some groups have reported increases in the number of apoptotic cells in DS brains [60, 151] and in Ts65Dn [89] and Ts1Cje [105] mice. In addition, changes in apoptotic regulatory proteins in different structures of DS brains have been found [152–156]. However, other studies have failed to find differences or have shown a reduced rate of apoptotic cell death in human and mouse trisomies [89, 157]. We have recently shown a downregulation of the antiapoptotic Bcl-Xl protein in the hippocampus of adult Ts65Dn mice, without changes in other pro- or antiapoptotic proteins in the cortex or the hippocampus [158]. Furthermore, we did not find any evidence of changes in molecular or cellular markers of apoptosis, suggesting that programmed cell death is not likely to play a role in the hypocellularity found in these mice brains.
4.4. Dendritic Hypotrophy
Altered synaptic plasticity is an additional mechanism that may underlie intellectual disability in DS individuals. Synaptic plasticity includes alterations in the number and the functional characteristics of synapses, which are mostly localized to dendrites and dendritic spines [159, 160].
Numerous studies have demonstrated impaired dendritic morphology in trisomic conditions. Although normal or even increased dendritic branching has been reported in DS fetuses and neonates [161–163], in DS children, neurons of the motor [164], visual [163, 165], and parietal cortex [166] show dendritic hypotrophy. These anomalies continue throughout the lifespan; in DS adults, the visual cortex, CA1, and CA3 are characterized by the presence of dendrites of shorter length and dendritic trees with reduced branching that progressively degenerate [161, 165, 167, 168]. Dendritic spines also show altered morphologies in the DS brain. Starting from infancy, spines are less numerous and smaller [168, 169], and their density is reduced to a greater extent in older DS individuals with AD [167, 170].
Mouse models also resemble the dendritic pathology of DS. In adult Ts65Dn mice, neocortical pyramidal neurons show a reduction in the length and arborization of dendrites and in the density of spines [171]. Spine density is also reduced in the granule cells of the DG in Ts65Dn, Ts1Cje, Ts2Cje, and Ts1Rhr mice [21, 48, 52, 172, 173]. In addition, in Ts65Dn, Ts1Cje, and Ts2Cje mice, these DG spines are characterized by several morphological anomalies, including an increase in the size of the heads and a decrease in the length of the necks [21, 52, 172]. Ts1Rhr mice also display enlarged spine heads, but no changes have been found in the morphology of spine necks of neurons in the cortex or the DG [174].
4.4.1. Therapies Targeting Dendrites and Dendritic Spines
These anomalies in dendritic tree arborization, spine density, and morphology lead to reduced density of synapses and compromised synaptic function in DS individuals. Several groups have tested the value of various therapeutic strategies for rescuing dendritic pathologies.
Based on the observation that fluoxetine favors dendritic development in normal animals [175], Bianchi et al. [54] reported that the early administration of fluoxetine (P3–P15) restored dendritic maturation and dendritic branching of DG neurons in Ts65Dn mice. As mentioned above, this drug also rescued neurogenesis and cognitive deficits in this mouse model.
The enhancement of dendritic arborization and spine density has been firmly established as one of the positive effects of environmental enrichment [138, 139, 176]. Because environmental enrichment has been shown to improve cognition in female but not male Ts65Dn mice [55], Dierssen et al. [171] tested the effect of this experimental protocol on dendritic morphology. The authors found that the enriched environment increased dendritic length and spine density in the basal dendritic trees of the neocortical pyramidal cells of euploid animals but had no effect on Ts65Dn mice [171]. Thus, enhancements in dendritic arborization and spine density do not appear to be the mechanism by which enrichment improves cognition in Ts65Dn females.
4.5. Synaptic Pathology
As predicted by the reduced length and number of branches of dendrites and density of spines, the trisomic condition is characterized by a reduced number of synaptic contacts and alterations in synaptic plasticity. Ts65Dn mice show a reduction in synaptic density in the neocortex and CA1 at P9 [67] and in the hippocampal DG, CA1, and CA3 regions in adulthood [88]. However, the size of presynaptic boutons and the average length of synaptic clefts are increased in the cortex and hippocampus of Ts65Dn and Ts1Cje mice [52, 69, 172].
Not only the number and characteristics of synapses but also the relative distribution of different types of synapses is altered in trisomic mice, leaving the balance between excitatory and inhibitory synapses shifted toward increased inhibition in the trisomic brain. Ts65Dn mice have less excitatory (asymmetric) synapses in the temporal cortex, DG, CA1, and CA3 [88, 177], and glutamatergic synapses are reduced in the hippocampus of Ts65Dn mice [90]. An increased number of inhibitory synapse markers [172] have been reported in the DG of these mice, but no changes in the number of symmetric or asymmetric synapses were found in the fascia dentata of Ts65Dn mice [69]. Ts65Dn and Ts1Cje brains also show a redistribution of inhibitory synapses, with a relative decrease in inputs to the dendritic shafts and an increase in inputs on spine necks [52, 172]. An increased number of GABAergic interneurons in the somatosensory cortex of Ts65Dn mice have been reported [178], which implies an enhancement of inhibitory synapses. Finally, Chakrabarti et al. [103] have found enhanced neurogenesis of inhibitory neurons in the forebrain of Ts65Dn mice, which led to an increased inhibitory drive.
Overall, these morphological and functional disturbances compromise the physiological properties of synapses, possibly leading to cognitive impairments in DS and trisomic mice.
5. Electrophysiological Alterations in DS and Mouse Models of DS
DS individuals present small electroencephalographic (EEG) abnormalities. EEG alpha activity is relatively preserved in young individuals with DS, but older patients with dementia present abnormal activity [179]. EEG coherence differences [180] and alterations in event-related brain potentials (ERPs) have also been reported in DS individuals [181].
In mouse models of DS, altered synaptic plasticity in the hippocampus has been consistently reported. Hippocampal long-term potentiation (LTP) is considered to be the electrophysiological substrate of learning. Ts65Dn mice display reduced LTP in the hippocampal CA1 and DG regions [52, 182–186]. Similarly, Ts1Cje, Ts1Rhr, Dp(16)1Yey/+, and Tc1 mice show reduced hippocampal LTP [28, 30, 31, 48, 52]. However, Dep(17)1Yey/+ mice showed enhanced LTP [28].
It has been proposed that altered synaptic plasticity in the hippocampus of DS mouse models results from increased inhibition due to unbalanced excitatory and inhibitory neurotransmission [172, 186, 187]. Reduced activation of NMDA receptors is thought to hinder LTP induction in trisomic mice [30, 48, 52, 186]. Enhanced hippocampal long-term depression (LTD) has also been reported in the Ts65Dn mouse [183]. Scott-McKean and Costa [188] have demonstrated that increased cerebellar LTD, mediated by exaggerated NMDAR-dependent mechanisms, could be rescued by the administration of the NMDA receptor antagonist memantine.
In addition, overinhibition in the hippocampus of Ts65Dn mice has also been shown to be dependent on GABAA receptors [172] because the GABAA antagonist picrotoxin rescued the reduction in LTP induced by theta-burst stimulation (TBS) in these mice [184]. Furthermore, Kleschevnikov et al. [189] have shown that both GABAA and GABAB receptor-mediated components of evoked inhibitory postsynaptic currents (IPSCs) were significantly higher in Ts65Dn mice, suggesting an increase in presynaptic release of GABA. Thus, both GABAA and GABAB receptors are implicated in the reduced synaptic efficiency found in the DG of Ts65Dn mice.
The G-protein-activated inwardly rectifying potassium channel 2 (Girk2) gene is overexpressed in DS individuals. Girk channels generate a GABAB receptor-dependent slow inhibitory postsynaptic potential in hippocampal neurons [190]. It has been proposed that an increase in Girk2 gene expression may produce overinhibition in hippocampal neurons and contribute to LTP failure in the trisomic condition [191].
5.1. Therapies Targeting Overinhibition
Because overinhibition in the trisomic brain appears to underlie LTP impairments and, therefore, alter learning and memory processes, a number of studies have tested drugs that reduce GABA-mediated inhibition in an attempt to rescue the electrophysiological substrates of cognition.
It is well established that the GABAA receptor system plays an important role in cognition. Nonselective positive modulators of the GABAA receptor disrupt learning and memory processes [192–194], while nonselective negative modulators improve cognitive processes [195–197]. Reducing inhibition in the Ts65Dn brain by administering the GABAA antagonists picrotoxin (PTX), bilobalide (BB), or pentylenetetrazole (PTZ) restored LTP and cognition in the object recognition test in these mice [185]. Rueda et al. [198] confirmed that chronic PTZ treatment also rescued Ts65Dn mice performance in the Morris water maze.
However, non-selective GABAA negative modulators cannot be safely used to improve cognition due to their anxiogenic and proconvulsant effects [199]. Among the different GABAA receptor subtypes, GABAA α5 subunit-containing receptors are known to facilitate cognition in hippocampal-dependent tasks [200, 201]. Moreover, selective GABAA α5 negative allosteric modulators, also called inverse agonists, have cognition-enhancing effects without anxiogenic or proconvulsant side effects [202–204]. A functionally selective GABAA α5 inverse agonist, α5IA, has been shown to rescue learning and memory deficits in TS mice without inducing anxiogenic and convulsant side effects [205].
Further support for the efficacy of reducing GABA-mediated overinhibition to improve cognition in trisomic mice comes from a recent report that demonstrated that environmental enrichment reduced the release of GABA in the hippocampus and visual cortex of Ts65Dn mice while rescuing spatial learning and hippocampal LTP [206].
6. Altered Neurotransmission and Receptors
Alterations in several neurotransmitters and changes in the expression and function of their receptors, in both DS individuals and mouse models of this condition have been demonstrated. These impairments may be responsible for other phenotypes found in trisomic conditions, such as defects in neurogenesis, synaptic transmission, and cognition. Dopamine, taurine, and histamine levels have been shown to be altered in the brains of DS fetuses and adults [97, 98, 207–209]. The main neurotransmitter and receptor alterations in DS and in the Ts65Dn mouse model are summarized in Table 4.
Table 4.
Neurotransmitter and receptor alterations in DS and in the Ts65Dn mouse model of Down syndrome.
| DS | Ts65Dn | |
|---|---|---|
| GABA | (i) Reduced in fetuses | (i) Increased number of GABAergic interneurons |
|
| ||
| Excitatory transmitters | (i) Decreased levels of glutamate and aspartate in adults | (i) Alterations in the composition of the AMPA and NMDA receptor (ii) Alterations in the signalling mechanisms downstream the NMDA receptor |
|
| ||
| 5-HT | (i) Deficits of 5-HT in the frontal cortex (ii) Reduced levels of the 5-HT1A receptor |
(i) Unchanged levels of 5-HT (ii) Reduced levels of the 5-HT1A receptor |
|
| ||
| Ach | (i) Deficits in the cholinergic system and in ChAT activity | (i) Reduced levels of markers for Ach (ii) Increased ChAT activity |
|
| ||
| NA | (i) Reduced levels in adult brains (ii) Altered β-adrenoceptor function in aged brains |
(i) Loss of locus coeruleus neurons starting at 6 months of age (ii) Altered β-adrenoceptor function |
|
| ||
| Neurotrophins | ||
| (i) BDNF (ii) NT3 (iii) NGF |
(i) Reduced levels in fetuses | (i) Reduced levels (ii) Increased levels (iii) Reduced levels |
6.1. GABA
GABA is reduced in DS fetuses [207]. However, as predicted from the enhanced inhibition of the trisomic brain, an increase in the number of inhibitory neurons has been found in Ts65Dn mice due to the overexpression of the Olig1 and Olig2 genes (see above) [103, 178]. Furthermore, it has been suggested that enhanced presynaptic GABA release may be responsible for the increased hippocampal inhibitory postsynaptic potentials (IPSPs) observed in these mice [189].
A number of alterations have been reported in the expression of various GABA receptor subunits. In neurospheres from fetuses with DS, upregulation of the α2 and downregulation of the α5 and β3 subunits of the GABAA receptor have been reported [210]. In the hippocampus of Ts65Dn mice, reductions in the number of β2 and β3 subunits of the GABAA receptor were found [69]. Brain synaptosomes of Ts65Dn mice show a reduction in GABAA α1 receptor expression [211]. Changes in the R1 subunit of the GABAB receptor have also been reported in Ts65Dn mice [69]. However, Kleschevnikov et al. [189] did not find changes in the levels of GABAA or GABAB receptor subunits by western blot analysis.
GABAA activity is known to regulate neuronal proliferation, migration, differentiation, and integration of newly generated neurons [212–214]. The enhanced GABAA-mediated inhibition shown by Ts65Dn mice could, therefore, be implicated in the alterations in neuronal proliferation and survival found in these mice.
6.2. Excitatory Transmitters
Increased inhibition in the trisomic condition is also caused by alterations in excitatory transmission. Although similar levels of glutamate [215] are found in fetuses with and without DS, decreased levels of aspartate and glutamate have been found in several areas of the adult DS brain [97, 98, 208].
As detailed above, altered hippocampal LTP in trisomic mice suggests disturbances in NMDA receptor signaling. In Ts65Dn mice, a reduction of the GluR1 subunit of the AMPA receptor [69] and of the NR2A and NR2B subunits of the NMDA receptor [216] has been reported. However, other studies failed to find changes in the GluR1 subunit in brain homogenates or changes in the NR2A and NR2B subunits in synaptosomes of these mice [211].
Ts65Dn and Ts1Cje mice exhibit hypersensitivity to the locomotor stimulatory effect of MK-801, an NMDA receptor channel blocker [217].
Alterations in the signaling mechanisms downstream of the NMDA receptor have also been reported; the hippocampi of Ts65Dn mice show disturbances in the calcium/calmodulin-dependent protein kinase II (CaMKII), phosphatidylinositol 3-kinase (PIP3K)/Akt, extracellular signal-regulated kinase (ERK), protein kinase A (PKA), and protein kinase C (PKC), all of which have been shown to be involved in synaptic plasticity [218].
One of the targets of the NMDA receptor is the protein phosphatase calcineurin (CaN). The DSCR1 gene encodes a protein that inhibits CaN, and this gene is overexpressed in the Ts65Dn brain [219]. The inhibition of CaN activity increases the mean open time and opening probability of the NMDA receptor [220]. Memantine, a partial agonist of the NMDA receptor, often prescribed for the treatment of AD-dementia, acts as an open-channel blocker and has been proposed to mimic the actions of CaN and restore the function of the NMDA receptor. Costa et al. [221] demonstrated that the acute administration of memantine improved contextual fear conditioning in Ts65Dn mice. Chronic treatment with memantine also improved Ts65Dn mice performance in the Morris water maze [90] and in the novel object recognition test and water radial arm maze [222]. Memantine slightly reduced brain APP levels and normalized the levels of hippocampal excitatory synapses in Ts65Dn mice [90]. However, memantine did not rescue Ts65Dn morphological alterations, as the number of hippocampal granule [90], basal forebrain cholinergic, and locus coeruleus neurons [222] remained low in memantine-treated Ts65Dn mice. Nevertheless, these mice showed increased BDNF levels in the hippocampus and the prefrontal cortex.
In spite of the rescue induced by memantine of several DS-relevant phenotypes in the Ts65Dn mouse, a recent randomized double-blind clinical trial failed to find any benefit of memantine administration for 52 weeks on cognitive impairment and dementia in DS individuals over 40 years of age [223].
6.3. Serotonin
Deficits in serotonin (5-HT) have been reported in the frontal cortex of DS fetuses [207] and in adult DS brains [97, 98, 209]. However, Ts65Dn mice show unchanged levels of 5-HT in the hippocampus [54], and no alterations were found in the histological analysis of serotonergic neurons of the dorsal and medial raphe nuclei of these mice [224]. 5-HT has a role in neurogenesis, neuronal differentiation, dendritic development, axon myelination, and synaptogenesis [225]. Thus, the reduction of this transmitter in DS fetal and adult brains may underlie a number of altered neuromorphological and cognitive phenotypes.
The 5-HT1A receptor has also been implicated in the regulation of neurogenesis [113, 226, 227]. Reduced levels of the 5-HT1A receptor have been reported in the DS brain at birth [228], in hippocampal neurospheres, and in the hippocampus of newborn Ts65Dn mice [54]. Thus, reduced 5-HT1A receptor expression may underlie the defective neurogenesis found in Ts65Dn mice [54]. Moreover, treatment with the 5-HT1A reuptake inhibitor fluoxetine rescued the expression levels of this receptor in Ts65Dn mice, suggesting that this effect may underlie the rescue of proliferation produced by this drug, as previously mentioned.
6.4. Acetylcholine
One of the anatomical substrates of learning and attention is the septohippocampal cholinergic system [95, 96]. A number of studies have demonstrated alterations of this system in the trisomic condition. Deficits in the cholinergic system have been found in DS fetuses [207], and choline acetyltransferase (ChAT) activity is reduced in the brains of adults with DS [97, 98].
Although a normal number of cholinergic neurons is found in young Ts65Dn mice, basal forebrain cholinergic neurons (BFCNs) degenerate with age in these mice [99–102]. However, ChAT activity is increased in the cortex and hippocampus of 10-month-old Ts65Dn mice, likely in an attempt to compensate for the reduced number of cholinergic neurons [82, 100, 229, 230].
6.5. Noradrenaline
The levels of noradrenaline (NA) are normal in DS fetuses [207] but are reduced in adult DS brains [97, 98], likely as a consequence of the neurodegeneration of the locus coeruleus [231, 232]. Ts65Dn mice also show a loss of locus coeruleus neurons starting at 6 months of age [53].
Ts65Dn mice show unchanged numbers of β-adrenoceptors in the cortex and hippocampus; however, their function is altered. Basal production of cyclic AMP in the hippocampus of TS mice was impaired. In addition, the responses of adenylyl cyclase to the stimulation of β-adrenoceptors with isoprenaline and of the catalytic subunit with forskolin were both severely depressed [233, 234]. Aging DS brains also show a dramatic reduction in basal and stimulated cAMP production [235].
NA has been shown to play a role in neurogenesis, as neuron proliferation is enhanced or impaired following increases or reductions in NA transmission, respectively [236]. Therefore, altered NA transmission in the trisomic condition may also play a role in the impairment of adult hippocampal neurogenesis. Furthermore, a link between noradrenergic afferents from the locus coeruleus to hippocampal neurons and contextual learning has been demonstrated [237]. This hippocampal-dependent cognitive process is impaired in individuals with DS [6] and in Ts65Dn mice [53]. A recent study by Salehi et al. [53] demonstrated that enhancing NA transmission through the administration of L-Threo-3, 4-dihydroxyphenylserine (L-DOPS), a synthetic amino acid that is metabolized by NA-containing neurons to produce NA, or xamoterol, a ß1-adrenergic receptor partial agonist, rescued contextual learning in Ts65Dn mice. These authors hypothesized that, given the finding that NA can activate or inhibit GABAergic neurons and that GABA can increase the release of NA, there could be an overlap in the mechanisms by which GABAA antagonists and NA-enhancing drugs improve learning in Ts65Dn mice.
6.6. Neurotrophins
The role of neurotrophins (NT) in neuronal survival, differentiation, migration, and synaptic plasticity is well documented [238–240]. Consequently, alterations in their expression may alter many aspects of neurodevelopment.
The reduced expression of BDNF has been observed in the hippocampus of DS fetuses [241], and the reduced expression of both BDNF and the tyrosine kinase receptor TrkB has been observed in the cerebral cortex of DS fetuses [242]. Young Ts65Dn mice also show reduced BDNF levels in the hippocampus [54, 243] and in the frontal cortex during adult stages [244]. Because BDNF has a role in neuronal survival and differentiation [213, 215], it is a natural target for several treatments to restore neurogenesis in the trisomic brain. In Ts65Dn mice, fluoxetine restored BDNF expression, survival of newborn cells, differentiation, and granule cell number.
NT-3 is increased in the hippocampus of newborn and adult Ts65Dn mice [245], potentially in an attempt to compensate for the neuronal loss found in these mice.
Nerve growth factor (NGF) is generated in the hippocampus and retrogradely transported to the soma of BFCNs [239]. NGF levels are reduced in the hippocampus of young Ts65Dn mice [50], and the retrograde transport of NGF to the basal forebrain is hindered in older Ts65Dn and Ts1Cje mice [50, 100]. NGF enhances the survival, differentiation, and maintenance of neurons, including BFCNs [239]. The administration of NGF to Ts65Dn mice rescued the altered size and number of BFCNs [100].
Peptide 6, an active region of ciliary neurotrophic factor (CNTF), modulates the CNTF pathway by inhibiting the antineurogenic activity of leukemia inhibitory factor, thereby increasing neurogenesis [246]. Administration of peptide 6 to Ts65Dn mice reduced learning and memory deficits, enhanced the pool of neural progenitor cells in the hippocampus, and increased the level of synaptic proteins crucial for synaptic plasticity [247].
Considering the role of Dyrk1A in neuronal progenitor proliferation, neurogenesis, and neurodifferentiation, it has been suggested that molecules targeting this gene could provide therapeutic benefits to DS phenotypes. Epigallocatechin gallate (EGCG), an antioxidant extracted from green tea, is an inhibitor of the protein kinase DYRK1A [248].
The chronic administration of EGCG from conception to adulthood rescued BDNF levels in the hippocampus of Dyrk1a transgenic mice [241]. Concomitant to this neurotrophic factor normalization, these mice presented an increase in brain volume and improved cognitive performance. Other studies have demonstrated that the acute administration of EGCG normalizes hippocampal LTP in Ts65Dn mice [249]. However, EGCG affects a wide array of signal transduction pathways including the MAPK, PI3K/AKT, Wnt, and Notch pathways [250], which are altered in Ts65Dn mice [135]; thus, its beneficial effects could be mediated by mechanisms different from Dyrk1A inhibition.
Finally, Fukuda et al. [243] have recently demonstrated that the chronic administration of the analgesic neurotrophin to Ts65Dn mice prevents the age-dependent decline in hippocampal BDNF expression. This treatment also enhanced the performance of these mice in the radial arm maze. It has been proposed that the analgesic action of neurotrophin is mediated by the noradrenergic and GABAergic systems [251]; therefore, the cognitive-enhancing effects could also be determined by improvements in the function of these transmitter systems.
7. Neurodegeneration
Although neurodevelopmental alterations occurring from early embryonic stages are likely to cause intellectual disability, there are a number of neurodegenerative mechanisms in DS that complicate this scenario. Atrophy of a number of structures, including the hippocampus, amygdala [71, 252], the corpus callosum, and the parietal, frontal, and occipital cortices [77, 78], has been reported in nondemented adult DS brains. Furthermore, neuroinflammation, increased oxidative stress, and the development of AD neuropathology are hallmarks of DS (Table 5).
Table 5.
Neurodegenerative processes in DS and in the Ts65Dn mouse.
| DS | Ts65Dn | |
|---|---|---|
| Neuroinflammation | (i) Activated microglia and increased levels of proinflammatory cytokines | (i) Activated microglia |
|
| ||
| Oxidative stress | (i) Increased | (i) Increased |
|
| ||
| AD neuropathology | (i) Cholinergic neuron loss, plaques, and tangles | (i) Cholinergic neuron degeneration, increased APP and β-amyloid levels |
7.1. Neuroinflammation
DS and AD brains are characterized by activated microglia, and increased levels of proinflammatory cytokines that lead to neuroinflammation are likely involved in neurodegeneration [253, 254]. The activation of microglia may play a role in the loss of basal forebrain cholinergic neurons in Ts65Dn mice.
Minocycline is a semisynthetic tetracycline that inhibits neuronal death and reduces inflammatory activity by blocking inflammatory mediators [255]. The chronic administration of minocycline to adult Ts65Dn mice inhibits microglia activation in the basal forebrain and hippocampus, prevents the loss of cholinergic neurons in the medial septal nucleus, attenuates the loss of hippocampal calbindin-positive neurons, and improves working and reference memory in these mice [102].
7.2. Neuropeptides
Vasoactive intestinal peptide (VIP) is neuroprotective, as it promotes the release of several survival factors from astrocytes and regulates neuropeptide release from glial cells, including activity-dependent neuroprotective protein (ADNP) and activity-dependent neurotrophic factor (ADNF) [256]. The active peptide fragments of ADNP and ADNF, NAPVSIPQ (NAP), and SALLRSIPA (SAL) have been shown to protect neurons from oxidative stress and limit the severity of traumatic head injuries, stroke, and the toxicity associated with the Aβ peptide [257, 258].
In cultures of DS cortical neurons, treatment with SAL or NAP increases neuronal survival [259]. In Ts65Dn mice, prenatal treatment with these two peptides rescued the acquisition of neurodevelopmental milestones [260], increased the reduced levels of ADNP, and normalized the levels of the NMDA receptor subunits NR2A, NR2B, and the GABAA receptor subunit β3 [216]. Furthermore, subchronic treatments with D-NAP and D-SAL to adult Ts65Dn mice rescued learning and memory and ADNP and NRD2 levels [256].
7.3. Oxidative Stress
In DS individuals and in the Ts65Dn mouse, there is an overexpression of SOD1, the gene responsible for the formation of superoxide dismutase, an enzyme that modifies oxygen free radicals into hydrogen peroxide. The overproduction of hydrogen peroxide leads to the overproduction of highly reactive oxygen free radicals, which damage cell membranes, including the mitochondrial membrane, and deteriorate lipids, proteins, and mitochondrial DNA. This set of alterations is called oxidative stress. Evidence for increased mitochondrial superoxide production in DS individuals has been repeatedly demonstrated [261, 262]. Therefore, in this condition, some cells are under the permanent threat of oxidative stress with mitochondrial damage, which deteriorates cell life, facilitating aging, and death. This increase in oxidative stress occurs during pre- and postnatal development. Increased oxidative stress in the fetal stages can modify processes such as neurogenesis, differentiation, migration and net connexion, as well as survival [261, 263–265].
In an attempt to reduce oxidative stress-induced neurodegeneration, several groups have tested the efficacy of various antioxidants to reduce the altered phenotypes in Ts65Dn mice. SGS-111, an analogue of the nootropic piracetam, has been shown to increase neuronal survival and prevent the accumulation of intracellular free radicals, peroxidative damage, and the development of neurodegenerative changes in both normal and DS cultured neurons [265]. However, the chronic administration of SGS-111 to Ts65Dn mice from conception to adulthood did not rescue their cognitive alterations [266]. Conversely, the administration of another antioxidant, vitamin E, to Ts65Dn mice during adult stages [267] or from conception throughout their entire life [268] reduced markers of oxidative stress, improved cognitive performance, reduced cholinergic neuron pathology in the basal forebrain, and increased cell density in the DG.
A recent report [269] revealed a positive effect of folinic acid on developmental age in children with DS. Folinic acid has an antioxidant effect and is known to be involved in CNS development. Folate deficiency causes neurological, psychiatric, and cognitive disorders, and DS probably involves either folate deficiency or defective folate use [270]. However, in a randomized controlled trial, Ellis et al. [271] failed to find any efficacy of antioxidants and folinic acid supplementation in children with DS cognitive development. In addition, a number of studies on the effects of antioxidant supplementation in children and adults with DS did not find any benefit of this treatment on cognition. Salman [272] performed a systematic review of eleven randomized controlled trials on the effects of dietary supplements (vitamins and/or minerals) on cognitive function in subjects with DS. None of these trials reported cognitive enhancing effects in individuals with DS. Moreover, in a two-year randomized, double blind, placebo-controlled trial daily oral antioxidant supplementation (α-tocopherol, ascorbic acid, and the mitochondrial cofactor: α-lipoic acid) did not produce any improvement in cognitive functioning nor a stabilization of cognitive decline in adults with DS [273].
7.4. Estrogens
Because estrogens maintain the function of basal forebrain cholinergic neurons, it has been proposed that the administration of estrogens may be useful in reducing the loss of these neurons in AD and DS individuals [274].
The chronic administration of estrogens to aged female Ts65Dn mice enhanced cognition, increased the size and number of cholinergic neurons, increased the levels of NGF in the medial septum [275], restored the number of cholinergic terminals in hippocampus, and restored the levels of the dendritic marker Map2 [276].
7.5. AD Neuropathology
One of the genes triplicated in the trisomic condition is APP. In DS individuals, the increased expression of this gene leads to the increased production of β-amyloid, which is thought to be responsible for the amyloid plaque pathology and degeneration of BFCNs found in 100% of DS individuals over 40 years of age. Ts65Dn mice also show age-related elevations in the levels of the APP protein [277] and the β-amyloid peptide [278] in the cortex and hippocampus. In these mice, the overexpression of APP has also been implicated in the degeneration of both the cholinergic and noradrenergic neurons that provide strong modulatory inputs to the hippocampus [279]. Thus, this age-related noradrenergic and cholinergic deafferentation is likely compromising hippocampal function.
To test the effect of β-amyloid reductions on the Ts65Dn mice altered phenotypes, Netzer et al. [278] administered the gamma secretase inhibitor DAPT (N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester). This treatment reduced β-amyloid levels and rescued spatial learning in these mice. Because β-amyloid is a regulator of the glutamatergic system, the authors proposed that the cognitive enhancing effects of DAPT could be mediated by an enhancement and/or a regulation of excitatory synaptic transmission.
Given the role of the cholinergic system in cognition and the degeneration of this system in AD and DS individuals, it has been proposed that pharmacological enhancements of this system could help diminish cognitive deterioration in these conditions. Donepezil is an acetylcholinesterase inhibitor that is widely prescribed to enhance cholinergic transmission in the treatment of AD dementia. However, the chronic administration of donepezil did not improve learning and memory in Ts65Dn mice [198]. Similarly, donepezil administration to young adult individuals with DS has produced ambiguous results [280–283].
Piracetam is a drug that shows cognitive-enhancing effects in patients with a number of cognitive disorders and dementia [284] and in several animal models. Although the mechanisms underlying these effects are not known, it has been proposed that piracetam may be enhancing cholinergic and modulating glutamatergic transmission [284]. However, piracetam treatment did not improve cognitive impairments in children with DS [285] or in the Ts65Dn mouse [286].
8. Concluding Remarks
The first partial trisomic models, the Ts65Dn and Ts1Cje models, demonstrated that DS phenotypes could be recapitulated in mice. More recently, knockout and transgenic mice for individual genes and new animals that are trisomic for different regions of orthologues of Hsa21 regions are helping to identify dosage-sensitive genes involved in DS phenotypes. Although some of these triplicated genes may play a role individually, it appears that DS phenotypes arise from the complex effects of groups of Hsa21 genes.
In the last 20 years, the characterization of these animal models of DS, particularly the Ts65Dn mouse, has been enormously useful to understand of the neurobiological basis of intellectual disability. Several mechanisms have been proposed to underlie this altered cognition, including impaired neurogenesis leading to hypocellularity in the cortex, hippocampus, and cerebellum, altered dendritic morphology, altered synapses, increased inhibition and neurodegeneration. The new knowledge of the pathogenic mechanisms in DS individuals has been applied to the development of new pharmacotherapies. Several drugs have been shown to rescue neurogenesis, hypocellularity, electrophysiological deficits, and cognitive alterations in the Ts65Dn mouse. These studies provide the basis for developing clinical trials in DS individuals and sustain the hope that some of these drugs will be useful in rescuing intellectual disability in DS individuals.
Acknowledgments
This work was supported by the Jerome Lejeune Foundation and the Spanish Ministry of Innovation and Science (BFU2008-04397 and BFU2011-24755).
References
- 1.Shin M, Besser LM, Kucik JE, Lu C, Siffel C, Correa A. Prevalence of Down syndrome among children and adolescents in 10 regions of the United States. Pediatrics. 2009;124(6):1565–1571. doi: 10.1542/peds.2009-0745. [DOI] [PubMed] [Google Scholar]
- 2.Vicari S. Memory development and intellectual disabilities. Acta Paediatrica. 2004;93(445):60–64. doi: 10.1111/j.1651-2227.2004.tb03059.x. [DOI] [PubMed] [Google Scholar]
- 3.Vicari S. Motor development and neuropsychological patterns in persons with Down syndrome. Behavior Genetics. 2006;36(3):355–364. doi: 10.1007/s10519-006-9057-8. [DOI] [PubMed] [Google Scholar]
- 4.Chapman RS, Hesketh LJ. Behavioral phenotype of individuals with down syndrome. Mental Retardation and Developmental Disabilities Research Reviews. 2000;6(2):84–95. doi: 10.1002/1098-2779(2000)6:2<84::AID-MRDD2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 5.Carlesimo GA, Marotta L, Vicari S. Long-term memory in mental retardation: evidence for a specific impairment in subjects with Down’s syndrome. Neuropsychologia. 1997;35(1):71–79. doi: 10.1016/s0028-3932(96)00055-3. [DOI] [PubMed] [Google Scholar]
- 6.Nadel L. Down’s syndrome: a genetic disorder in biobehavioral perspective. Genes, Brain and Behavior. 2003;2(3):156–166. doi: 10.1034/j.1601-183x.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- 7.Cenini G, Dowling ALS, Beckett TL, et al. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochimica et Biophysica Acta. 2012;1822(2):130–138. doi: 10.1016/j.bbadis.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabbagh MN, Fleisher A, Chen K, et al. Positron emission tomography and neuropathologic estimates of fibrillar amyloid-β in a patient with down syndrome and Alzheimer disease. Archives of Neurology. 2011;68(11):1461–1466. doi: 10.1001/archneurol.2011.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapiro BL. Amplified developmental instability in Down’s syndrome. Annals of Human Genetics. 1975;38(4):429–437. doi: 10.1111/j.1469-1809.1975.tb00632.x. [DOI] [PubMed] [Google Scholar]
- 10.Delabar JM, Theophile D, Rahmani Z, et al. Molecular mapping of twenty-four features of Down syndrome on chromosome 21. European Journal of Human Genetics. 1993;1(2):114–124. doi: 10.1159/000472398. [DOI] [PubMed] [Google Scholar]
- 11.Aït Yahya-Graison E, Aubert J, Dauphinot L, et al. Classification of human chromosome 21 gene-expression variations in down syndrome: impact on disease phenotypes. American Journal of Human Genetics. 2007;81(3):475–491. doi: 10.1086/520000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyle R, Béna F, Gagos S, et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. European Journal of Human Genetics. 2009;17(4):454–466. doi: 10.1038/ejhg.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lana-Elola E, Watson-Scales SD, Fisher EMC, Tybulewicz VLJ. Down syndrome: Searching for the genetic culprits. DMM Disease Models and Mechanisms. 2011;4(5):586–595. doi: 10.1242/dmm.008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bourin M, Petit-Demoulière B, Nic Dhonnchadha B, Hascöet M. Animal models of anxiety in mice. Fundamental and Clinical Pharmacology. 2007;21(6):567–574. doi: 10.1111/j.1472-8206.2007.00526.x. [DOI] [PubMed] [Google Scholar]
- 15.Crawley JN. Designing mouse behavioral tasks relevant to autistic-like behaviors. Mental Retardation and Developmental Disabilities Research Reviews. 2004;10(4):248–258. doi: 10.1002/mrdd.20039. [DOI] [PubMed] [Google Scholar]
- 16.Sturgeon X, Gardiner KJ. Transcript catalogs of human chromosome 21 and orthologous chimpanzee and mouse regions. Mammalian Genome. 2011;22:261–271. doi: 10.1007/s00335-011-9321-y. [DOI] [PubMed] [Google Scholar]
- 17.Gropp A, Kolbus U, Giers D. Systematic approach to the study of trisomy in the mouse. II. Cytogenetics and Cell Genetics. 1975;14(1):42–62. doi: 10.1159/000130318. [DOI] [PubMed] [Google Scholar]
- 18.Davisson MT, Schmidt C, Reeves RH, et al. Segmental trisomy as a mouse model for Down syndrome. Progress in clinical and biological research. 1993;384:117–133. [PubMed] [Google Scholar]
- 19.Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling down syndrome. Mammalian Genome. 2011;22(11-12):674–684. doi: 10.1007/s00335-011-9356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korbel JO, Tirosh-Wagner T, Urban AE, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(29):12031–12036. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villar AJ, Belichenko PV, Gillespie AM, Kozy HM, Mobley WC, Epstein CJ. Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between T(1716)65Dn and mouse Chromosome 12. Mammalian Genome. 2005;16(2):79–90. doi: 10.1007/s00335-004-2428-7. [DOI] [PubMed] [Google Scholar]
- 22.Sago H, Carlson EJ, Smith DJ, et al. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6256–6261. doi: 10.1073/pnas.95.11.6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sago H, Carlson EJ, Smith DJ, et al. Genetic dissection of region associated with behavioral abnormalities in mouse models for Down syndrome. Pediatric Research. 2000;48(5):606–613. doi: 10.1203/00006450-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 24.Olson LE, Richtsmeier JT, Leszl J, Reeves RH. A chromosome 21 critical region does not cause specific down syndrome phenotypes. Science. 2004;306(5696):687–690. doi: 10.1126/science.1098992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z, Yu T, Morishima M, et al. Duplication of the entire 22.9 Mb human chromosome 21 causes cariovascular and gastrointestinal abnormalities. American Journal of Mental Retardation. 2007;96:189–201. doi: 10.1093/hmg/ddm086. [DOI] [PubMed] [Google Scholar]
- 26.Pereira PL, Magnol L, Sahún I, et al. A new mouse model for the trisomy of the Abcg1-U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Human Molecular Genetics. 2009;18(24):4756–4769. doi: 10.1093/hmg/ddp438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu T, Li Z, Jia Z, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Human Molecular Genetics. 2010;19(14):2780–2791. doi: 10.1093/hmg/ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu T, Liu C, Belichenko P, et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Research. 2010;1366:162–171. doi: 10.1016/j.brainres.2010.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vacík T, Ort M, Gregorová S, et al. Segmental trisomy of chromosome 17: a mouse model of human aneuploidy syndromes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(12):4500–4505. doi: 10.1073/pnas.0500802102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Doherty A, Ruf S, Mulligan C, et al. Genetics: an aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309(5743):2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morice E, Andreae LC, Cooke SF, et al. Preservation of long-term memory and synaptic plasticity despite short-term impairments in the Tcl mouse model of down syndrome. Learning and Memory. 2008;15(7):492–500. doi: 10.1101/lm.969608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galante M, Jani H, Vanes L, et al. Impairments in motor coordination without major changes in cerebellar plasticity in the Tc1 mouse model of Down syndrome. Human Molecular Genetics. 2009;18(8):1449–1463. doi: 10.1093/hmg/ddp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis WE, Kelso JAS. Analysis of ’invariant characteristics’ in the motor control of Down’s syndrome and normal subjects. Journal of Motor Behavior. 1982;14(3):194–211. doi: 10.1080/00222895.1982.10735273. [DOI] [PubMed] [Google Scholar]
- 34.Latash ML, Corcos DM. Kinematic and electromyographic characteristics of single-joint movements of individuals with Down syndrome. American Journal on Mental Retardation. 1991;96(2):189–201. [PubMed] [Google Scholar]
- 35.Shumway-Cook A, Woollacott MH. Dynamics of postural control in the child with Down syndrome. Physical Therapy. 1985;65(9):1315–1322. doi: 10.1093/ptj/65.9.1315. [DOI] [PubMed] [Google Scholar]
- 36.Escorihuela RM, Fernández-Teruel A, Vallina IF, et al. A behavioral assessment of Ts65Dn mice: a putative Down syndrome model. Neuroscience Letters. 1995;199(2):143–146. doi: 10.1016/0304-3940(95)12052-6. [DOI] [PubMed] [Google Scholar]
- 37.Baxter LL, Moran TH, Richtsmeier JT, Troncoso J, Reeves RH. Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Human Molecular Genetics. 2000;9(2):195–202. doi: 10.1093/hmg/9.2.195. [DOI] [PubMed] [Google Scholar]
- 38.Costa ACS, Walsh K, Davisson MT. Motor dysfunction in a mouse model for Down syndrome. Physiology and Behavior. 1999;68(1-2):211–220. doi: 10.1016/s0031-9384(99)00178-x. [DOI] [PubMed] [Google Scholar]
- 39.Holtzman DM, Santucci D, Kilbridge J, et al. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(23):13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reeves RH, Irving NG, Moran TH, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genetics. 1995;11(2):177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 41.Coussons-Read ME, Crnic LS. Behavioral assessment of the Ts65Dn mouse, a model for Down syndrome: altered behavior in the elevated plus maze and open field. Behavior Genetics. 1996;26(1):7–13. doi: 10.1007/BF02361154. [DOI] [PubMed] [Google Scholar]
- 42.Martínez-Cué C, Rueda N, García E, Davisson MT, Schmidt C, Flórez J. Behavioral, cognitive and biochemical responses to different environmental conditions in male Ts65Dn mice, a model of Down syndrome. Behavioural Brain Research. 2005;163(2):174–185. doi: 10.1016/j.bbr.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 43.Stewart LS, Persinger MA, Cortez MA, Snead OC. Chronobiometry of behavioral activity in the Ts65Dn model of Down syndrome. Behavior Genetics. 2007;37(2):388–398. doi: 10.1007/s10519-006-9119-y. [DOI] [PubMed] [Google Scholar]
- 44.Crnic LS, Pennington BF. Down syndrome: neuropsychology and animal models. Progress in Infancy Research. 2000;1:69–111. [Google Scholar]
- 45.Escorihuela RM, Vallina IF, Martínez-Cué C, et al. Impaired short- and long-term memory in Ts65Dn mice, a model for Down syndrome. Neuroscience Letters. 1998;247(2-3):171–174. doi: 10.1016/s0304-3940(98)00317-6. [DOI] [PubMed] [Google Scholar]
- 46.Martínez-Cué C, Rueda N, García E, Flórez J. Anxiety and panic responses to a predator in male and female Ts65Dn mice, a model for Down syndrome. Genes, Brain and Behavior. 2006;5(5):413–422. doi: 10.1111/j.1601-183X.2005.00175.x. [DOI] [PubMed] [Google Scholar]
- 47.Driscoll LL, Carroll JC, Moon J, Crnic LS, Levitsky DA, Strupp BJ. Impaired sustained attention and error-induced stereotypy in the aged Ts65Dn mouse: a mouse model of Down syndrome and Alzheimer’s disease. Behavioral Neuroscience. 2004;118(6):1196–1205. doi: 10.1037/0735-7044.118.6.1196. [DOI] [PubMed] [Google Scholar]
- 48.Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC. The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. Journal of Neuroscience. 2009;29(18):5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernandez F, Garner CC. Episodic-like memory in Ts65Dn, a mouse model of Down syndrome. Behavioural Brain Research. 2008;188(1):233–237. doi: 10.1016/j.bbr.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salehi A, Delcroix JD, Belichenko PV, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 51.Demas GE, Nelson RJ, Krueger BK, Yarowsky PJ. Spatial memory deficits in segmental trisomic Ts65Dn mice. Behavioural Brain Research. 1996;82(1):85–92. doi: 10.1016/s0166-4328(97)81111-4. [DOI] [PubMed] [Google Scholar]
- 52.Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. Journal of Comparative Neurology. 2007;504(4):329–345. doi: 10.1002/cne.21433. [DOI] [PubMed] [Google Scholar]
- 53.Salehi A, Faizi M, Colas D, et al. Restoration of norepinephrine-modulated contextual memory in a mouse model of Down syndrome. Science translational medicine. 2009;1(7):7–17. doi: 10.1126/scitranslmed.3000258. [DOI] [PubMed] [Google Scholar]
- 54.Bianchi P, Ciani E, Guidi S, et al. Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for down syndrome. Journal of Neuroscience. 2010;30(26):8769–8779. doi: 10.1523/JNEUROSCI.0534-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martínez-Cué C, Baamonde C, Lumbreras M, et al. Differential effects of environmental enrichment on behavior and learning of male and female Ts65Dn mice, a model for Down syndrome. Behavioural Brain Research. 2002;134(1-2):185–200. doi: 10.1016/s0166-4328(02)00026-8. [DOI] [PubMed] [Google Scholar]
- 56.Wenger GR, Schmidt C, Davisson MT. Operant conditioning in the Ts65Dn mouse: learning. Behavior Genetics. 2004;34(1):105–119. doi: 10.1023/B:BEGE.0000009480.79586.ee. [DOI] [PubMed] [Google Scholar]
- 57.Olson LE, Roper RJ, Sengstaken CL, et al. Trisomy for the Down syndrome ’critical region’ is necessary but not sufficient for brain phenotypes of trisomic mice. Human Molecular Genetics. 2007;16(7):774–782. doi: 10.1093/hmg/ddm022. [DOI] [PubMed] [Google Scholar]
- 58.Winter TC, Ostrovsky AA, Komamiski CA, Uhrich SB. Cerebellar and frontal lobe hypoplasia in fetuses with trisomy 21: usefulness as combined US markers. Radiology. 2000;214(2):533–538. doi: 10.1148/radiology.214.2.r00fe40533. [DOI] [PubMed] [Google Scholar]
- 59.Guihard-Costa A-M, Khung S, Delbecque K, Ménez F, Delezoide A-L. Biometry of face and brain in fetuses with trisomy 21. Pediatric Research. 2006;59(1):33–38. doi: 10.1203/01.pdr.0000190580.88391.9a. [DOI] [PubMed] [Google Scholar]
- 60.Guidi S, Bonasoni P, Ceccarelli C, et al. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathology. 2008;18(2):180–197. doi: 10.1111/j.1750-3639.2007.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guidi S, Ciani E, Bonasoni P, Santini D, Bartesaghi R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with down syndrome. Brain Pathology. 2011;21(4):361–373. doi: 10.1111/j.1750-3639.2010.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Golden JA, Hyman BT. Development of the superior temporal neocortex is anomalous in trisomy 21. Journal of Neuropathology and Experimental Neurology. 1994;53(5):513–520. doi: 10.1097/00005072-199409000-00011. [DOI] [PubMed] [Google Scholar]
- 63.Jernigan TL, Bellugi U, Sowell E, Doherty S, Hesselink JR. Cerebral morphologic distinctions between Williams and Down syndromes. Archives of Neurology. 1993;50(2):186–191. doi: 10.1001/archneur.1993.00540020062019. [DOI] [PubMed] [Google Scholar]
- 64.Pinter JD, Eliez S, Schmitt JE, Capone GT, Reiss AL. Neuroanatomy of Down’s syndrome: a high-resolution MRI study. American Journal of Psychiatry. 2001;158(10):1659–1665. doi: 10.1176/appi.ajp.158.10.1659. [DOI] [PubMed] [Google Scholar]
- 65.Teipel SJ, Hampel H. Neuroanatomy of Down syndrome in vivo: a model of preclinical Alzheimer’s disease. Behavior Genetics. 2006;36(3):405–415. doi: 10.1007/s10519-006-9047-x. [DOI] [PubMed] [Google Scholar]
- 66.Aldridge K, Reeves RH, Olson LE, Richtsmeier JT. Differential effects of trisomy on brain shape and volume in related aneuploid mouse models. American Journal of Medical Genetics A. 2007;143(10):1060–1070. doi: 10.1002/ajmg.a.31721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chakrabarti L, Galdzicki Z, Haydar TF. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. Journal of Neuroscience. 2007;27(43):11483–11495. doi: 10.1523/JNEUROSCI.3406-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ishihara K, Amano K, Takaki E, et al. Enlarged brain ventricles and impaired neurogenesis in the Ts1Cje and Ts2Cje mouse models of down syndrome. Cerebral Cortex. 2010;20(5):1131–1143. doi: 10.1093/cercor/bhp176. [DOI] [PubMed] [Google Scholar]
- 69.Belichenko PV, Kleschevnikov AM, Masliah E, et al. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. Journal of Comparative Neurology. 2009;512(4):453–466. doi: 10.1002/cne.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wisniewski KE, Laure-Kamionowska M, Wisniewski HM. Evidence of arrest of neurogenesis and synaptogenesis in brains of patients with Down’s syndrome. New England Journal of Medicine. 1984;311(18):1187–1188. doi: 10.1056/NEJM198411013111818. [DOI] [PubMed] [Google Scholar]
- 71.Kesslak JP, Nagata SF, Lott I, Nalcioglu O. Magnetic resonance imaging analysis of age-related changes in the brains of individuals with Down’s syndrome. Neurology. 1994;44(6):1039–1045. doi: 10.1212/wnl.44.6.1039. [DOI] [PubMed] [Google Scholar]
- 72.Raz N, Torres IJ, Briggs SD, et al. Selective neuroanatomic abnormalities in Down’s syndrome and their cognitive correlates: evidence from MRI morphometry. Neurology. 1995;45(2):356–366. doi: 10.1212/wnl.45.2.356. [DOI] [PubMed] [Google Scholar]
- 73.Pine SS, Landing BH, Shankle WR. Reduced inferior olivary neuron number in early down syndrome. Pediatric Pathology and Laboratory Medicine. 1997;17(4):537–545. [PubMed] [Google Scholar]
- 74.Aylward EH, Habbak R, Warren AC, et al. Cerebellar volume in adults with Down syndrome. Archives of Neurology. 1997;54(2):209–212. doi: 10.1001/archneur.1997.00550140077016. [DOI] [PubMed] [Google Scholar]
- 75.Aylward EH, Li Q, Honeycutt NA, et al. MRI volumes of the hippocampus and amygdala in adults with Down's syndrome with and without dementia. American Journal of Psychiatry. 1999;156(4):564–568. doi: 10.1176/ajp.156.4.564. [DOI] [PubMed] [Google Scholar]
- 76.Pinter JD, Brown WE, Eliez S, Schmitt JE, Capone GT, Reiss AL. Amygdala and hippocampal volumes in children with Down syndrome: a high-resolution MRI study. Neurology. 2001;56(7):972–974. doi: 10.1212/wnl.56.7.972. [DOI] [PubMed] [Google Scholar]
- 77.Teipel SJ, Schapiro MB, Alexander GE, et al. Relation of corpus callosum and hippocampal size to age in nondemented adults with Down’s syndrome. American Journal of Psychiatry. 2003;160(10):1870–1878. doi: 10.1176/appi.ajp.160.10.1870. [DOI] [PubMed] [Google Scholar]
- 78.Teipel SJ, Alexander GE, Schapiro MB, Möller HJ, Rapoport SI, Hampel H. Age-related cortical grey matter reductions in non-demented Down’s syndrome adults determined by MRI with voxel-based morphometry. Brain. 2004;127(4):811–824. doi: 10.1093/brain/awh101. [DOI] [PubMed] [Google Scholar]
- 79.Morris RGM, Garrud P, Rawlins JNP, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297(5868):681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 80.Morris RGM, Schenk F, Tweedie F, Jarrard LE. Ibotenate lesions of hippocampus and/or subiculum: dissociating components of allocentric spatial learning. European Journal of Neuroscience. 1990;2(12):1016–1028. doi: 10.1111/j.1460-9568.1990.tb00014.x. [DOI] [PubMed] [Google Scholar]
- 81.Lorenzi HA, Reeves RH. Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Research. 2006;1104(1):153–159. doi: 10.1016/j.brainres.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 82.Contestabile A, Ciani E, Contestabile A. The place of choline acetyltransferase activity measurement in the “cholinergic hypothesis” of neurodegenerative diseases. Neurochemical Research. 2008;33(2):318–327. doi: 10.1007/s11064-007-9497-4. [DOI] [PubMed] [Google Scholar]
- 83.Llorens-Martín MV, Rueda N, Tejeda GS, Flórez J, Trejo JL, Martínez-Cué C. Effects of voluntary physical exercise on adult hippocampal neurogenesis and behavior of Ts65Dn mice, a model of Down syndrome. Neuroscience. 2010;171(4):1228–1240. doi: 10.1016/j.neuroscience.2010.09.043. [DOI] [PubMed] [Google Scholar]
- 84.Weitzdoerfer R, Dierssen M, Fountoulakis M, Lubec G. Fetal life in Down Syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure. Journal of Neural Transmission, Supplement. 2001;(61):59–70. doi: 10.1007/978-3-7091-6262-0_5. [DOI] [PubMed] [Google Scholar]
- 85.Larsen KB, Laursen H, Græm N, Samuelsen GB, Bogdanovic N, Pakkenberg B. Reduced cell number in the neocortical part of the human fetal brain in Down syndrome. Annals of Anatomy. 2008;190(5):421–427. doi: 10.1016/j.aanat.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 86.Gandolfi A, Horoupian DS, De Teresa RM. Pathology of the auditory system in autosomal trisomies with morphometric and quantitative study of the ventral cochlear nucleus. Journal of the Neurological Sciences. 1981;51(1):43–50. doi: 10.1016/0022-510x(81)90058-7. [DOI] [PubMed] [Google Scholar]
- 87.Insausti AM, Megías M, Crespo D, et al. Hippocampal volume and neuronal number in Ts65Dn mice: a murine model of Down syndrome. Neuroscience Letters. 1998;253(3):175–178. doi: 10.1016/s0304-3940(98)00641-7. [DOI] [PubMed] [Google Scholar]
- 88.Ayberk Kurt M, Ilker Kafa M, Dierssen M, Ceri Davies D. Deficits of neuronal density in CA1 and synaptic density in the dentate gyrus, CA3 and CA1, in a mouse model of Down syndrome. Brain Research. 2004;1022(1-2):101–109. doi: 10.1016/j.brainres.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 89.Contestabile A, Fila T, Ceccarelli C, et al. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with down syndrome and in Ts65Dn mice. Hippocampus. 2007;17(8):665–678. doi: 10.1002/hipo.20308. [DOI] [PubMed] [Google Scholar]
- 90.Rueda N, Llorens-Martín M, Flórez J, et al. Memantine normalizes several phenotypic features in the Ts65Dn mouse model of down syndrome. Journal of Alzheimer’s Disease. 2010;21(1):277–290. doi: 10.3233/JAD-2010-100240. [DOI] [PubMed] [Google Scholar]
- 91.Dickson PE, Rogers TD, Mar ND, et al. Behavioral flexibility in a mouse model of developmental cerebellar Purkinje cell loss. Neurobiology of Learning and Memory. 2010;94(2):220–228. doi: 10.1016/j.nlm.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Olson LE, Roper RJ, Baxter LL, Carlson EJ, Epstein CJ, Reeves RH. Down syndrome mouse models Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn exhibit variable severity of cerebellar phenotypes. Developmental Dynamics. 2004;230(3):581–589. doi: 10.1002/dvdy.20079. [DOI] [PubMed] [Google Scholar]
- 93.Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down’s syndrome mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(5):1452–1456. doi: 10.1073/pnas.0510750103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Contestabile A, Fila T, Cappellini A, Bartesaghi R, Ciani E. Widespread impairment of cell proliferation in the neonate Ts65Dn mouse, a model for Down syndrome. Cell Proliferation. 2009;42(2):171–181. doi: 10.1111/j.1365-2184.2009.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgoten a generation following the holinergic hypothesis. Experimental Neurology. 2000;163:495–529. doi: 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- 96.Baxter MG, Chiba AA. Cognitive functions of the basal forebrain. Current Opinion in Neurobiology. 1999;9(2):178–183. doi: 10.1016/s0959-4388(99)80024-5. [DOI] [PubMed] [Google Scholar]
- 97.Godridge H, Reynolds GP, Czudek C, Calcutt NA, Benton M. Alzheimer-like neurotransmitter deficits in adult Down’s syndrome brain tissue. Journal of Neurology, Neurosurgery & Psychiatry. 1987;50:775–778. doi: 10.1136/jnnp.50.6.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Risser D, Lubec G, Cairns N, Herrera-Marschitz M. Excitatory amino acids and monoamines in parahippocampal gyrus and frontal cortical pole of adults with Down syndrome. Life Sciences. 1997;60(15):1231–1237. doi: 10.1016/s0024-3205(97)00067-2. [DOI] [PubMed] [Google Scholar]
- 99.Granholm ACE, Sanders LA, Crnic LS. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of down’s syndrome. Experimental Neurology. 2000;161(2):647–663. doi: 10.1006/exnr.1999.7289. [DOI] [PubMed] [Google Scholar]
- 100.Cooper JD, Salehi A, Delcroix JD, et al. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(18):10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hunter CL, Bimonte HA, Granholm ACE. Behavioral comparison of 4 and 6 month-old Ts65Dn mice: age-related impairments in working and reference memory. Behavioural Brain Research. 2003;138(2):121–131. doi: 10.1016/s0166-4328(02)00275-9. [DOI] [PubMed] [Google Scholar]
- 102.Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down’s syndrome. Annals of Neurology. 2004;56(5):675–688. doi: 10.1002/ana.20250. [DOI] [PubMed] [Google Scholar]
- 103.Chakrabarti L, Best TK, Cramer NP, et al. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nature Neuroscience. 2010;13(8):927–934. doi: 10.1038/nn.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hewitt CA, Ling KH, Merson TD, et al. Gene network disruptions and neurogenesis defects in the adult Ts1Cje mouse model of down syndrome. PLoS One. 2010;5(7) doi: 10.1371/journal.pone.0011561. Article ID e11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moldrich RX, Dauphinot L, Laffaire J, et al. Proliferation deficits and gene expression dysregulation in Down’s syndrome (Ts1Cje) neural progenitor cells cultured from neurospheres. Journal of Neuroscience Research. 2009;87(14):3143–3152. doi: 10.1002/jnr.22131. [DOI] [PubMed] [Google Scholar]
- 106.Bahn S, Mimmack M, Ryan M, et al. Nuronal target genes of the neuron-restrictive silener factor in neurosperes derived from fetuses with Down’s syndrome: a gene xpression study. Lancet. 2002;359:310–315. doi: 10.1016/S0140-6736(02)07497-4. [DOI] [PubMed] [Google Scholar]
- 107.Esposito G, Imitola J, Lu J, et al. Genomic and functional profiling of human Down syndrome neural progenitors implicates S100B and aquaporin 4 in cell injury. Human Molecular Genetics. 2008;17(3):440–457. doi: 10.1093/hmg/ddm322. [DOI] [PubMed] [Google Scholar]
- 108.Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997;386(6624):493–495. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- 109.Kempermann G, Gast D, Gage FH. Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Annals of Neurology. 2002;52(2):135–143. doi: 10.1002/ana.10262. [DOI] [PubMed] [Google Scholar]
- 110.Van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nature Neuroscience. 1999;2(3):266–270. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- 111.Van Praag H, Schinder AF, Christle BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415(6875):1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12(5):578–584. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. Journal of Neuroscience. 2000;20(24):9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bianchi P, Ciani E, Contestabile A, Guidi S, Bartesaghi R. Lithium restores neurogenesis in the subventricular zone of the Ts65Dn mouse, a model for Down syndrome. Brain Pathology. 2010;20:106–118. doi: 10.1111/j.1750-3639.2008.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trazzi S, Mitrugno VM, Valli E, et al. APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Human Molecular Genetics. 2011;20(8):1560–1573. doi: 10.1093/hmg/ddr033. [DOI] [PubMed] [Google Scholar]
- 116.Clark S, Schwalbe J, Stasko MR, Yarowsky PJ, Costa ACS. Fluoxetine rescues deficient neurogenesis in hippocampus of the Ts65Dn mouse model for Down syndrome. Experimental Neurology. 2006;200(1):256–261. doi: 10.1016/j.expneurol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 117.Rueda N, Mostany R, Pazos A, Flórez J, Martínez-Cué C. Cell proliferation is reduced in the dentate gyrus of aged but not young Ts65Dn mice, a model of Down syndrome. Neuroscience Letters. 2005;380(1-2):197–201. doi: 10.1016/j.neulet.2005.01.039. [DOI] [PubMed] [Google Scholar]
- 118.Isihara K, Amano K, Takaki E, et al. Increased lipid peroxidation in Down’s syndrome mouse models. Journal of Neurochemistry. 2009;110:1965–1976. doi: 10.1111/j.1471-4159.2009.06294.x. [DOI] [PubMed] [Google Scholar]
- 119.Contestabile A, Fila T, Bartesaghi R, Ciani E. Cell cycle elongation impairs proliferation of cerebellar granule cell precursors in the Ts65Dn mouse, an animal model for down syndrome. Brain Pathology. 2009;19(2):224–237. doi: 10.1111/j.1750-3639.2008.00168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Laffaire J, Rivals I, Dauphinot L, et al. Gene expression signature of cerebellar hypoplasia in a mouse model of Down syndrome during postnatal development. BMC Genomics. 2009;10, article no. 138 doi: 10.1186/1471-2164-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guimera J, Casas C, Pucharcos C, et al. A human homologue of Drosophila minibrain (MNB) is expressed in the neuronal regions affected in Down syndrome and maps to the critical region. Human Molecular Genetics. 1996;5:1305–1310. doi: 10.1093/hmg/5.9.1305. [DOI] [PubMed] [Google Scholar]
- 122.Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. Journal of Biological Chemistry. 1998;273(40):25893–25902. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- 123.Tejedor F, Zhu XR, Kaltenbach E, et al. Minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14(2):287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- 124.Guimera J, Casas C, Estivill X, Pritchard M. Human minigrain homologue (MNBH/DYRK1): characterization, alterantive splicing, differential tissue expression, and overexpression in Down syndrome. Genomics. 1999;57:407–418. doi: 10.1006/geno.1999.5775. [DOI] [PubMed] [Google Scholar]
- 125.Kentrup H, Becker W, Heukelbach J, et al. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. Journal of Biological Chemistry. 1996;271(7):3488–3495. doi: 10.1074/jbc.271.7.3488. [DOI] [PubMed] [Google Scholar]
- 126.Yang EJ, Ahn YS, Chung KC. Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. Journal of Biological Chemistry. 2001;276(43):39819–39824. doi: 10.1074/jbc.M104091200. [DOI] [PubMed] [Google Scholar]
- 127.Hammerle B, Carnicero A, Elizalde C, Ceron J, Martinez S, Tejedor FJ. Expression patterns and subcellular localization of the Down syndrome candidate protein MNB/DYRK1A suggest a role in late neuronal diffferentiation. European Journal of Neuroscience. 2003;17:2277–2286. doi: 10.1046/j.1460-9568.2003.02665.x. [DOI] [PubMed] [Google Scholar]
- 128.Yabut O, Domagauer J, D’Arcangelo G. Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. The Journal of Neuroscience. 2010;30:4004–4014. doi: 10.1523/JNEUROSCI.4711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Park J, Oh Y, Yoo L, et al. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. Journal of Biological Chemistry. 2010;285(41):31895–31906. doi: 10.1074/jbc.M110.147520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ortiz-Abalia J, Sahún I, Altafaj X, et al. Targeting Dyrk1A with AAVshRNA attenuates motor alterations in TgDyrk1A, a mouse model of Down syndrome. American Journal of Human Genetics. 2008;83(4):479–488. doi: 10.1016/j.ajhg.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Takebayashi H, Yoshida S, Sugimori M, et al. Dynamic expression of basic helix-loop-helix Olig family members: implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mechanisms of Development. 2000;99(1-2):143–148. doi: 10.1016/s0925-4773(00)00466-4. [DOI] [PubMed] [Google Scholar]
- 132.Lu QR, Sun T, Zhu Z, et al. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109(1):75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- 133.Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109(1):61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
- 134.Morrison SJ, Perez SE, Qiao Z, et al. Transient notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell. 2000;101(5):499–510. doi: 10.1016/s0092-8674(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 135.Fischer DF, Van Dijk R, Sluijs JA, et al. Activation of the notch pathway in down syndrome: cross-talk of notch and APP. FASEB Journal. 2005;19(11):1451–1458. doi: 10.1096/fj.04-3395.com. [DOI] [PubMed] [Google Scholar]
- 136.Shankaran M, King C, Lee J, Busch R, Wolff M, Hellerstein MK. Discovery of novel hippocampal neurogenic agents by using an in vivo stable isotope labeling technique. Journal of Pharmacology and Experimental Therapeutics. 2006;319(3):1172–1181. doi: 10.1124/jpet.106.110510. [DOI] [PubMed] [Google Scholar]
- 137.Das I, Reeves RH. Society for Neuroscience Meeting Abstracts. 2010.
- 138.Greenough WT, Volkmar FR, Juraska JM. Effects of rearing complexity on dendritic branching in frontolateral and temporal cortex of the rat. Experimental Neurology. 1973;41(2):371–378. doi: 10.1016/0014-4886(73)90278-1. [DOI] [PubMed] [Google Scholar]
- 139.Nilsson M, Perfilieva E, Johansson U, Orwar O, Eriksson PS. Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. Journal of Neurobiology. 1999;39:569–578. doi: 10.1002/(sici)1097-4695(19990615)39:4<569::aid-neu10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 140.Van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(23):13427–13431. doi: 10.1073/pnas.96.23.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Paylor R, Morrison SK, Rudy JW, Waltrip LT, Wehner JM. Brief exposure to an enriched environment improves performance on the Morris water task and increases hippocampal cytosolic protein kinase C activity in young rats. Behavioural Brain Research. 1992;52(1):49–56. doi: 10.1016/s0166-4328(05)80324-9. [DOI] [PubMed] [Google Scholar]
- 142.Hargreaves EL, Boon F, Cain DP. Rats housed in a complex environment exhibit greater hilar LTP than individually housed littermates. Society for Neuroscience - Abstracts. 1992;18, abstract 344 [Google Scholar]
- 143.Schwartz S. Effect of neonatal cortical lesions and early environmental factors on adult rat behavior. Journal of Comparative & Physiological Psychology. 1964;57:72–77. doi: 10.1037/h0045518. [DOI] [PubMed] [Google Scholar]
- 144.Kolb B, Sutherland RJ, Wishaw IQ. A comparison of the contribution of the frontal and parietal association cortex to spatial localization in rats. Behavioral Neuroscience. 1983;97:13–27. doi: 10.1037//0735-7044.97.1.13. [DOI] [PubMed] [Google Scholar]
- 145.Boehm GW, Sherman GF, Hoplight BJ, et al. Learning and memory in the autoimmune BXSB mouse: effects of neocortical ectopias and environmental enrichment. Brain Research. 1996;726(1-2):11–22. [PubMed] [Google Scholar]
- 146.Chakrabarti L, Scafidi J, Gallo V, Haydar TF. Environmental enrichment rescues postnatal neurogenesis defect in the male and female Ts65Dn mouse model of Down syndrome. Developmental Neuroscience. 2011;33:428–441. doi: 10.1159/000329423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.O’Callaghan RM, Ohle R, Kelly AM. The effects of torced exercise on hippocampal plasticity in the rat: a comparison of LTP, spatial and non-spatial learning. Behavioural Brain Research. 2007;176:362–366. doi: 10.1016/j.bbr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 148.Van Praag H. Neurogenesis and exercise: past and future directions. NeuroMolecular Medicine. 2008;10(2):128–140. doi: 10.1007/s12017-008-8028-z. [DOI] [PubMed] [Google Scholar]
- 149.Chen L, Gong S, Shan LD, et al. Effects of exercise on neurogenesis in the dentate gyrus and ability of learning and memory after hippocampus lesion in adult rats. Neuroscience Bulletin. 2006;22(1):1–6. [PubMed] [Google Scholar]
- 150.Clark PJ, Brzezinska WJ, Thomas MW, Ryzhenko NA, Toshkov SA, Rhodes JS. Intact neurogenesis is required for benefits of exercise on spatial memory but not motor performance or contextual fear conditioning in C57BL/6J mice. Neuroscience. 2008;155(4):1048–1058. doi: 10.1016/j.neuroscience.2008.06.051. [DOI] [PubMed] [Google Scholar]
- 151.Anderson AJ, Stolzner S, Lai F, Su J, Nixon RA. Morphological and biochemical assesment of DNA damage and apoptosis in Down syndrome and Alzheirmer's disease, an effect of postmortem tissue archival on tunnel. Neurobiology of Aging. 2000;21:511–524. doi: 10.1016/s0197-4580(00)00126-3. [DOI] [PubMed] [Google Scholar]
- 152.Sawa A, Oyama F, Cairns NJ, Amano N, Matsushita M. Aberrant expression of bcl-2 gene family in Down’s syndrome brains. Molecular Brain Research. 1997;48(1):53–59. doi: 10.1016/s0169-328x(97)00078-8. [DOI] [PubMed] [Google Scholar]
- 153.Gulesserian T, Engidawork E, Yoo BC, Cairns N, Lubec G. Alteration of caspases and other apoptosis regulatory proteins in Down syndrome. Journal of Neural Transmission. 2001;61:163–179. doi: 10.1007/978-3-7091-6262-0_13. [DOI] [PubMed] [Google Scholar]
- 154.Gulesserian T, Engidawork E, Yoo BC, Cairns N, Lubec G. Comparable apoptotic pathways in frontal cortex and cerebellum of patients with Down syndrome. 2001 [Google Scholar]
- 155.Helguera P, Pelsman A, Pigino G, Wolvetang E, Head E, Busciglio J. Ets-2 pronotes the activation of a mitocondrial death pathway in Down's syndrome neurons. The Journal of Neuroscience. 2005;25:2295–2303. doi: 10.1523/JNEUROSCI.5107-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Engidawork E, Balic N, Juranville JF, Fountoulakis M, Dierssen M, Lubec G. Unaltered expression of Fas (CD95/APO-1), Caspase-3, Bcl-2 and annexins in brains of fetal Down syndrome: evidence against increased apoptosis. Journal of Neural Transmission. 2001;61:149–162. doi: 10.1007/978-3-7091-6262-0_12. [DOI] [PubMed] [Google Scholar]
- 157.Seidl R, Fang-Kircher S, Bidmon B, Cairns N, Lubec G. Apoptosis-associated proteins p53 and APO-1/Fas (CD95) in brains of adult patients with Down syndrome. Neuroscience Letters. 1999;260(1):9–12. doi: 10.1016/s0304-3940(98)00945-8. [DOI] [PubMed] [Google Scholar]
- 158.Rueda N, Flórez J, Martínez-Cué C. The Ts65Dn mouse model of Down syndrome shows reduced expression of the Bcl-XL antiapoptotic protein in the hippocampus not accompanied by changes in molecular or cellular markers of cell death. International Journal of Developmental Neuroscience. 2011;29:711–716. doi: 10.1016/j.ijdevneu.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 159.Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends in Neurosciences. 2003;26(7):360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- 160.Newpher TM, Ehlers MD. Spine microdomains for postsynaptic signaling and plasticity. Trends in Cell Biology. 2009;19(5):218–227. doi: 10.1016/j.tcb.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 161.Takashima S, Iida K, Mito T, Arima M. Dendritic and histochemical development and ageing in patients with Down’s syndrome. Journal of Intellectual Disability Research. 1994;38:265–273. doi: 10.1111/j.1365-2788.1994.tb00394.x. [DOI] [PubMed] [Google Scholar]
- 162.Vuksic M, Petanjek Z, Rasin MR, Kostovic I. Perinatal growth of prefrontal layer III pyramids in Dosn syndrome. Pediatric Neurology. 2002;27:36–38. doi: 10.1016/s0887-8994(02)00380-6. [DOI] [PubMed] [Google Scholar]
- 163.Becker LE, Armstrong DL, Chan F. Dendritic atrophy in children with Down’s syndrome. Annals of Neurology. 1986;20(4):520–526. doi: 10.1002/ana.410200413. [DOI] [PubMed] [Google Scholar]
- 164.Prinz M, Prinz B, Schulz E. The growth of non-pyramidal neurons in the primary motor cortex of man: a Golgi study. Histology and Histopathology. 1997;12:895–900. [PubMed] [Google Scholar]
- 165.Takashima S, Becker LE, Armstrong DL, Chan FW. Abnormal neuronal development in the visual cortex of the human fetus and infant with Down’s syndrome. A quantitative and qualitative Golgi study. Brain Research. 1981;225(1):1–21. doi: 10.1016/0006-8993(81)90314-0. [DOI] [PubMed] [Google Scholar]
- 166.Schulz E, Scholz B. Neurohistological findings in the parietal cortex of children with chromosome aberrations. Journal für Hirnforschung. 1992;33:37–62. [PubMed] [Google Scholar]
- 167.Takashima S, Ieshima A, Nakamura H, Becker LE. Dendrites, dementia and the Down syndrome. Brain and Development. 1989;11(2):131–133. doi: 10.1016/s0387-7604(89)80082-8. [DOI] [PubMed] [Google Scholar]
- 168.Suetsugu M, Mehraein P. Spine distribution along the apical dendrites of the pyramidal neurons in Down’s syndrome. A quantitative Golgi study. Acta Neuropathologica. 1980;50(3):207–210. doi: 10.1007/BF00688755. [DOI] [PubMed] [Google Scholar]
- 169.Marin Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down’s syndrome. A Golgi study. Journal of Comparative Neurology. 1976;167(1):63–81. doi: 10.1002/cne.901670105. [DOI] [PubMed] [Google Scholar]
- 170.Ferrer I, Gullotta F. Down’s syndrome and Alzheimer’s disease: dendritic spine counts in the hippocampus. Acta Neuropathologica. 1990;79(6):680–685. doi: 10.1007/BF00294247. [DOI] [PubMed] [Google Scholar]
- 171.Dierssen M, Benavide-Piccione R, Martinez-Cue C, et al. Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: effects of environmental enrichment. Cerebral Cortex. 2003;13:758–764. doi: 10.1093/cercor/13.7.758. [DOI] [PubMed] [Google Scholar]
- 172.Belichenko PV, Masliah E, Kleschevnikov AM, et al. Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. Journal of Comparative Neurology. 2004;480(3):281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- 173.Popov VI, Kleschevkikov AM, Klimenko OA, Stewart MG, Belichenko PV. Three-dimensional ultrastructure in the dentate gyrus and hippocampal area CA3 in the Ts65Dn mouse model of Down syndrome. The Journal of Comparative Neurology. 2011;519:1338–1354. doi: 10.1002/cne.22573. [DOI] [PubMed] [Google Scholar]
- 174.Belichenko PV, Wright EE, Belichenko NP, et al. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. The Journal of Comparative Neurology. 2009;514:240–258. doi: 10.1002/cne.22009. [DOI] [PubMed] [Google Scholar]
- 175.Wang JW, David DJ, Monckton JE, Battaglia F, Hen R. Chronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cells. Journal of Neuroscience. 2008;28(6):1374–1384. doi: 10.1523/JNEUROSCI.3632-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kempermann G, Brandon EP, Gage FH. Environmental stimulation of 129/SvJ mice causes increased cell proliferation and neurogenesis in the adult dentate gyrus. Current Biology. 1998;8(16):939–942. doi: 10.1016/s0960-9822(07)00377-6. [DOI] [PubMed] [Google Scholar]
- 177.Ayberk Kurt M, Ceri Davies D, Kidd M, Dierssen M, Flórez J. Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Research. 2000;858(1):191–197. doi: 10.1016/s0006-8993(00)01984-3. [DOI] [PubMed] [Google Scholar]
- 178.Perez-Cremades D, Hernandez S, Blasco-Ibañez JM, Crespo C, Nacher J, Varea E. Alteration in inhibitory circuits in the somatoseensory cortex of Ts65Dn mice, a model for Down’s syndrome. Journal of Neural Transmission. 2010;117:445–455. doi: 10.1007/s00702-010-0376-9. [DOI] [PubMed] [Google Scholar]
- 179.Devinsky O, Sato S, Conwit RA, Schapiro MB. Relation of EEG alpha backgroud to cognitive function, brain atrophy, and cerebral metabolism in Down’s sndrome. Age specific changes. Archives of Neurology. 1990;47:58–62. doi: 10.1001/archneur.1990.00530010072022. [DOI] [PubMed] [Google Scholar]
- 180.Schmid RG, Tirsch WS, Rappelsberger P, Weinmann HM, Poppl SJ. Comparative coherence studies in healthy volunteers and Down’s syndrome patients from childhood to adult age. Electroencephalography and Clinical Neurophysiology. 1992;83:112–123. doi: 10.1016/0013-4694(92)90024-c. [DOI] [PubMed] [Google Scholar]
- 181.Karrer JH, Karrer R, Bloom D, Chaney L, Davis R. Event-related brain potentials during na extended visual recognition memory task depict delayed development of cerebelar inhibitory processes among 6-month-old infants with Down synrome. International Journal of Psychophysiology. 1998;29:167–200. doi: 10.1016/s0167-8760(98)00015-4. [DOI] [PubMed] [Google Scholar]
- 182.Siarey RJ, Stoll J, Rapoport SI, Galdzicki Z. Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down Syndrome. Neuropharmacology. 1997;36(11-12):1549–1554. doi: 10.1016/s0028-3908(97)00157-3. [DOI] [PubMed] [Google Scholar]
- 183.Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, Galdzicki Z. Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology. 1999;38(12):1917–1920. doi: 10.1016/s0028-3908(99)00083-0. [DOI] [PubMed] [Google Scholar]
- 184.Costa ACS, Grybko MJ. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neuroscience Letters. 2005;382(3):317–322. doi: 10.1016/j.neulet.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 185.Fernandez F, Morishita W, Zuniga E, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nature Neuroscience. 2007;10(4):411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 186.Kleschevnicov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of down syndrome. Journal of Neuroscience. 2004;24(37):8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hanson JE, Blank M, Valenzuela RA, Garner CC, Madison DV. The functional nature of synaptic circuitry is altered in area CA3 of the hippocampus in a mouse model of Down’s syndrome. Journal of Physiology. 2007;579(1):53–67. doi: 10.1113/jphysiol.2006.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Scott-McKean JJ, Costa AC. Exaggerated NMDA mediated LTD in a mouse model of Down syndrome and pharmacological rescuing by memantine. Learning & Memory. 2011;18:774–778. doi: 10.1101/lm.024182.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kleschevnikov AM, Belichenko PV, Gall J, et al. Increased efficiency of the GABAA and GABAB receptor-mediated neurotransmission in the Ts65Dn mouse model of Down syndrome. Neurobiology of Disease. 2012;45(2):683–691. doi: 10.1016/j.nbd.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19(3):687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- 191.Best TK, Siarey RJ, Galdzicki Z. Ts65Dn, a mouse model of Down syndrome, exibits increaed GABAB-induced potasium current. Journal of Neurophysiology. 2007;97:892–900. doi: 10.1152/jn.00626.2006. [DOI] [PubMed] [Google Scholar]
- 192.Cole SO. Effects of benzodiazepines on acquisition and performance: a critical assessment. Neuroscience and Biobehavioral Reviews. 1986;10(3):265–272. doi: 10.1016/0149-7634(86)90013-8. [DOI] [PubMed] [Google Scholar]
- 193.Lister RG. The amnesic actions of benzodiazepines in man. Neuroscience and Biobehavioral Reviews. 1985;9(1):87–94. doi: 10.1016/0149-7634(85)90034-x. [DOI] [PubMed] [Google Scholar]
- 194.Ghoneim MM, Mewaldt SP. Benzodiazepines and human memory: a review. Anesthesiology. 1990;72(5):926–938. [PubMed] [Google Scholar]
- 195.Sarter M, Bruno JP, Berntson GG. Psychotogenic properties of benzodiazepine receptor inverse agonists. Psychopharmacology. 2001;156(1):1–13. doi: 10.1007/s002130100756. [DOI] [PubMed] [Google Scholar]
- 196.Venault P, Chapouthier G, de Carvalho LP, et al. Benzodiazepine impairs and beta-carboline enhances performance in learning and memory tasks. Nature. 1986;2:864–866. doi: 10.1038/321864a0. [DOI] [PubMed] [Google Scholar]
- 197.Venault P, Chapouthier G. From the behavioral pharmacology of beta-carbolines to seizures, anxiety, and memory. TheScientificWorldJournal. 2007;7:204–223. doi: 10.1100/tsw.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rueda N, Florez J, Martinez-Cue C. Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neuroscience Letters. 2008;433:22–27. doi: 10.1016/j.neulet.2007.12.039. [DOI] [PubMed] [Google Scholar]
- 199.Dorow R, Horowski R, Pascheke G, Amin M, Braestrup C. Severe anxiety induced by FG7142, a beta-carboline ligand for benzodiazepine receptors. Lancet. 1983;2:98–99. doi: 10.1016/s0140-6736(83)90076-4. [DOI] [PubMed] [Google Scholar]
- 200.Collinson N, Kuenzi FM, Jarolimek W, et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the α5 subunit of the GABAA receptor. Journal of Neuroscience. 2002;22(13):5572–5580. doi: 10.1523/JNEUROSCI.22-13-05572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Crestani F, Kesist R, Frischy JM, et al. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Atack JR, Bayley PJ, Seabrook GR, Watford KA, McKernan RM, Dawson GR. L-655,708 enhances cognition in rats but is not proconvulsant at a dose selective for alfa5-containing GABAA receptors. Neuropharmacology. 2006;51:1023–1029. doi: 10.1016/j.neuropharm.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 203.Dawson GR, Mauback KA, Collison N, et al. An inverse agonist selective for α5 subunit-containing GABAA receptors enhances cognition. Journal of Pharmacology and Experimental Therapeutics. 2006;316:1335–1345. doi: 10.1124/jpet.105.092320. [DOI] [PubMed] [Google Scholar]
- 204.Ballard TM, Knoflach F, Prinssen E, et al. RO4938581, a novel cognitive enhancer acting at GABAA α5 subunit-containing receptors. Psychopharmacology. 2009;202(1–3):207–223. doi: 10.1007/s00213-008-1357-7. [DOI] [PubMed] [Google Scholar]
- 205.Bradeau J, Delatour B, Duchon A, et al. Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. Psychopharmacology. 2011;25(8):1030–1042. doi: 10.1177/0269881111405366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Begenisic T, Spolidoro M, Braschi C, et al. Environmental enrichment decreases GABAergic inhibition and improves cognitive abilities, synaptic plasticity, and visual functions in a mouse model of Down syndrome. Frontiers in Cellular Neuroscience. 2011;5, article 29 doi: 10.3389/fncel.2011.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Whittle N, Sartori SB, Dierssen M, Lubec G, Singewald N. Fetal Down syndrome brains exhibit aberrant levels of neurotransmitters critical for normal brain development. Pediatrics. 2007;120(6):e1465–e1471. doi: 10.1542/peds.2006-3448. [DOI] [PubMed] [Google Scholar]
- 208.Schneider C, Risser D, Kirchner L, et al. Similar deficits of central histaminergic system in patients with Down syndrome and Alzheimer disease. Neuroscience Letters. 1997;222(3):183–186. doi: 10.1016/s0304-3940(97)13379-1. [DOI] [PubMed] [Google Scholar]
- 209.Yates CM, Simpson J, Gordon A. Regional brain 5-hydroxytryptamine levels are reduced in senile Down’s syndrome as in Alzheimer’s disease. Neuroscience Letters. 1986;65(2):189–192. doi: 10.1016/0304-3940(86)90302-2. [DOI] [PubMed] [Google Scholar]
- 210.Bhattacharyya A, McMillan E, Chen SI, Wallace K, Svendsen CN. A critical period in cortical interneuron neurogenesis in down syndrome revealed by human neural progenitor cells. Developmental Neuroscience. 2009;31(6):497–510. doi: 10.1159/000236899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Fernandez F, Trinidad JC, Blank M, Feng DD, Burlingame AL, Garner CC. Normal protein composition of synapses in Ts65Dn mice: a mouse model of Down syndrome. Journal of Neurochemistry. 2009;110(1):157–169. doi: 10.1111/j.1471-4159.2009.06110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tozuka Y, Fukuda S, Namba T, Seki T, Hisatsune T. GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron. 2005;47(6):803–815. doi: 10.1016/j.neuron.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 213.Ge S, Goh ELK, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439(7076):589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Earnheart JC, Schweizer C, Crestani F, et al. GABA-ergic control of adult hippoampal neurogenesis in relation to behavior indicative of trait anxiety and depression states. The Journal of Neuroscience. 2007;27:3845–3854. doi: 10.1523/JNEUROSCI.3609-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sairanen M, Lucas G, Ernfors P, Castrén M, Castrén E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. Journal of Neuroscience. 2005;25(5):1089–1094. doi: 10.1523/JNEUROSCI.3741-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Vink J, Incerti M, Toso L, Robretson R, Abebe D, Spong CY. Prenatal NAP+SAL prevents developmental delay in a mouse model of Down syndrome through effects on N-methyl-D aspartic acid and gamma-aminobutyric acid receptors. American Journal of Obstetrics & Gynecology. 2009;200:521–524. doi: 10.1016/j.ajog.2009.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Siddiqui A, Lacroix T, Stasko MR, Scott-McKean JJ, Costa ACS, Gardiner KJ. Molecular responses of the Ts65Dn and Ts1Cje mouse models of Down syndrome to MK-801. Genes, Brain and Behavior. 2008;7(7):810–820. doi: 10.1111/j.1601-183X.2008.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Siarey RJ, Kline-Burgess A, Cho M, et al. Altered signaling pathways underlying abnormal hippocampal synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Journal of Neurochemistry. 2006;98(4):1266–1277. doi: 10.1111/j.1471-4159.2006.03971.x. [DOI] [PubMed] [Google Scholar]
- 219.Kahlem P, Sultan M, Herwig R, et al. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of Down syndrome. Genome Research. 2004;14(7):1258–1267. doi: 10.1101/gr.1951304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature. 1994;369(6477):235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- 221.Costa ACS, Scott-McKean JJ, Stasko MR. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology. 2008;33(7):1624–1632. doi: 10.1038/sj.npp.1301535. [DOI] [PubMed] [Google Scholar]
- 222.Lockrow J, Boger H, Bimonte-Nelson H, Granholm AC. Effecs of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behavioural Brain Research. 2011;221:610–622. doi: 10.1016/j.bbr.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hanney M, Prasher V, Williams N, et al. Memantine for dementia in adults older than 40 years with Down’s syndrome (MEADOWS): a randomised, double-blind placebo-controlled trial. The Lancet. 2012;6736:61676–67180. doi: 10.1016/S0140-6736(11)61676-0. [DOI] [PubMed] [Google Scholar]
- 224.Megías M, Verduga R, Dierssen M, Flórez J, Insausti R, Crespo D. Cholinergic, serotonergic and catecholaminergic neurons are not affected in Ts65Dn mice. NeuroReport. 1997;8(16):3475–3478. doi: 10.1097/00001756-199711100-00012. [DOI] [PubMed] [Google Scholar]
- 225.Whitaker-Azmitia PM. Serotonin and brain development: role in human developmental diseases. Brain Research Bulletin. 2001;56(5):479–485. doi: 10.1016/s0361-9230(01)00615-3. [DOI] [PubMed] [Google Scholar]
- 226.Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301(5634):805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 227.Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(21):8233–8238. doi: 10.1073/pnas.0601992103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bar-Peled O, Gross-Isseroff R, Ben-Hur H, Hoskins I, Groner Y, Biegon A. Fetal human brain exhibits a prenatal peak in the density of serotonin 5-HT(1A) receptors. Neuroscience Letters. 1991;127(2):173–176. doi: 10.1016/0304-3940(91)90787-t. [DOI] [PubMed] [Google Scholar]
- 229.Chen Y, Dyakin VV, Branch CA, et al. In vivo MRI identifies cholinergic circuitry deficits in a Down syndrome model. Neurobiology of Aging. 2008;30:1453–1465. doi: 10.1016/j.neurobiolaging.2007.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Contestabile A, Fila T, Bartesaghi R, Contestabile A, Ciani E. Choline acetyltransferase activity at different ages in brain of Ts65Dn mice, an animal model for Down’s syndrome and related neurodegenerative diseases. Journal of Neurochemistry. 2006;97(2):515–526. doi: 10.1111/j.1471-4159.2006.03769.x. [DOI] [PubMed] [Google Scholar]
- 231.Mann DM, Yates PO, Marcyniuk B, Ravindra CR. Pathological evidence for neurotransmitter deficits in Down’s syndrome of middle age. Journal of Mental Deficiency Research. 1985;29:125–135. doi: 10.1111/j.1365-2788.1985.tb00320.x. [DOI] [PubMed] [Google Scholar]
- 232.Coyle JT, Oster-Granite ML, Gearhart JD. The neurobiologic consequences of Down syndrome. Brain Research Bulletin. 1986;16(6):773–787. doi: 10.1016/0361-9230(86)90074-2. [DOI] [PubMed] [Google Scholar]
- 233.Dierssen M, Vallina IF, Baamonde C, et al. Impaired cyclic AMP production in the hippocampus of a Down syndrome murine model. Developmental Brain Research. 1996;95(1):122–124. doi: 10.1016/0165-3806(96)00071-5. [DOI] [PubMed] [Google Scholar]
- 234.Dierssen M, Vallina IF, Baamonde C, García-Calatayud S, Lumbreras MA, Flórez J. Alterations of central noradrenergic transmission in Ts65Dn mouse, a model for Down syndrome. Brain Research. 1997;749(2):238–244. doi: 10.1016/s0006-8993(96)01173-0. [DOI] [PubMed] [Google Scholar]
- 235.Lumbreras M, Baamonde C, Martínez-Cué C, et al. Brain G protein-dependent signaling pathways in Down syndrome and Alzheimer’s disease. Amino Acids. 2006;31(4):449–456. doi: 10.1007/s00726-005-0272-z. [DOI] [PubMed] [Google Scholar]
- 236.Duman RS, Nakagawa S, Malberg J. Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology. 2001;25(6):836–844. doi: 10.1016/S0893-133X(01)00358-X. [DOI] [PubMed] [Google Scholar]
- 237.Murchison CF, Zhang XY, Zhang WP, Ouyang M, Lee A, Thomas SA. A distinct role for norepinephrine in memory retrieval. Cell. 2004;117(1):131–143. doi: 10.1016/s0092-8674(04)00259-4. [DOI] [PubMed] [Google Scholar]
- 238.Chao MY, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clinical Science. 2006;110(2):167–173. doi: 10.1042/CS20050163. [DOI] [PubMed] [Google Scholar]
- 239.Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annual Review of Neuroscience. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- 240.Campenot RB, McInnis BI. Retrograde transport of neurotrophins: fact and function. Journal of Neurobiology. 2004;58:217–229. doi: 10.1002/neu.10322. [DOI] [PubMed] [Google Scholar]
- 241.Guedj F, Sebrie C, Rivals I, et al. Green tea polyphenols rescue brain defects induced by overexpression of DYRK1A. PLoS One. 2009;4, article e4606 doi: 10.1371/journal.pone.0004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Toiber D, Azkona G, Ben-Ari S, Toran N, Soreq H, Dierssen M. Engineering DYRK! A overdosage yields Down syndrome-characteristic cortical splicing aberrations. Neurobiology of Disease. 2010;40:348–359. doi: 10.1016/j.nbd.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 243.Fukuda Y, Berry TL, Nelson M, et al. Stimulated neuronal expression of brain-derived neurotrophic factor by Neurotropin. Molecular and Cellular Neuroscience. 2010;45(3):226–233. doi: 10.1016/j.mcn.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 244.Bimonte-Nelson HA, Hunter CL, Nelson ME, Granholm ACE. Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behavioural Brain Research. 2003;139(1-2):47–57. doi: 10.1016/s0166-4328(02)00082-7. [DOI] [PubMed] [Google Scholar]
- 245.Pollonini G, Gao V, Rabe A, Palminiello S, Albertini G, Alberini CM. Abnormal expression of synaptic proteins and neurotrophin-3 in the down syndrome mouse model Ts65Dn. Neuroscience. 2008;156(1):99–106. doi: 10.1016/j.neuroscience.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Chohan MO, Li B, Blanchard J, et al. Enhancement of dentate gyrus neurogenesis, dendritic and synaptic plasticity and memory by a neurotrophic peptide. Neurobiology of Aging. 2011;32(8):1420–1434. doi: 10.1016/j.neurobiolaging.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 247.Blanchard J, Bolognin S, Chohan MO, Rabe A, Iqbal K, Grundke-Iqbal I. Rescue of synaptic failure and alleviation of learning and memory impairments in a trisomic mouse model of down syndrome. Journal of Neuropathology & Experimental Neurology. 2011;70:1070–1079. doi: 10.1097/NEN.0b013e318236e9ad. [DOI] [PubMed] [Google Scholar]
- 248.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochemical Journal. 2003;371(1):199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Xie W, Ramakrishna N, Wieraszko A, Hwang YW. Promotion of neuronal plasticity by (-)-epigallocatechin-3-gallate. Neurochemical Research. 2008;33(5):776–783. doi: 10.1007/s11064-007-9494-7. [DOI] [PubMed] [Google Scholar]
- 250.Singha B, Shankarb S, Srivastavaa RK. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical Biochemical applications. Pharmacology. 2011;82:1807–1821. doi: 10.1016/j.bcp.2011.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Hata T, Kita T, Itoh E, Oyama R, Kawabata A. Mechanism of the analgesic effect of neurotropin. Japanese Journal of Pharmacology. 1988;48(2):165–173. doi: 10.1254/jjp.48.165. [DOI] [PubMed] [Google Scholar]
- 252.Krasuski JS, Alexander GE, Horwitz B, Rapoport SI, Schapiro MB. Relation of medial temporal lobe volumes to age and memory function in nondemented adults with Down’s syndrome: implications for the prodromal phase of Alzheimer’s disease. American Journal of Psychiatry. 2002;159(1):74–81. doi: 10.1176/appi.ajp.159.1.74. [DOI] [PubMed] [Google Scholar]
- 253.Griffin WST, Stanley LC, Ling C, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(19):7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wenk GL, McGann K, Mencarelli A, Hauss-Wegrzyniak B, Del Doldato P, Fiorucci S. Mechanisms to prevent the toxicity of chronic neuroinflammtion on forebrain cholinergic neurons. European Journal of Pharmacology. 2000;402:77–85. doi: 10.1016/s0014-2999(00)00523-9. [DOI] [PubMed] [Google Scholar]
- 255.Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behavioural Brain Research. 2009;196(2):168–179. doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 256.Incerti M, Toso L, Vink J, et al. Prevention of learning deficit in a Down syndrome model. Obstetrics & Gynecology. 2011;117:354–361. doi: 10.1097/AOG.0b013e3182051ca5. [DOI] [PubMed] [Google Scholar]
- 257.Gozes I, Morimoto BH, Tiong J, et al. NAP: search and development of a peptide derived from activit-deendent neuroprotective protein (ADNP) CNS Drug Reviews. 2005;11:353–368. doi: 10.1111/j.1527-3458.2005.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Gozes I, Divinski I, Piltzer I. NAP and D-SAL: neuroprotection against the β amyloid peptide (1-42) BMC Neuroscience. 2008;9(3, article no. S3) doi: 10.1186/1471-2202-9-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Busciglio J, Pelsman A, Helguera P, et al. NAP and ADNF-9 protect normal and Down’s syndrome cortical neurons from oxidative damage and apoptosis. Current Pharmaceutical Design. 2007;13(11):1091–1098. doi: 10.2174/138161207780618957. [DOI] [PubMed] [Google Scholar]
- 260.Toso L, Carmeroni I, Roberson R, Abebe D, Bissell S, Spong CY. Prevention of developmental delays in a Down syndrome mouse model. Obstetrics & Gynecology. 2008;112:1242–1251. doi: 10.1097/AOG.0b013e31818c91dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Capone G, Kim P, Jovanovich S, et al. Evidence for increased mitochondrial superoxide production in Down syndrome. Life Sciences. 2002;70(24):2885–2895. doi: 10.1016/s0024-3205(02)01538-2. [DOI] [PubMed] [Google Scholar]
- 262.Jovanovic SV, Clements D, MacLeod K. Biomarkers of oxidative stress are significantly elevated in Down syndrome. Free Radical Biology and Medicine. 1998;25(9):1044–1048. doi: 10.1016/s0891-5849(98)00137-3. [DOI] [PubMed] [Google Scholar]
- 263.Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature. 1995;378(6559):776–779. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 264.Kim SH, Yoo BC, Broers JL, Cairns N, Lubec G. Neuroendocrine-specific protein C, a marker of neuronal differentiation, is reduced in brain of patients with Down syndrome and Alzheimer’s disease. Biochemical and Biophysical Research Communications. 2000;276:329–334. doi: 10.1006/bbrc.2000.3464. [DOI] [PubMed] [Google Scholar]
- 265.Pelsman A, Hoyo-Vadillo C, Gudasheva TA, Seredenin SB, Ostrovskaya RU, Busciglio J. GVS-111 prevents oxidative damage and apoptosis in normal and Down’s syndrome human cortical neurons. International Journal of Developmental Neuroscience. 2003;21(3):117–124. doi: 10.1016/s0736-5748(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 266.Rueda N, Florez J, Martinez-Cue C. Effects of chronic administration of SGS-111 during adulthood and during the pre- and post-natal periods on the cognitive deficits of Ts65Dn mice, a model of Down syndrome. Behavioural Brain Research. 2008;188:355–367. doi: 10.1016/j.bbr.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 267.Lockrow J, Prakasam A, Huang P, Bimonte-Nelson H, Sambamurti K, Granholm AC. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Experimental Neurology. 2009;216(2):278–289. doi: 10.1016/j.expneurol.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Shichiri M, Yoshida Y, Ishida N, et al. alpha-Tocopherol suppreses lipid peroxiation and behavioural and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radical Biology and Medicine. 2011;15:1801–1811. doi: 10.1016/j.freeradbiomed.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 269.Blehaut H, Mircher C, Ravel A, et al. Effect of leucovorin (folinic acid) on the developmental quotient of children with Down’s syndrome (trisomy 21) and influence of thyroid status. PLoS One. 2010;5(1) doi: 10.1371/journal.pone.0008394. Article ID e8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Coppedè F. The complex relationship between folate/homocysteine metabolism and risk of Down syndrome. Mutation Research. 2009;682(1):54–70. doi: 10.1016/j.mrrev.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 271.Ellis JM, Tan HK, Gilbert RE, et al. Supplementation with antioxidants and folinic acid for children with Down’s syndrome: randomized controlled trial. British Medical Journal. 2008;336:594–597. doi: 10.1136/bmj.39465.544028.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Salman MS. Systematic review of the effect of therapeutic dietary supplements and drugs on cognitive function in subjects with Down syndrome. European Journal of Paediatric Neurology. 2002;6(4):213–219. doi: 10.1053/ejpn.2002.0596. [DOI] [PubMed] [Google Scholar]
- 273.Lott IT, Doran E, Nguyen VQ, Tournay A, Head E, Gillen DL. Down syndrome and dementia: a randomized, controlled trial of antioxidant supplementation. American Journal of Medical Genetics A. 2011;155(8):1939–1948. doi: 10.1002/ajmg.a.34114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Inestrosa NC, Marzolo MP, Bonnefont AB. Cellular and molecular basis of estrogen’s neuroprotection: potential relevance for Alzheimer’s disease. Molecular Neurobiology. 1998;17(1–3):73–86. doi: 10.1007/BF02802025. [DOI] [PubMed] [Google Scholar]
- 275.Granholm AC, Ford KA, Hyde LA, et al. Estrogen restores cognition and cholinergic phenotype in an animal model of Down syndrome. Physiology & Behavior. 2002;77:371–385. doi: 10.1016/s0031-9384(02)00884-3. [DOI] [PubMed] [Google Scholar]
- 276.Granholm AC, Sanders L, Seo H, Lin L, Ford K, Isacson O. Estrogen alters amyloid precursor protein as well as dendritic and cholinergic markers in a mouse model of down syndrome. Hippocampus. 2003;13(8):905–914. doi: 10.1002/hipo.10130. [DOI] [PubMed] [Google Scholar]
- 277.Seo H, Isacson O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down’s model mice. Experimental Neurology. 2005;193(2):469–480. doi: 10.1016/j.expneurol.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 278.Netzer WJ, Powell C, Nong Y, et al. Lowering beta-amyloid levels rescues learning and memory in a Down syndrome mouse model. PLoS One. 2010;5(6) doi: 10.1371/journal.pone.0010943. Article ID e10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Millan Sanchez M, Heyn SN, Das D, Moghadam S, Martin KJ, Salehi A. Neurobiological elements of cognitive dysfunction in Down syndrome: exploring the role of APP. Biological Psychiatry. 2012;71(5):403–409. doi: 10.1016/j.biopsych.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 280.Heller JH, Spiridigliozzi GA, Sullivan JA, Doraiswamy PM, Krishnan RR, Kishnani PS. Donepezil for the tratment of language deficits in adults swith Down syndrome: A preliminary 24-week open trial. American Journal of Medical Genetics. 2003;116:111–116. doi: 10.1002/ajmg.a.10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Heller JH, Spiridigliozzi GA, Doraiswamy PM, Sullivan JA, Crissman BG, Kishnani PS. Donepezil effects on language in children with down syndrome: results of the first 22-week pilot clinical trial. American Journal of Medical Genetics. 2004;130(3):325–326. doi: 10.1002/ajmg.a.30184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kishnani PS, Sommer BR, Handen BL, et al. The efficacy, safety, and tolerability of donepezil for the treatment of young adults with Down syndrome’. American Journal of Medical Genetics A. 2009;149(8):1641–1654. doi: 10.1002/ajmg.a.32953. [DOI] [PubMed] [Google Scholar]
- 283.Kishnani PS, Heller JH, Spiridigliozzi GA, et al. Donepezil for treatment of cognitive dysfunction in children with Down syndrome aged 10–17. American Journal of Medical Genetics A. 2010;152(12):3028–3035. doi: 10.1002/ajmg.a.33730. [DOI] [PubMed] [Google Scholar]
- 284.Winblad B. Piracetam: a review of pharmacological properties and clinical uses. CNS Drug Reviews. 2005;11(2):169–182. doi: 10.1111/j.1527-3458.2005.tb00268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Lobaugh NJ, Karaskov V, Tombough V, et al. Piracetam therapy does not enhance cognitive function in children with Down syndrome. Archives of Pediatrics & Adolescent Medicine. 2001;155:442–448. doi: 10.1001/archpedi.155.4.442. [DOI] [PubMed] [Google Scholar]
- 286.Moran TH, Capone GT, Knipp S, Davisson MT, Reeves RH, Gearhart JD. The effects of piracetam on cognitive performance in a mouse model of Down’s syndrome. Physiology & Behavior. 2002;77:403–409. doi: 10.1016/s0031-9384(02)00873-9. [DOI] [PubMed] [Google Scholar]