

Abstract
In the amygdala, GABAergic neurons in the intercalated medial paracapsular cluster (Imp) have been suggested to play a key role in fear learning and extinction. These neurons project to the central (CE) amygdaloid nucleus and to other areas within and outside the amygdala. In addition, they give rise to local collaterals that innervate other neurons in the Imp. Several drugs, including benzodiazepines (BZ), are allosteric modulators of GABAA receptors. BZ has both anxiolytic and sedative actions, which are mediated through GABAA receptors containing α2/α3 and α1 subunits, respectively. To establish whether α1 or α2/α3 subunits are expressed at Imp cell synapses, we used paired recordings of anatomically identified Imp neurons and high resolution immunocytochemistry in the mouse. We observed that a selective α3 subunit agonist, TP003 (100 nM), significantly increased the decay time constant of the unitary IPSCs. A similar effect was also induced by zolpidem (10 μM) or by diazepam (1 μM). In contrast, lower doses of zolpidem (0.1–1 μM) did not significantly alter the kinetics of the unitary IPSCs. Accordingly, immunocytochemical experiments established that the α2 and α3, but not the α1 subunits of the GABAA receptors, were present at Imp cell synapses of the mouse amygdala. These results define, for the first time, some of the functional GABAA receptor subunits expressed at synapses of Imp cells. The data also provide an additional rationale to prompt the search of GABAA receptor α3 selective ligands as improved anxiolytic drugs.
Keywords: intercalated cells, synaptic transmission, GABAA receptor, benzodiazepine, amygdala, anxiety
Introduction
GABAergic cells of the amygdala participate in distinct aspects of fear learning and memory (Ehrlich et al., 2009; Herry et al., 2010; Pape and Pare, 2010). Amongst them intercalated medial paracapsular (Imp) neurons, located between the basolateral (BL) complex and the central (CE) nucleus (Millhouse, 1986; Nitecka and Ben-Ari, 1987; McDonald and Augustine, 1993; Pare and Smith, 1993), represent one of the key cell populations mediating fear learning and extinction (Likhtik et al., 2008; Amano et al., 2010). The Imp neurons provide feed-forward inhibition to neurons of the CE nucleus (Royer et al., 1999, 2000; Royer and Pare, 2002), and are under cortical control (Berretta et al., 2005; Li et al., 2011). However, their axonal fields target not only the CE nucleus, but also segregate within the intermediate capsule or project to the ansa lenticularis, resulting in three Imp cell types (Busti et al., 2011). Therefore, Imp neurons are likely to influence the activity of several cell populations. When presynaptic Imp neurons are stimulated at physiologically relevant frequencies (range: 0.1–10 Hz), unitary inhibitory postsynaptic currents (uIPSCs) that display short-term facilitation, short-term depression or that remain constant during the stimulation are detected (Geracitano et al., 2007). Since this short-term plasticity is not cell target-specific (Geracitano et al., 2007), it should occur in virtually all the cell types synaptically coupled to the Imp cells.
The uIPSCs evoked by Imp neurons are mediated by GABAA receptors (Geracitano et al., 2007). These are pentameric ligand-gated anion channels consisting of several subunits heterogeneously distributed in the brain and in different subcellular domains (Farrant and Nusser, 2005). Typically, two α subunits, two β subunits, and one γ subunit co-assemble to form a functional receptor. The subunit composition of GABAA receptors affects ligand affinity, channel gating, and modulation, thereby shaping the kinetics of the resulting inhibitory potentials and their functional impact on postsynaptic neurons (Capogna and Pearce, 2011). Notably, GABAA receptors are modulated by clinically important drugs including benzodiazepines (BZ) (Farrant and Nusser, 2005). BZs increase the affinity of the receptor for GABA, and their broad spectrum effects include: anticonvulsion, sedation, anxiolysis, myorelaxation, and anterograde amnesia (Rudolph and Knoflach, 2011; Smith and Rudolph, 2012). GABAA receptor subtypes containing the α2 and/or α3 subunits appear to mediate primarily the anxiolytic action of BZs (Rudolph and Mohler, 2006; Whiting, 2006; Smith and Rudolph, 2012). One of the best evidence for this involvement is the result that a novel imidazopyridine compound, namely TP003, with full and selective agonistic activity at α3-containing receptors, reduces anxiety, without inducing sedation (Dias et al., 2005).
In summary, the available data suggest a prominent role of Imp cells of amygdala in fear learning, and the α2 and/or α3 subunit of GABAA receptors appears to be a novel molecular target for anxiolytic drugs (Likhtik et al., 2008; Rudolph and Knoflach, 2011). Therefore, we aimed to define whether these subunit-containing GABAA receptors are functionally present at synapses between Imp neurons. To address this issue, we have studied the modulatory actions of TP003, zolpidem and diazepam (a broad spectrum BZ agonist) on synaptic inhibition mediated by Imp neurons, and tested the presence of α3, α2, and α1 subunits at these synapses.
Materials and methods
Animals
Acute coronal slices were prepared from 2 to 3 weeks-old glutamate decarboxylase 65 (GAD65)-GFP transgenic mice (Lopez-Bendito et al., 2004; Geracitano et al., 2007), whereas all immunocytochemical studies were carried out using adult male C57Bl/6N mice (25–30 g; Charles River). All procedures involving animals were performed according to methods approved by the UK Home Office or by the Austrian Animal Experimentation Ethics Board in compliance with both the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (ETS no. 123) and the European Communities Council Directive of 24 November 1986 (86/609/EEC). The authors further attest that all efforts were made to minimize animal suffering and the number of animals used.
Preparation of acute slices
Two–three-weeks-old GAD65-GFP transgenic mice were deeply anaesthetized with isoflurane in oxygenated air and then decapitated. The brain was rapidly removed and placed in semi-frozen sucrose artificial cerebro-spinal fluid (ACSF) cutting solution containing, (in mM) 75 sucrose, 87 NaCl, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, pH 7.3, and bubbled with 95%O2, 5%CO2. Slices (330 μm) were cut (Leica VT 1000S, Leica Microsystems GmbH, Nussloch, Germany) and transferred to a nylon mesh where they were maintained in a chamber containing sucrose ACSF at 37°C for 30 min before returning to room temperature (24–26°C) for another 30 min. During this one hour time period, sucrose ACSF was substituted with normal ACSF at a rate of 1–2 ml/min.
Electrophysiology and analysis
Acute slices were secured under a nylon mesh, submerged, and superfused (at 1–2 ml/min and at 34 ± 1°C) with ACSF, containing (in mM) 130 NaCl, 3.5 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.25 NaH2PO4, 24 NaHCO3, 10 glucose (all from VWR International), pH 7.4 (bubbled with 95%O2, 5%CO2), in a 2 ml chamber mounted on the stage of an upright microscope (Axioskop or Axioskop 2 FS, Zeiss, Jena, Germany). Slices were visualized with 10×/0.3 NA or 40×/0.8 NA (Zeiss) water-immersion objectives coupled with infrared and differential interference contrast (DIC) optics linked to a video camera (Newvicon C2400, Hamamatsu, Hamamatsu City, Japan), and a 100 W mercury vapour short-arc lamp (N HBO 103, Zeiss) connected to an epifluorescence system to visualize the GFP-expressing neurons. Somatic whole-cell patch clamp recordings were performed from visually identified cells using borosilicate glass capillaries (GC120F, 1.2 mm o.d., Clarke Electromedical Instruments, Reading, UK, 4–6 MΩ), pulled on a DMZ puller (Zeitz-instrumente GmbH, Munich, Germany) and filled with a filtered intracellular solution consisting of (in mM): 126 K-gluconate, 4 KCl, 4 Mg-ATP, 0.3 Na-GTP, 10 Na2-phosphocreatine, 10 Hepes, and 0.5%w/v biocytin (all from Sigma-Aldrich Co. Ltd., Poole, UK), osmolarity 270–280 mOsm without biocytin, pH 7.3 with KOH. Biocytin was added to allow post-hoc visualization of the recorded neurons. Cells were only accepted if the initial seal resistance was greater than 1 GΩ. The series resistance (Rs) was compensated online by 50–70% in voltage clamp mode to reduce voltage errors, and cells were only accepted for analysis if the initial Rs did not change by more than 25% throughout the recording period. The electrophysiological signals were amplified (10 mV/pA, EPC9/2 amplifier HEKA Electronik, Lambrecht, Germany, PULSE™ software), filtered at 2.9 kHz, and digitized at 5 kHz. Currents/voltages were acquired online with Pulse software (HEKA) and analyzed offline with IGOR Pro 5 software (Wavemetrics Inc., Oregon, USA). The peak amplitude, latency, 20–80% rise time and decay time (fitted with a single exponential) of unitary events were analyzed with a user-defined programme in IGOR. Data throughout the text are presented as mean ± SEM. Non-parametric two-tailed Wilcoxon-signed ranks test was used in pre-drug/drug comparison. Non-parametric two-tailed Mann–Whitney U-test and parametric two-tailed independent sample t-test were used to compare the effects of zolpidem (10 μM) to those of diazepam (1 μM) on uIPSCs.
Histological procedures
After electrophysiological recordings, slices were sandwiched between two filter papers (cellulose nitrate membrane filters, 0.45 μm, Whatman International Ltd., Maidstone, UK) and immersed in a fixative of 4% paraformaldehyde and ~0.2% picric acid in phosphate buffer (PB, 0.1 M, pH 7.4) for at least 24 h. Then, slices were embedded in a block of gelatin and re-sectioned into 50–60 μm slices with a Leica VT 1000 S vibratome (Leica Microsystems, Vienna, Austria). Sections were washed in tris-buffered saline (TBS; 0.9% NaCl, 0.05 M tris, pH 7.4) and incubated overnight at 4°C in a 1:100 solution of avidin-biotinylated horseradish peroxidase (HRP) complex (Vector labs, Burlingame, CA, USA) in TBS + 0.1% Triton X-100 (VWR International). Sections were further washed in TBS and Tris buffer (TB, 0.05 M, pH 7.4) before incubation in 0.5 mg/ml diaminobenzidine (DAB, Sigma) in TB. Hydrogen peroxide (0.003%) was the electron donor for the peroxidase reaction, which was carried out in TB. Sections were rinsed in TB, then PB, and subsequently mounted on gelatin-coated slides and left to air-dry overnight. Sections were then hydrated, counterstained with 0.5% cresyl violet acetate (Sigma), dehydrated in graded ethanol (50%, 70%, 90%, 95%, and 100% ×2), immersed in n-butyl acetate (Merck Sharp & Dohme, Hertfordshire, UK) and permanently mounted on slides. All the recorded pairs included in the study were identified as Imp neurons because their somata and dendrites were located in the intermediate capsule. Immunofluorescent experiments were carried out as following. Briefly, free-floating sections were pre-incubated for 1 h in blocking solution, composed of 20% normal goat serum (NGS), 0.1% Triton X-100 in TBS. The sections were then incubated in rabbit polyclonal anti-GFP (diluted 1:2500, Molecular Probes, no. A11122), made up in TBS, 0.1% Triton X-100, and 1% NGS for approximately 48 h (4°C). After extensive washes in TBS, sections were incubated overnight (4°C) with a donkey antirabbit Alexa 488 (1:1000, Molecular Probes). Biocytin was visualized with streptavidin-Cy3 (1:1000; Vector). Sections were then washed and mounted onto gelatin-coated slides in Vectashield (Vector). Immunofluorescence was studied using a Zeiss Axioplan 2 microscope with epifluorescence illumination. Images were analyzed and displayed using the Openlab software (version 5.5.0; Improvision, Coventry, UK). Brightness and contrast were adjusted for the whole frame and no part of a frame was modified in any way.
Immunocytochemical procedures
For immunocytochemical light and pre-embedding immunoelectron microscopy experiments, six C57Bl/6N adult male mice were deeply anaesthetized by i.p. injection of 0.15 ml/mouse of Thiopental (50 mg/ml; Sandoz, Kundl, Austria) and perfused with 0.9% saline for 30 s, followed by ice-chilled fixative made up of 4% paraformaldehyde w/v (PFA; Agar Scientific, Stansted, UK) and 15% of a saturated picric acid solution v/v in phosphate buffer (PB 0.1 M, pH 7.4) for 15 min. For electron microscopy experiments, glutaraldehyde was added to the fixative at a final dilution of 0.05% v/v just before the perfusion. Brains were immediately removed from the skull washed in 0.1 M PB and sliced coronally in 40 μm (for light microscopy) or 70 μm (for electron microscopy) thick sections, unless otherwise specified, on a Leica VT1000S vibratome. Sections were stored in 0.1 M PB containing 0.05% NaN3 at 6°C until immunocytochemical experiments were performed.
Pre-embedding immunocytochemistry for electron microscopy
Pre-embedding immunocytochemistry experiments were carried out according to previously published procedures with minor modifications (Sreepathi and Ferraguti, 2012). Briefly, free-floating sections were washed three times in 0.1 M PB, cryoprotected in 20% sucrose made in 0.1 M PB overnight at 6°C. After removal of the sucrose, the sections were freeze-thawed twice to allow antibody penetration and then incubated in 20% NGS in TBS for 2 h at RT. After blocking, sections were exposed for ~72 h at 6°C to primary antibodies (see Table 1) made up in a solution containing 2% NGS in TBS. After three washes in TBS, sections were incubated overnight at 6°C with the appropriate secondary antibodies (Table 1). Antigen–antibody complexes were visualized either by HRP or by nanogold-silver-enhanced reaction. After several washes with TBS, sections were incubated with secondary antibodies either biotinylated (Vector) or coupled to nanogold (1.4 nm; Nanoprobes Inc., Stony Brook, NY, USA) overnight at 6°C. Silver enhancement of the gold particles was carried out using the HQ kit (Nanoprobes) for ~10–15 min. Sections were then washed extensively in milliQ water and then with TB. Sections processed for the HRP reaction were incubated in ABC complex (diluted 1:100; Vector) made up in TB either overnight at 6°C or at RT for 2 h and then washed in TB several times before the antigen/antibody complex was visualized by means of the DAB-H2O2 reaction. Sections were subsequently washed with 0.1 M PB and treated with 2% OsO4 in 0.1 M PB for 40 min at RT. After several washes with 0.1 M PB and then with milliQ water, sections were contrasted with 1% uranyl-acetate in 50% ethanol for 30 min at RT, making sure they were protected from light. Sections were washed with milliQ water followed by graded ethanol and propylene oxide at RT. Sections were then quickly transferred into weighting boats containing epoxy resin (Durcupan ACM-Fluka, Sigma, Gillingham, UK) and kept overnight at RT. The following day, the sections were transferred onto siliconized slides, coverslipped with ACLAR®–film coverslips (Ted Pella, Inc., Redding, CA), and incubated for three days at 60°C. Blocks containing the Imp were cut under a stereomicroscope and re-embedded in epoxy resin. Ultrathin sections (70 nm) were cut using a diamond knife (Diatome, Biel, Switzerland) on an ultramicrotome (EM UC7, Leica, Vienna, Austria), collected on copper slot grids coated with pioloform (Agar, Stansted, England) and analyzed with a transmission electron microscope (Philips CM120).
Table 1.
List of the concentrations and combinations of primary and secondary antibodies.
| Primary antibody | Source | Antigen | Species | Dilution | Secondary antibody | Dilution | Reaction |
|---|---|---|---|---|---|---|---|
| μ-opioid receptor | ImmunoStar; catalog #24216; lot #607102 | Synthetic peptide corresponding to amino acids 384–398 predicted from cloned rat MOR1 | Rabbit | 1:2000 | Gt anti-Rb Biotinylated (Vector) | 1:500 | a |
| 1:1000 | Gt anti-Rb Immunogold conjugate (BBI) | 1:30 | λ ν | ||||
| α1 GABAA receptor subunit | Kindly provided by Prof. Shigemoto (NIPS, Okazaki, Japan) | Amino acidic sequence 328–382 in the intracellular loop domain | Guinea Pig | 1:200 | Gt anti-Gp Biotinylated (Vector) | 1:500 | b |
| 1:100 | Gt anti-Gp nanogold (Nanoprobes) | 1:100 | A | ||||
| Rat | 1:50 | Gt anti-Rt Immunogold conjugate (BBI) | 1:30 | λ ν σ | |||
| α2 GABAA receptor subunit | Kindly provided by Prof. Sieghart (Medical University of Vienna, Vienna, Austria) | Amino acidic sequence 322–357 in the intracellular loop domain | Rabbit | 1:1000 | Gt anti-Rb Biotinylated (Vector) | 1:500 | c |
| 1:500 | Gt anti-Rb Immunogold conjugate (BBI) | 1:30 | σ τ | ||||
| 1:500 | Gt anti-Rb nanogold (Nanoprobes) | 1:100 | B | ||||
| α3 GABAA receptor subunit | Kindly provided by Prof. Sieghart (Medical University of Vienna, Vienna, Austria) | Amino acidic sequence 1–11 in the N-terminal domain | Rabbit | 1:1500 | Gt anti-Rb Biotinylated (Vector) | 1:500 | d |
| 1:1000 | Gt anti-Rb Biotinylated (Vector) | 1:100 | C | ||||
| 1:750 | Gt anti-Rb nanogold (Nanoprobes) | 1:100 | D | ||||
| β3 GABAA receptor subunit | Kindly provided by Prof. Shigemoto (NIPS, Okazaki, Japan) | Amino acidic sequence 345–408 in the intracellular loop domain | Guinea Pig | 1:50 | Gt anti-Gp Immunogold conjugate (BBI) | 1:30 | ν τ |
Reactions performed for light microscopy are indicated by small letters, those for pre-embedding electron microscopy by capital letters, whereas symbols identify reactions for SDS-FRL.
SDS-digested freeze-fracture replica labeling (SDS-FRL)
SDS-Freeze-Fracture Replica Labeling (FRL) was performed according to previously published procedures with minor modifications (Kasugai et al., 2010). The brains of adult mice (C57/Bl6N mice) were perfusion-fixed with PB (0.1 M, pH 7.4) containing 1% formaldehyde, and 15% of a saturated solution of picric acid. Blocks containing the amygdala were cut into 150 μm coronal sections by a Leica VT1000S Vibratome and the Imp was dissected under a stereomicroscope using an ophthalmic knife. Tissue blocks of the Imp were cryoprotected with 30% glycerol in 0.1 M PB overnight at 4°C, then frozen by use of a high-pressure freezing machine (HPM 010; Bal-Tec, Balzers, Liechtenstein) and fractured by double replica method in a freeze-etching device (BAF 060; Bal-Tec). Fractured faces were replicated by rotary deposition of carbon (5 nm) evaporated with an electron beam gun positioned at a 90° angle, shadowed unidirectionally by platinum-carbon (2 nm) with the gun positioned at a 60° angle, followed by an additional carbon layer (15 nm) applied from a 90° angle. Tissue was solubilized in a solution containing 2.5% SDS and 20% sucrose made up in 15 mM Tris buffer, pH 8.3, at 80°C on a shaking platform for 18 h. Replicas were kept in the same solution at room temperature until processed further. On the day of immunolabeling, replicas were washed in 25 mM TBS containing 0.05% BSA and incubated in a blocking solution containing 5% BSA in 25 mM TBS for 1 h. Subsequently, the replicas were incubated in primary antibodies (see Table 1). After several washes, the replicas were reacted overnight at room temperature with a mixture of gold-coupled secondary antibodies (Table 1) made up in 25 mM TBS (1:30) and containing 5% BSA. They were then washed, picked up on 100-line copper grids and analyzed using the same TEM imaging system (CM120 TEM, Morada CCD camera) as described before. To test for cross reactivity of gold-conjugated secondary antibodies in colocalization studies, control replicas were reacted with one of the primary antibodies and a mixture of secondary antibodies conjugated to different sizes of gold and directed against different species. No non-specific cross reactivity of gold-conjugated secondary antibodies was observed.
Chemicals and drugs
All drugs were superfused to the slices through the bath solution. Salts used for the patch pipette solution and ACSF were obtained from either VWR International or Sigma. Zolpidem (Tocris Cookson Inc, Avonmouth, UK) was initially dissolved in dimethyl sulfoxide (DMSO) before addition to the ACSF. For 1 μM zolpidem, the final concentration of DMSO in the bath was 1:10,000–1:100,000. Drugs were superfused at the following concentrations and duration: SR 95531 hydrobromide (gabazine, 2 μM), zolpidem (100 nM, 1 μM, and 10 μM) for 13.7 ± 2 min, diazepam (1 μM) for 12.5 ± 1.5 min, TP003 (100 nM) for 14.3 ± 0.7 min. The α3 subunit selective agonist TP003 was supplied by and used with the permission of Merck Research Laboratory (NJ, USA).
Results
Actions of zolpidem and diazepam on uIPSCs mediated by Imp neurons of amygdala
We have performed paired recordings between visually identified Imp neurons in slices obtained from GAD65-GFP mice. The Imp neurons were observed as clusters of densely packed cells or as thin strands of cells present between the BL and CE nuclei. Presynaptic action potentials resulted in uIPSCs in neurons recorded at the holding potential (VH) = −50 mv, in approximately one out of 10 recorded pairs. An action current was evoked in the presynaptic Imp neuron at 0.1 Hz and a uIPSC was recorded in a postsynaptic Imp neuron. The events were entirely mediated by GABAA receptors, since they were abolished by the application of 2 μM gabazine (n = 3, not shown). The mean peak amplitude of the uIPSCs was −21.7 ± 3.9 pA, the mean 20–80% rise time was 1.3 ± 0.09 ms, and the mean decay time constant was 15.8 ± 1.4 ms (n = 16). These values are similar to the ones we previously reported for uIPSCs evoked by Imp cells (Geracitano et al., 2007). The α1 subunit is the most commonly expressed GABAA receptor subunit in the brain including the amygdala (Pirker et al., 2000; Rudolph and Knoflach, 2011). Zolpidem, an imidazopyridine BZ site ligand, applied at 100 nM, is a selective agonist for this subunit (Pritchett and Seeburg, 1990). Bath application of zolpidem at this concentration did not affect the amplitude and the kinetics of the uIPSCs (p > 0.5, Figure 1, Table 2). When applied at 1 μM, zolpidem is likely to have some agonistic action on α2 and α3 subunit-containing receptors. Applied at this concentration, zolpidem did not modify the peak amplitude or the rise time of the uIPSCs, but slightly increased the decay time constant of the events, although this effect did not reach statistical significance (p > 0.05, Figure 1, Table 2). When applied at 10 μM, a concentration that is likely to fully activate α2 and/or α3 subunits-containing receptors, zolpidem significantly increased the 20–80% rise time and the decay time constant of the uIPSCs (p < 0.05) without affecting their peak amplitude (Figure 1, Table 2). These results were confirmed by testing the non-specific BZ agonist diazepam (1 μM) in a limited number of experiments. The bath application of this drug mimicked the action of high concentration of zolpidem (p > 0.1, independent sample t-test or p > 0.5, Mann–Whitney U-test, for the comparison of the effects on uIPSC kinetics induced by 1 μM diazepam vs. 10 μM zolpidem). Specifically, diazepam increased the 20–80% rise time and the decay time constant of the uIPSCs without altering their peak amplitude (data not shown). On average, the uIPSC peak amplitude was −22.2 ± 6.3 and −23.2 ± 4.6 pA, the 20–80% rise time was 1.5 ± 0.2 and 2.2 ± 0.4 ms, and the decay time constant was 16.3 ± 3.9 and 24.3 ± 4.2, before and during diazepam (n = 3).
Figure 1.

High, but not low, concentration of zolpidem increases the decay time constant of Imp-mediated uIPSCs. Upper traces: whole-cell patch clamp recording of two-synaptically coupled Imp neurons from an acute slice of amygdala. A short (3 ms) voltage step to + 20 mV elicited an action current in the presynaptic cell that evoked a uIPSC in the postsynaptic cell. Each trace is the average of 10 consecutive sweeps (0.1 Hz), recorded with a K-gluconate-based solution at the holding potential of −55 mV. In the experiment shown, the decay of the uIPSCs, fitted with one exponential, was: 32 ms (control), 32.4 ms (zolpidem 100 nM), 38 ms (zolpidem 1 μM), 49 ms (zolpidem 10 μM). Lower graph: summary histograms of normalized peak amplitude, 20–80% rise time and decay time constant of the uIPSCs in control (n = 6), 100 nM (n = 6), 1 μM (n = 6) and 10 μM (n = 6) zolpidem, and wash out (n = 3); *Refers to p < 0.05, Wilcoxon-signed ranks test.
Table 2.
Effects of zolpidem (100 nM, 1 μM, and 10 μM) on uIPSCs recorded from Imp paired recordings experiments.
| uIPSCs | Control (n = 6) | zolpidem (100 nM, n = 6) | zolpidem (1 μM, n = 6) | zolpidem (10 μM, n = 6) | wash (n = 3) |
|---|---|---|---|---|---|
| peak amplitude (pA) | −27.1 ± 9.5 | −27.3 ± 9.5 | −25.1 ± 7.9 | −27.5 ± 9.0 | −29.0 ± 17.4 |
| Rise time | 1.3 ± 0.2 | 1.3 ± 0.1 | 1.4 ± 0.2 | 1.8 ± 0.3* | 1.1 ± 0.3 |
| Decay time (ms) | 18.6 ± 2.9 | 19.7 ± 2.6 | 21.0 ± 2.8 | 29.3 ± 4.4 | 18.2 ± 1.8 |
Data are expressed as mean ± SEM; n = number of recorded pairs; two-tailed Wilcoxon-signed ranks test was used for comparison between pre- and post-zolpidem values (
P < 0.05).
Actions of TP003 on uIPSCs mediated by Imp neurons of amygdala
Next, we tested more directly the involvement of the α3 subunit at Imp cell synapses by using the selective α3 agonist TP003. Consistent with the data reported above, the application of 100 nM TP003 significantly increased the decay time constant of the uIPSCs (p < 0.05) but did not have a significant effect neither on the 20–80% rise time nor on the peak amplitude of the events (p > 0.5) (Figure 2, Table 3).
Figure 2.

The α3 subunit selective agonist TP003 increases the decay time constant of Imp-mediated uIPSCs. All experimental conditions are as in Figure 1. Upper traces: in the case illustrated, the decay of the uIPSCs, fitted with one exponential, increased from 14.6 ms to 19.3 ms during the application of 100 nM TP003. The peak amplitude was 11.8 pA before and 9.9 pA during TP003; the rise time was 1.3 ms before and 1.1 ms during TP003. Lower graph: summary histograms of normalized peak amplitude, 20–80% rise time and decay time constant of the uIPSCs in control (n = 7), 100 nM TP003 (n = 7), and wash out (n = 3); *Refers to p < 0.05, Wilcoxon-signed ranks test.
Table 3.
Effects of TP003 (100 nM) on uIPSCs recorded from Imp paired recordings experiments.
| uIPSCs | Control (n = 6) | TP003 (100 nM, n = 7) | wash (n = 3) | |
|---|---|---|---|---|
| peak amplitude (pA) | −16 ± 1.6 | −14.5 ± 1.2 | −16.4 ± 0.8 | |
| Rise time | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.3 ± 0.1 | |
| Decay time (ms) | 13.1 ± 0.9 | 16.5 ± 1.8* | 16.6 ± 1.8 |
Data are expressed as mean ± SEM; n = number of recorded pairs; two-tailed Wilcoxon-signed ranks test was used for comparison between pre- and post-TP003 values (
P < 0.05).
Expression of α1, α2, α3 subunits of GABAA receptor by Imp neurons
The functional data presented above indicate the presence of the α2 and α3 and the absence of the α1 GABAA receptor subunit at Imp cell inhibitory synapses. Next, we have investigated the GABAA receptor subunits expressed by Imp cells by using highly specific antibodies. Intercalated cell masses of the amygdala are characterized by an intense expression of μ-opioid receptors, and we have used this labeling to selectively identify processes belonging to these cells (Figure 3A). Immunocytochemistry revealed a lack of detectable α1 immunoreactivity in the Imp, with the notable exception of a few medium-large dendrites coursing across the cluster and probably belonging to large neurons with the soma located at the edge of the cluster (Figure 3B). Conversely, both α2 and α3 appeared highly enriched in the Imp (Figures 3C,D). The presence of GABAA receptors containing α2 or α3 subunits at Imp synapses was then confirmed by pre-embedding immunoelectron microscopy (Figure 4). To reveal the α3 subunit, we carried out both immunoperoxidase and silver-intensified immunogold reactions. As the α3 antibody recognized an extracellular epitope, the peroxidase reaction end product was observed primarily associated with the plasma membrane of the postsynaptic specialization of symmetric synapses (Figure 4A). However, due to its diffusion, the presynaptic active zone and some intracellular organelles located immediately underneath the postsynaptic membrane appeared also labeled (Figure 4A). On the other hand, silver-gold particles were directly observed at the postsynaptic membrane within the main body of symmetric synapses thus demonstrating a restricted localization of the α3 subunit to postsynaptic elements (Figure 4B). Silver-intensified immunogold reaction for the α2 subunit revealed intense labeling of symmetric synapses (Figures 4C,D). Conversely, no detectable labeling for the α1 subunit could be observed (not shown). To further demonstrate a lack of α1 subunits in Imp neurons, we draw on the SDS-FRL technique. SDS-FRL is a highly sensitive immunocytochemical method that was shown to allow the detection of GABAA receptor subunits at the protoplasmic face (P-face) of plasma membranes (Kasugai et al., 2010). Dendrites of Imp neurons were identified in SDS-FRL by their expression of μ-opioid receptors and presence of numerous spines (Figure 5A). In double labeling experiments, the μ-opioid receptor and the α1 subunit were revealed by 10 and 5 nm gold particle-coupled antibodies, respectively. Whereas large gold particles identifying μ-opioid receptors were densely distributed throughout the extrasynaptic area of Imp dendrites, only a few 5 nm gold particles could be detected that were not associated with synapses and showed a density comparable to the non-specific labeling of the secondary antibody (Figure 5A). In a subsequent triple-labeling experiment, we indeed showed the lack of detectable α1 subunits in GABA synapses on μ-opioid receptor-positive dendrites (Figure 5B). GABA synapses were clearly identified by the presence of numerous gold particles for the GABAA β3 subunit on clusters of intramembrane particles (Figure 5B). As shown in Figure 5C, β3 and α2 subunits coexist in the same synapse on Imp neurons. Moreover, we demonstrate that α1 subunits are enriched in GABA synapses on pyramidal-like neurons of the basal nucleus of the amydgala, where they coexist with α2 subunits (Figure 5D), thus excluding a technical limitation in their identification. The α3 subunit could not be investigated by SDS-FRL as in our experimental conditions the antibody did not give specific labeling on replica.
Figure 3.
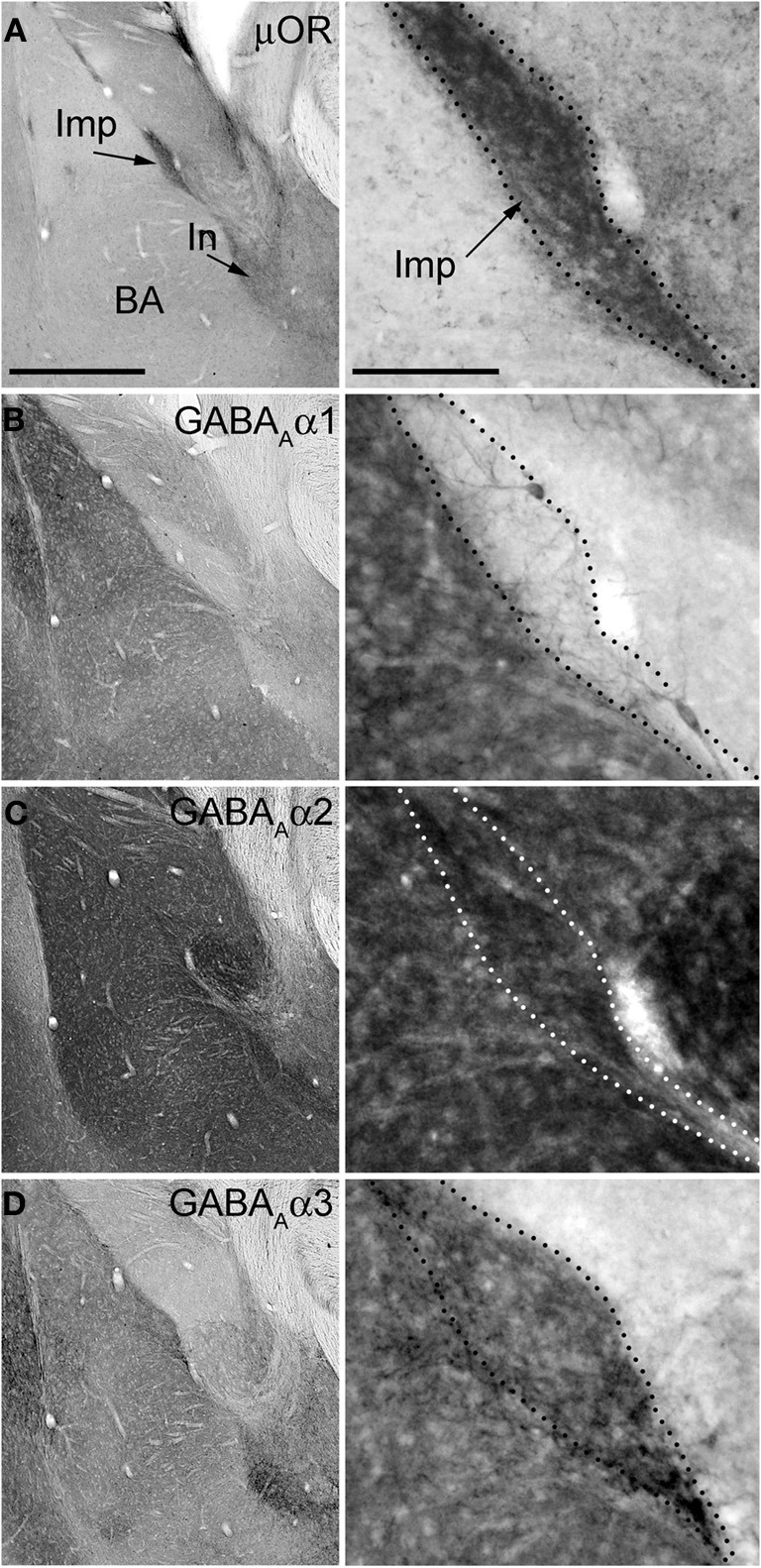
Immunostainings for μ-opioid receptor and GABAA receptor subunits α1–3 in consecutive sections of the mouse amygdala. Panels on the left show low magnification images of the basolateral (BL) complex and intercalated cell masses, whereas the panels on the right show a higher magnification of the Imp cluster. (A) Immunoreactivity for μOR is particularly enriched in the intercalated cell masses such as the Imp and In (as indicated by the arrows). (B) Immunoreactivity for the α1 subunit is very intense in the BL complex whereas it is undetectable in the Imp, although a few dendrites can be observed coursing across it and probably belonging to the strongly labeled large neurons located at the edges of the cluster. (C) The expression of the α2 subunit appears particularly strong in both the BL complex and intercalated cell masses, including the Imp. (D) Immunoreactivity for the α3 subunit can be observed throughout the amygdala although the intercalated cell masses display a stronger signal compared to the BL complex and central nucleus. Abbreviations: BA, basal nucleus of the amygdala; Imp, medial paracapsular intercalated cluster; In, main intercalated nucleus; μ-opioid receptor, μOR. Scale bars: left panel, 500 μm; right panel, 100 μm.
Figure 4.

Electron micrographs of the immunolocalization of the α2 and α3 subunits in Imp synapses. (A) A symmetric synapse (indicated by the open arrowhead) on an Imp dendrite shows electron dense peroxidase reaction end product for α3 enriched at the plasma membrane of the postsynaptic specialization. Because the antibody recognizes an extracellular epitope, also the presynaptic active zone and some intracellular organelles located immediately underneath the postsynaptic membrane appear labeled due to the diffusion of the HRP-immunoreaction product. Nearby an immunonegative excitatory synapse (closed arrowhead) made with a small spine(s) can be observed. (B) Immunometal particles visualizing α3 subunits can be seen directly located at the postsynaptic membrane of an inhibitory synapse in the Imp. (C and D) Numerous immunometal particles identifying the GABAA α2 subunit can be observed leaning on the cytoplasmic portion of the postsynaptic plasma membrane of a symmetrical inhibitory synapse (open arrowheads) in the Imp. Abbreviations: at, axon terminal; d, dendrite; s, spine. Scale bars: A, 500 nm; B, 200 nm; C, 250 nm; D, 200 nm.
Figure 5.

SDS-FRL images show the localization of μ-opioid (μOR) receptor and GABAA receptor subunits α1, α2, and β3 on the P-face of Imp dendrites. (A) Double labeling for μOR (10 nm) and α1 subunit (5 nm gold particle, indicated by red arrows) shows strong expression of μOR in extrasynaptic areas of Imp dendrites. Only background labeling levels were observed for the α1 subunit. Insert shows an enlarged view of a single 5 nm immunogold particle. (B) Triple-labeling for μOR (5 nm), α1 (15 nm), and β3 (10 nm) shows the lack of detectable α1 labeling both in a GABAergic synapse identified by concentrated immunogold particles for the β3 subunit (indicated by red arrows) on a distinct cluster of intramembrane particles, and in the extrasynaptic area consistent with the lack or very low density of the α1 subunit at Imp dendrites. Black arrows show immunogold particles for μOR located in extrasynaptic areas. (C) Double labeling for α2 (5 nm) and β3 (10 nm) subunits showing their colocalization in a single GABAergic synapse of an Imp dendrite. (D) Double-labeling for α1 (10 nm) and α2 (5 nm) showing the colocalization of these two subunits in a GABAergic synapse of a pyramidal cell dendrite of the basal nucleus of the amygdala. Scale bars: A, 500 nm; Insert, 250 nm; B, −500 nm; C and D, 200 nm.
Discussion
Taken together, our electrophysiological and immunocytochemical data demonstrate that the α2 and α3, but not the α1, GABAA subunits are expressed at synapses of Imp neurons of amygdala and underlie the reciprocal synaptic inhibition mediated by this cell population. This conclusion relies on the selective potency of zolpidem and TP003 as well as on the specificity of the antibodies for the α subunits of the GABAA receptor. In particular, zolpidem has a high affinity for α1-containing GABAA receptors, a 20-fold lower affinity for α2/α3-containing GABAA receptors, and no affinity for α5-containing GABAA receptors (Pritchett and Seeburg, 1990). The recently synthesized compound TP003 is a selective agonist of GABAA receptor containing the α3 subunit (Dias et al., 2005) (but see below). Our data are consistent with previous reports showing that the α3 subunit, but not the α1 subunit, is enriched in the intercalated cell masses of the mouse amygdala (Marowsky et al., 2004; Fritschy, 2008). Positive controls in the present experiments strengthen the interpretation of a lack or very low level of expression of the α1 subunit at Imp synapses. Namely, zolpidem affected the decay of the uIPSCs only when applied at micromolar concentration. Moreover, in the immunohistochemical experiments, the same antibody for the α1 subunit that was not detectable in medium-spiny intercalated neurons, produced a strong labeling in other areas of the amygdala.
While the drugs prolonged the decay of the uIPSCs, they did not enhance the peak amplitude of the events. At other synapses, when zolpidem increased the decay of evoked spontaneous and quantal (miniature) IPSCs also enhanced the amplitude of the events (De Koninck and Mody, 1994; Perrais and Ropert, 1999; Hajos et al., 2000; Cope et al., 2005). The degree of potentiation of the IPSC amplitude depends on the degree of saturation of the receptors during synaptic transmission (Frerking et al., 1995). Therefore, the variability in the response to various BZ ligands in different types of inhibitory synapses in some areas of the brain has been attributed to different degrees of receptor occupancy (Nusser et al., 1997; Hajos et al., 2000). Our data suggest that the GABAA receptors present at Imp cell synapses are saturated under baseline transmission. This contrasts with inhibitory synapses in the BL nucleus, where diazepam largely enhances both evoked and mIPSCs indicating that single quanta are not saturating GABAA receptors in this part of the amygdala (Marowsky et al., 2004). The drugs enhanced the duration, but not amplitude, of the uIPSCs. At the network level, the prolongation of individual inhibitory currents is likely to result into changes of the power and/or frequency of rhythmic activity (Cope et al., 2005). Our data predict that selective ligands for the α3 subunit of the GABAA receptor could change the power and/or the frequency of network oscillations detected in the amygdala during behavioral challenges (Pare et al., 2002).
Experimental evidence suggests the involvement of Imp cells in fear learning and extinction expression. Indeed, the pharmacological ablation of Imp cells causes a deficit in the expression of fear extinction (Likhtik et al., 2008), and a potentiation of Imp cells-mediated synaptic inhibition of output neurons of the CE occurs during fear extinction (Amano et al., 2010). Consistent with this scenario, sub-populations of Imp cells are also activated during fear conditioning, and this generates the inhibition of the main intercalated nucleus and of the CE (central/lateral subdivisions), leading to the disinhibition of CE (medial subdivision) output neurons (Busti et al., 2011; Manko et al., 2011). Thus, Imp neurons may represent a promising target for the pharmacological treatment of anxiety disorders. It is important to keep in mind, however, that fear learning and anxiety are not the same event, and they can be dissociated (Lissek et al., 2005). Indeed, fear learning is a phasic, apprehensive arousal to an explicit threat, whereas anxiety is a longer-term state elicited by more diffuse threat cues (Davis, 1998).
It is remarkable that individual GABAA receptor subtypes involved in anxiolytic actions appear to be different from those involved in sedation (Smith and Rudolph, 2012). Since BZs have sedative-hypnotic but also anxiolytic effects, two actions that would be very useful to dissociate, understanding receptor subtype specificity in the amygdala, could provide a rationale for the development of non-sedating BZ anxiolytics (Rudolph and Knoflach, 2011). Experimentally it was found that GABAA receptors expressing the α2 subunit mediate the anxiolytic-like action of diazepam (Low et al., 2000), whereas GABAA receptors expressing the α1 subunit mediate the sedative action of this drug (Rudolph et al., 1999; McKernan et al., 2000). Furthermore, the selective α3 ligand TP003 is anxiolytic in the rodent elevated plus-maze and in stress-induced-hyperthermia models (Dias et al., 2005), as well as in the primate conditioned model (Fischer et al., 2011). It is important to note that TP003 has also been demonstrated to have a selective efficacy at recombinant α3-containing GABAA receptor in vitro (Dias et al., 2005), but its selectivity has not been tested in vivo.
Using the SDS-FRL technique, we also revealed that μORs are localized extrasynaptically in Imp neurons. Our work confirms the enrichment of these receptors in intercalated cell masses (Likhtik et al., 2008; Busti et al., 2011; Pinard et al., 2012).
In conclusion, GABAA receptors containing the α2 and α3 subunits are enriched in synapses of the Imp of the mouse amygdala. In contrast, GABAA receptors containing the α1 subunit were below detection in this area of the mouse amygdala. Consistent with this, drugs able to activate the α2/α3 subunits prolonged the duration, but not the amplitude, of uIPSCs mediated by Imp neurons. Our data strengthen the idea that Imp neurons of the amygdala constitute a promising target for the pharmacological treatment of anxiety disorders, and specifically for a new generation of anxiolytic drugs without sedative effects.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Medical Research Council UK (Medical Research Council award U138197106) to Marco Capogna, and by the Austrian Science Fund FWF (grant No. P22969) to Francesco Ferraguti. Raffaella Geracitano was supported by Marie Curie Intra-European Fellowship within the sixth European Community Framework Programme. We thank the Merck Research Laboratory (NJ, USA) for the gift of TP003, and Prof. W. Sieghart and Prof. R. Shigemoto for providing antibodies against GABAAα and β subunits.
References
- Amano T., Unal C. T., Pare D. (2010). Synaptic correlates of fear extinction in the amygdala. Nat. Neurosci. 13, 489–494 10.1038/nn.2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta S., Pantazopoulos H., Caldera M., Pantazopoulos P., Pare D. (2005). Infralimbic cortex activation increases c-Fos expression in intercalated neurons of the amygdala. Neuroscience 132, 943–953 10.1016/j.neuroscience.2005.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busti D., Geracitano R., Whittle N., Dalezios Y., Manko M., Kaufmann W., Satzler K., Singewald N., Capogna M., Ferraguti F. (2011). Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. J. Neurosci. 31, 5131–5144 10.1523/JNEUROSCI.6100-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M., Pearce R. A. (2011). GABAA,slow: causes and consequences. Trends Neurosci. 34, 101–112 10.1016/j.tins.2010.10.005 [DOI] [PubMed] [Google Scholar]
- Cope D. W., Halbsguth C., Karayannis T., Wulff P., Ferraguti F., Hoeger H., Leppa E., Linden A. M., Oberto A., Ogris W., Korpi E. R., Sieghart W., Somogyi P., Wisden W., Capogna M. (2005). Loss of zolpidem efficacy in the hippocampus of mice with the GABAA receptor gamma2 F77I point mutation. Eur. J. Neurosci. 21, 3002–3016 10.1111/j.1460-9568.2005.04127.x [DOI] [PubMed] [Google Scholar]
- Davis M. (1998). Are different parts of the extended amygdala involved in fear versus anxiety? Biol. Psychiatry 44, 1239–1247 [DOI] [PubMed] [Google Scholar]
- De Koninck Y., Mody I. (1994). Noise analysis of miniature IPSCs in adult rat brain slices: properties and modulation of synaptic GABAA receptor channels. J. Neurophysiol. 71, 1318–1335 [DOI] [PubMed] [Google Scholar]
- Dias R., Sheppard W. F., Fradley R. L., Garrett E. M., Stanley J. L., Tye S. J., Goodacre S., Lincoln R. J., Cook S. M., Conley R., Hallett D., Humphries A. C., Thompson S. A., Wafford K. A., Street L. J., Castro J. L., Whiting P. J., Rosahl T. W., Atack J. R., Mckernan R. M., Dawson G. R., Reynolds D. S. (2005). Evidence for a significant role of alpha 3-containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J. Neurosci. 25, 10682–10688 10.1523/JNEUROSCI.1166-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I., Humeau Y., Grenier F., Ciocchi S., Herry C., Luthi A. (2009). Amygdala inhibitory circuits and the control of fear memory. Neuron 62, 757–771 10.1016/j.neuron.2009.05.026 [DOI] [PubMed] [Google Scholar]
- Farrant M., Nusser Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6, 215–229 10.1038/nrn1625 [DOI] [PubMed] [Google Scholar]
- Fischer B. D., Atack J. R., Platt D. M., Reynolds D. S., Dawson G. R., Rowlett J. K. (2011). Contribution of GABA(A) receptors containing alpha3 subunits to the therapeutic-related and side effects of benzodiazepine-type drugs in monkeys. Psychopharmacology (Berl.) 215, 311–319 10.1007/s00213-010-2142-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M., Borges S., Wilson M. (1995). Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron 15, 885–895 [DOI] [PubMed] [Google Scholar]
- Fritschy J. M. (2008). Is my antibody-staining specific? How to deal with pitfalls of immunohistochemistry. Eur. J. Neurosci. 28, 2365–2370 10.1111/j.1460-9568.2008.06552.x [DOI] [PubMed] [Google Scholar]
- Geracitano R., Kaufmann W. A., Szabo G., Ferraguti F., Capogna M. (2007). Synaptic heterogeneity between mouse paracapsular intercalated neurons of the amygdala. J. Physiol. 585, 117–134 10.1113/jphysiol.2007.142570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N., Nusser Z., Rancz E. A., Freund T. F., Mody I. (2000). Cell type- and synapse-specific variability in synaptic GABAA receptor occupancy. Eur. J. Neurosci. 12, 810–818 10.1046/j.1460-9568.2000.00964.x [DOI] [PubMed] [Google Scholar]
- Herry C., Ferraguti F., Singewald N., Letzkus J. J., Ehrlich I., Luthi A. (2010). Neuronal circuits of fear extinction. Eur. J. Neurosci. 31, 599–612 10.1111/j.1460-9568.2010.07101.x [DOI] [PubMed] [Google Scholar]
- Kasugai Y., Swinny J. D., Roberts J. D., Dalezios Y., Fukazawa Y., Sieghart W., Shigemoto R., Somogyi P. (2010). Quantitative localisation of synaptic and extrasynaptic GABAA receptor subunits on hippocampal pyramidal cells by freeze-fracture replica immunolabelling. Eur. J. Neurosci. 32, 1868–1888 10.1111/j.1460-9568.2010.07473.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Amano T., Pare D., Nair S. S. (2011). Impact of infralimbic inputs on intercalated amygdala neurons: a biophysical modeling study. Learn. Mem. 18, 226–240 10.1101/lm.1938011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E., Popa D., Apergis-Schoute J., Fidacaro G. A., Pare D. (2008). Amygdala intercalated neurons are required for expression of fear extinction. Nature 454, 642–645 10.1038/nature07167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissek S., Powers A. S., McClure E. B., Phelps E. A., Woldehawariat G., Grillon C., Pine D. S. (2005). Classical fear conditioning in the anxiety disorders: a meta-analysis. Behav. Res. Ther. 43, 1391–1424 10.1016/j.brat.2004.10.007 [DOI] [PubMed] [Google Scholar]
- Lopez-Bendito G., Sturgess K., Erdelyi F., Szabo G., Molnar Z., Paulsen O. (2004). Preferential origin and layer destination of GAD65-GFP cortical interneurons. Cereb. Cortex 14, 1122–1133 10.1093/cercor/bhh072 [DOI] [PubMed] [Google Scholar]
- Low K., Crestani F., Keist R., Benke D., Brunig I., Benson J. A., Fritschy J. M., Rulicke T., Bluethmann H., Mohler H., Rudolph U. (2000). Molecular and neuronal substrate for the selective attenuation of anxiety. Science 290, 131–134 [DOI] [PubMed] [Google Scholar]
- Manko M., Geracitano R., Capogna M. (2011). Functional connectivity of the main intercalated nucleus of the mouse amygdala. J. Physiol. 589, 1911–1925 10.1113/jphysiol.2010.201475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marowsky A., Fritschy J. M., Vogt K. E. (2004). Functional mapping of GABAA receptor subtypes in the amygdala. Eur. J. Neurosci. 20, 1281–1289 10.1111/j.1460-9568.2004.03574.x [DOI] [PubMed] [Google Scholar]
- McDonald A. J., Augustine J. R. (1993). Localization of GABA-like immunoreactivity in the monkey amygdala. Neuroscience 52, 281–294 10.1016/0306-4522(93)90156-A [DOI] [PubMed] [Google Scholar]
- McKernan R. M., Rosahl T. W., Reynolds D. S., Sur C., Wafford K. A., Atack J. R., Farrar S., Myers J., Cook G., Ferris P., Garrett L., Bristow L., Marshall G., Macaulay A., Brown N., Howell O., Moore K. W., Carling R. W., Street L. J., Castro J. L., Ragan C. I., Dawson G. R., Whiting P. J. (2000). Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat. Neurosci. 3, 587–592 10.1038/75761 [DOI] [PubMed] [Google Scholar]
- Millhouse O. E. (1986). The intercalated cells of the amygdala. J. Comp. Neurol. 247, 246–271 10.1002/cne.902470209 [DOI] [PubMed] [Google Scholar]
- Nitecka L., Ben-Ari Y. (1987). Distribution of GABA-like immunoreactivity in the rat amygdaloid complex. J. Comp. Neurol. 266, 45–55 10.1002/cne.902660105 [DOI] [PubMed] [Google Scholar]
- Nusser Z., Cull-Candy S., Farrant M. (1997). Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron 19, 697–709 10.1016/S0896-6273(00)80382-7 [DOI] [PubMed] [Google Scholar]
- Pape H. C., Pare D. (2010). Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol. Rev. 90, 419–463 10.1152/physrev.00037.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D., Collins D. R., Pelletier J. G. (2002). Amygdala oscillations and the consolidation of emotional memories. Trends Cogn. Sci. 6, 306–314 10.1016/S1364-6613(02)01924-1 [DOI] [PubMed] [Google Scholar]
- Pare D., Smith Y. (1993). Distribution of GABA immunoreactivity in the amygdaloid complex of the cat. Neuroscience 57, 1061–1076 10.1016/0306-4522(93)90049-L [DOI] [PubMed] [Google Scholar]
- Perrais D., Ropert N. (1999). Effect of zolpidem on miniature IPSCs and occupancy of postsynaptic GABAA receptors in central synapses. J. Neurosci. 19, 578–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinard C. R., Mascagni F., McDonald A. J. (2012). Medial prefrontal cortical innervation of the intercalated nuclear region of the amygdala. Neuroscience 205, 112–124 10.1016/j.neuroscience.2011.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S., Schwarzer C., Wieselthaler A., Sieghart W., Sperk G. (2000). GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101, 815–850 10.1016/S0306-4522(00)00442-5 [DOI] [PubMed] [Google Scholar]
- Pritchett D. B., Seeburg P. H. (1990). Gamma-aminobutyric acidA receptor alpha 5-subunit creates novel type II benzodiazepine receptor pharmacology. J. Neurochem. 54, 1802–1804 [DOI] [PubMed] [Google Scholar]
- Royer S., Martina M., Pare D. (1999). An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J. Neurosci. 19, 10575–10583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S., Martina M., Pare D. (2000). Polarized synaptic interactions between intercalated neurons of the amygdala. J. Neurophysiol. 83, 3509–3518 [DOI] [PubMed] [Google Scholar]
- Royer S., Pare D. (2002). Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 115, 455–462 [DOI] [PubMed] [Google Scholar]
- Rudolph U., Crestani F., Benke D., Brunig I., Benson J. A., Fritschy J. M., Martin J. R., Bluethmann H., Mohler H. (1999). Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature 401, 796–800 10.1038/44579 [DOI] [PubMed] [Google Scholar]
- Rudolph U., Knoflach F. (2011). Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 10, 685–697 10.1038/nrd3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U., Mohler H. (2006). GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr. Opin. Pharmacol. 6, 18–23 10.1016/j.coph.2005.10.003 [DOI] [PubMed] [Google Scholar]
- Smith K. S., Rudolph U. (2012). Anxiety and depression: mouse genetics and pharmacological approaches to the role of GABA(A) receptor subtypes. Neuropharmacology 62, 54–62 10.1016/j.neuropharm.2011.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreepathi H. K., Ferraguti F. (2012). Subpopulations of neurokinin 1 receptor-expressing neurons in the rat lateral amygdala display a differential pattern of innervation from distinct glutamatergic afferents. Neuroscience 203, 59–77 10.1016/j.neuroscience.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting P. J. (2006). GABA-A receptors: a viable target for novel anxiolytics? Curr. Opin. Pharmacol. 6, 24–29 10.1016/j.coph.2005.08.005 [DOI] [PubMed] [Google Scholar]