

Abstract
Context:
Anti-neutrophil p-ANCA antibodies are directed against antigens in the peripheral cytoplasm of both neutrophilic granulocytes and monocytes. They are detected in several autoimmune disorders and are particularly associated with systemic vasculitis.
Case Report:
We report a case of a 54-year-old female presenting with a pruritic rash, including purpura and diffuse erythema. A biopsy with hematoxylin and eosin (H & E) analysis, direct immunofluorescence (DIF), immunohistochemistry (IHC) and enzyme-linked immunosorbent assays for ANCAs were performed. The H & E staining demonstrated leukocytoclastic vasculitis, with focal vascular fibrinoid necrosis. The DIF revealed evidence of vasculitis, the presence of p-ANCAs and neutrophil extracellular traps (NETs). The IHC displayed autoreactivity to myeloperoxidase within the vessels. The IHC aided in ruling out any intrinsic autofluorescence of the vessels.
Conclusions:
By observing the deposition of neutrophil extracellular traps and myeloperoxidase in inflamed skin vessels, biopsy analysis may alert physicians for rapid therapeutic intervention in patients presenting with possible vasculitides.
Keywords: Vasculitis, p-ANCA, myeloperoxidase, neutrophil extracellular traps (NETs), IgD, skin
Introduction
Anti-neutrophil cytoplasmic antibodies (ANCAs) are a group of predominately IgG antibodies, directed against antigens in the cytoplasm of both neutrophilic granulocytes and monocytes[1]. They are detected in a number of autoimmune disorders, but are most often associated with forms of systemic vasculitis, the ANCA-associated vasculitides[1]. ANCAs were first divided into two major classes, c-ANCAs (cytoplasmic) and p-ANCAs (or perinuclear), based on their pattern of staining inside the affected cells[1]. ANCA titers are classically measured using enzyme-linked immunosorbent assays (ELISAs), and by indirect immunofluorescence (IIF)[1]. As noted, the p-ANCAs display a perinuclear staining pattern. The perinuclear pattern occurs because the vast majority of the antigens targeted by p-ANCAs are strongly cationic (positively charged) at pH 7.00[1–3]. During fixation, the more cationic antigens migrate within the cell cytoplasm and congregate around the nucleus, attracted by its negatively charged deoxyribonucleic acid (DNA) content[1–3].
Immunofluorescence antibody staining then results in fluorescence of the region around the nucleus that reveals the classic p-ANCA perinuclear pattern. The most frequent p-ANCA target is myeloperoxidase (MPO), a neutrophil granule protein whose primary role in classic metabolic processes is generation of oxygen radicals. ANCAs will, less commonly, form against alternative antigens, that may also result in the p-ANCA pattern. These antigens include lactoferrin, elastase, and cathepsin G. The presence of p-ANCAs is a sensitive, but not specific test for ulcerative colitis[1–3]. In contradistinction, c-ANCAs show a diffusely granular, cytoplasmic staining pattern. The c-ANCA pattern results from the binding of ANCAs to antigen targets throughout the neutrophil cytoplasm, and the most common protein targets at proteinase 3 (PR3). The PR3 c-ANCA pattern is often encountered in patients with Wegener's granulomatosis. Other antigens may also occasionally result in a c-ANCA pattern.
Other types of ANCAs that develop against antigens other than myeloperoxidase or proteinase 3 will sporadically result in patchy staining when visualized by immunofluorescence[2]. The resultant pattern is commonly called the “snowdrift” prototype, and classically occurs in patients with non-vasculitic diseases that result in ANCA formation. It is not clear how these ANCAs are developed, although several hypotheses have been suggested. There is probably a genetic contribution (predominantly in genes controlling the level of the immune response); however, genetic susceptibility is likely to be linked to an environmental factor, as well as probable factors including vaccination or exposure to silicates[2].
Case Report
A 54-year-old female presented with a clinical history of a rapidly appearing, prutitic rash on her scalp, neck, hands, chest, abdomen, buttocks and back. A physical exam of the patient revealed an erythematous, macular rash in these areas, with lesions concentrated on the legs. The leg lesions included palpable purpura. No associated systemic symptoms were noted; lung and kidney function were within normal limits. No known concurrent medical conditions were present, and the patient was also not under treatment with any medications or herbal supplements. No paresthesias were present; urinary examination revealed no microhematuria or other abnormalities. A punch biopsy of the skin was performed for hematoxylin and eosin (H & E) analysis, as well as for multicolor direct immunofluorescence (DIF) and for immunohistochemistry (IHC).
Direct immunofluorescence (DIF): In brief, four um thickness skin cryosections were partially fixed in 3% paraformaldehyde, and then incubated with secondary antibodies, all conjugated with fluorescein isothiocyanate (FITC). The following anti-human rabbit antibodies were utilized: anti-human IgG (γ chain), anti-human IgA (α chain), anti-human IgM (μ-chain), anti-complement 3c, anti-fibrinogen, anti-albumin from (all at either 1:20 to 1:40 dilutions; all from Dako, Carpinteria, California, USA). In addition, we utilized goat anti-human IgE-antiserum, conjugated with FITC from Vector Laboratories, Bridgeport New Jersey, USA, as well as goat-anti-human complement C1q antiserum from Southern Biotech, Birmingham, Alabama, USA. The slides were then counterstained with either 4’, 6-diamidino-2-phenylindole (DAPI) (Pierce, Rockford, Illinois, USA), or with the carbocyanine monomer TO-PRO-3 dye (Molecular Probes, Invitrogen, Carlsbad, California, USA), washed, and coverslipped. The results are summarized in Fig. 1.
Fig. 1.

Presence of neutrophil extracellular traps and antineutrophil cytoplasmic antibodies associated with vasculitides using DIF and IHC.
a through f, and i, DIF. a. Area of vasculitis, showing deposits of FITC-conjugated anti-human fibrinogen within dermal vessels (red arrows). b. Intra-cytoplasmic deposits of FITC-conjugated antihuman C1q within epidermal cells (red arrow). c. Positive p-ANCAs (yellow arrow) using FITC-conjugated anti-human IgG. The nuclei are counterstained in red with TO-PRO-3 dye (blue arrow); vessel autoreactivity is displayed in green utilizing the FITC-conjugated anti-human IgG (white arrow). d. Positive p-ANCAs (yellow arrows) in the dermis utilizing FITC-conjugated anti-human IgG antibodies. The nuclei are counterstained in red with TO-PRO-3 (blue arrows). e, Same as d, but at higher magnification, and without nuclear counterstaining. f. Shows a detail of the p-ANCA antigen(s) in the epidermis and the NETs (yellow arrows). g IHC staining with collagen IV shows strong reactivity to the BMZ, as well as around the superficial dermal vessels (brown stain) (aqua arrows). h. H & E staining demonstrating leukocytoclastic vasculitis with fibrinoid necrosis (blue arrow). .i, DIF, and g, deposits of nuclear dust (yellow) on some superficial vessels utilizing FITC-conjugated anti-human IgA antibodies (yellow arrows). The blue arrows show the nuclei counterstained with TO-PRO-3. j An IHC stain using anti-CD34 on the vascular area in i, and showing strong, fragmented endothelial expression of this molecule (aqua arrows). k, IHC stain utilizing myeloperoxidase (red arrows). l. IHC CD31 antibody shows fragmented endothelial cells (red arrows).
Hematoxylin and Eosin (H&E) stain: The H&E staining displayed histologic features of mild, diffuse epidermal spongiosis, and a mild, superficial dermal inflammatory process involving capillaries and small venules. A mild, superficial, perivascular dermal infiltrate of lymphocytes, histiocytes and neutrophils was present, with perivascular leukocytoclastic debris and focal vascular fibrinoid necrosis also appreciated (Fig. 1). The H &E displayed other findings, including focal, perivascular extravasation of erythrocytes into the dermal interstitial tissue; occasional neutrophils were also noted to be present within the epidermal stratum spinosum (Fig. 1). Direct immunofluorescence (DIF) displayed the following results: anti-human IgG, IgA, and albumin (all++), and anti-human fibrinogen (+++) deposits were noted, all in a linear pattern along the BMZ, and surrounding the superficial dermal blood vessels. Significant p-ANCAs were positive within selected neutrophils when utilizing anti-human IgG, IgA and IgM. In addition, by DIF we detected the presence of autoreactivity to intracytoplasmic keratinocytes with FITC-conjugated anti human C1q (Fig. 1).
Immunohistochemistry (IHC): We performed IHC to confirm the DIF data, and to be able to distinguish between real, pathologic autoreactivity detected by direct immunofluorescence, versus non-pathologic, incidental autofluorescence of the dermal blood vessels. We performed IHC by using a dual endogenous peroxidase blockage, according to the Dako instructions, with the addition of an Envision dual link. We applied 3, 3 diaminobenzidine (DAB) as a chromogen marker, and then counterstained with hematoxylin. The samples were run in a Dako Autostainer Universal Staining System. We stained for rabbit anti-human IgA, IgM, IgD, IgE, C1q, fibrinogen, albumin, and the blood vessel vascular endothelial antigens CD31 (PECAM) and CD 34; in addition, we utilized D2-40 to highlight dermal lymphatic vessel endothelia. Anti-myeloperoxidase (MPO) antibody from Dako was also utilized in the IHC studies. Our tests confirmed the autoreactivity detected by DIF to the basement membrane zone (BMZ) area of the skin, as well as to the superficial and intermediate dermal vessels (Fig. 2). In addition, the IHC analysis revealed positive staining with myeloperoxidase (MPO) antibodies in the same areas where the p-ANCAs were observed in the epidermis and dermis by DIF. The patient was treated immediately with topical corticosteroids, as well as with one injection of intramuscular steroids (60 mg IM). One and a half weeks later, the patient was tested for 1) blood complement C3, and 2) an ANCA panel for vasculitis including myeloperoxidase (MPO), proteinase 3 antibodies and immunoglobulin E antibodies. Each of these test parameters were reported within normal limits, and the clinical lesions exhibited clearance at this time.
Fig. 2.
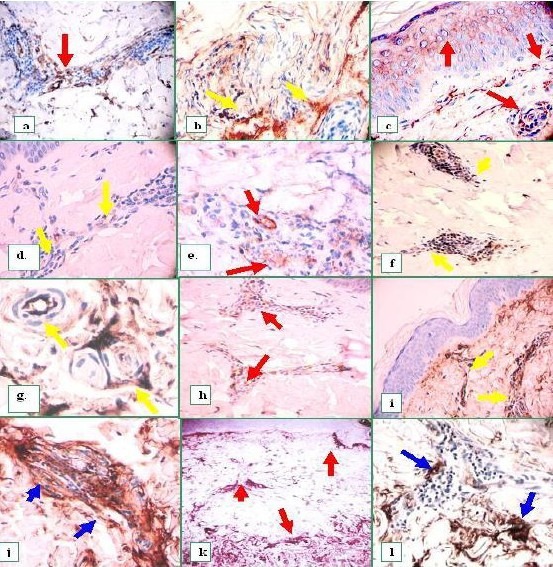
Real immunoreactivity versus intrinsic, non-pathologic background immunoreactivity in the dermal blood vessels using IHC.
Our results confirmed our previous DIF findings. In panel a, note fragmented positivity for CD 34 (red arrow). In b, autoreactivity to intermediate vessels using C1q (yellow arrows). c. Again using C1q, 1) an intracytoplasmic epidermal stratum spinosum reactivity is noted [(also shown with the DIF], as well as 2) a reactivity to the superficial plexus dermal vessels, and 3) to the intermediate dermal vessels (red arrows), (also IHC colocalized with both anti-CD31/PECAM and CD34 antibodies). d. Interestingly, some of these vessels also demonstrated positive findings utilizing anti-human IgD (yellow arrows) e, (red arrows). In f, Dermal perivascular reactivity was also noted utilizing anti-IgE (yellow arrows) and utilizing IgA (data not shown). g. Autoreactivity to the vessels utilizing anti-IgM (yellow arrows), as well as in h, to IgG (red arrows). In (i) note positive autoreactivity to the basal BMZ area, and the superficial vascular plexus (yellow arrows), and, in j, to intermediate dermal vessels (blue arrows). Finally, in k and l, we note compartmentalization or “tight cuffing” of the observed in the inflammatory areas utilizing anti-human fibrinogen (red arrows), and anti-human albumin (blue arrows), respectively.
Discussion
Small-vessel vasculitides are autoimmune disorders, often associated with antineutrophil cytoplasmic autoantibodies (ANCAs)[1]. The c-ANCAs are detected serum testing, as well as by direct and indirect immunofluorescence in a number of autoimmune disorders[1]. ANCAs were originally divided into two primary classes, c-ANCAs and p-ANCAs, based on their pattern of staining in neutrophils. ANCA associated vasculitides classically affect the small vessels, and can present diverse clinical manifestations[1–3]. The primary clinical treatment of these disorders is systemic corticosteroid therapy, with or without other immunosuppressants.
Attempts to explain and model the presence of ANCAs have included the theory of molecular mimicry. For instance, microbial superantigens are molecules expressed by bacteria and other microrganisms that have the power to stimulate a strong immune response by activation of T lymphocytes. These molecules generally have regions that resemble self-antigens; thus, the origin of the theory of molecular mimicry. Staphylococcal and streptococcal superantigens have been characterized in several autoimmune diseases. The classic example of such superantigen pathophysiology is found in group A Streptococcal rheumatic heart disease, where there is similarity between the M proteins of Streptococcus pyogenes and those of cardiac myosin and laminin. It has also been shown that up to 70% of patients with Wegener's granulomatosis are chronic nasal carriers of Staphylococcus aureus, with carriers having an eight times increased risk of disease presentation[3]. In addition, an alternative theory of defective apoptosis has been suggested as a possible explanation of ANCA pathophysiology. Neutrophil apoptosis, or programmed cell death, is critical in controlling the duration of the early inflammatory response, thus reducing injury to normal tissues by the neutrophils. ANCAs may be developed secondary to either 1) ineffective apoptosis or 2) ineffective removal of apoptotic cell fragments, leading to the exposure of the immune system to molecules normally sequestered inside the cells. The apoptosis theory solves the seeming pathophysiologic contradiction of how it could be possible for ANCAs to be raised against intracellular antigenic targets[3].
The role of ANCAs in disease has been linked to three primary diseases that are consistently associated with ANCAs. The most commonly associated disorders are Wegener's granulomatosis, microscopic polyangiitis and glomerulonephritis[4–8]. The antibodies are assumed to be involved in the generation and/or progression of the clinical lesions. Traditionally, c-ANCAs are associated with Wegener's granulomatosis; p-ANCAs are associated with both microscopic polyangiitis and focal necrotizing and crescentic glomerulonephritis[4–8]. However, in recent years, ANCAs targeted against other autoantigens have been identified, including those involved in Churg-Strauss syndrome. Patients with a number of other diseases, such as ulcerative colitis and ankylosing spondylitis, will frequently display ANCAs as well[4–8]. However, in these cases, there is classically no associated vasculitis, and the ANCAs are thought to be incidental or epiphenomena rather than part of the primary disease itself. Churg-Strauss syndrome is associated with p-ANCAs directed against MPO[5]. It is unclear what the role of ANCAs may be Wegener's granulomatosis, glomerulonephritis, and Churg Strauss syndrome; they may be markers of the diseases, or, alternatively, may play some part in the pathogenic process. In Wegener's granulomatosis, there is positive correlation between ANCA titer and disease activity; further, in vitro studies have shown that ANCAs cause activation of primed neutrophils and react with endothelial cells expressing PR3[4–8]. ANCAs may thus act by causing release of lytic enzymes from the white blood cells, causing vasculitis in the nearby blood vessel walls.
In our case, the clinical diagnosis of cutaneous vasculitis is supported by biopsy confirmation. Within the biopsy, microscopic findings of acute vasculitis such as fibrinoid necrosis, chronic signs such as endarteritis obliterans, and distant chronic signs such as acellular scarring were identified. Our findings did not favor the diagnosis of microscopic polyangiitis due to the following: 1) no additional indicators of granulomatous inflammation were present (e.g., evidence of lung cavities or infiltrates present for over one month duration); and 2) no biopsy findings of either a) glomerulonephritis, nor b) involvement of more than one organ system existed (as documented by a biopsy or surrogate marker). The patient was not taking any medications at the time of biopsy. By definition, ANCA related vasculitides are usually “pauci-immune”, that is, they present few and weak vascular immunoreactants by DIF .However, for more than fifty years, dermatopathologists and immunodermatologists were utilizing only monochromatic FITC DIF, and not multicolor immunofluorescence as we did in our case. Thus, the sensitivity and specificity of our analysis is increased by utilizing these techniques. We also utilized IHC; this technique was employed to rule out nonpathologic, intense autofluorescence of the vessels (i.e., to be able to distinguish between real pathologic reactivity versus intrinsic self fluorescence of the vessels).
Our case also displays acute findings, and highlights the difficulty of making a specific diagnosis of vasculitis (especially due to the presence of IgA, IgD, vascular deposits and p-ANCAs). Specifically, we believe our case may represent a brief, largely subclinical presentation of Henoch Schonlein purpura, or, alternatively, an ANCA-positive small vessel vasculitis, i.e., polyarteritis nodosa[10–12]. Other diseases where the combined, simultaneous findings of vasculitis and ANCAs can be found include Goodpasture's syndrome/anti-GBM-positive renal disease, and eleven per cent of cases of giant-cell arteritis. Other rare, less likely diseases that should be considered include Felty's syndrome, atypical pneumonia, post-Streptococcal glomerulonephritis, systemic lupus erythematosus, mixed connective tissue disease, and Wegener's granulomatosis. Finally, it should be noted that many non-MPO p-ANCAs exist, including 1) those directed at other polycytoplasmic enzymes (elastase, lactoferrin, lactoperoxidase, lysozyme, azurocidin, or cathepsin G), as well as 2) ANAs, and 3) granulocyte-specific ANAs (GS-ANAs)[10–12]. Based on the clinical, histologic and DIF findings in our case, a complete ANCA panel was ordered for the patient. One of the primary clinical problems encountered when evaluating patients with vasculitis is the fact that patients often present during an acute flare of the disease process.
It is thus difficult for us to compare our case to other studies regarding treatment of early small vessel vasculitis. Specifically, we found reports where early treatment of MPO-p-ANCA - associated small vessel vasculitis was felt to be crucial for the patient's positive prognosis (patients were treated with oral corticosteroid and cyclophosphamide therapy, in conjunction with plasmapheresis)[10–12]. Notably, one of the major problems physicians have to face when treating patients with early or chronic phase vasculitis is that patients may not respond to standard therapy, or may require unacceptably high doses of steroids or immunosuppressants[10–13]. In our case, prompt medical intervention resulted in the clearance of the clinical as well as normalization of the laboratory findings in our patient. Finally, specific predictors of treatment resistance and relapse of ANCA-associated small-vessel vasculitis have been recently described by utilizing clinical, pathologic and serologic predictors of treatment resistance and relapse in a community-based cohort of patients with ANCA-associated vasculitides[10–13]. The presence of IgD has been also reported in association with allergic vasculitis[14]. Finally, we were also were able to demonstrate evidence of the p-ANCA antigen(s) in the epidermis. These likely represent chromatin fibers, or so-called neutrophil extracellular traps (NETs), released by ANCA-stimulated neutrophils; these antigens may contain the targeted autoantigens PR3 and MPO[15]. We conclude that in these cases, early H&E and multicolor DIF diagnosis and rapid treatment (with systemic corticosteroid therapy, with or without other immunosuppressants) may contribute to both a favorable prognosis, and to a lack of lesional clinical recurrence .
Acknowledgement
There are no conflicts of interest.
References
- 1.Radice A, Sinico RA. Antineutrophil cytoplasmic antibodies (ANCA) Autoimmunity. 2005;38:93–103. doi: 10.1080/08916930400022673. [DOI] [PubMed] [Google Scholar]
- 2.Carlson JA, Ng BT, Chen KR. Cutaneous vasculitis update: diagnostic criteria, classification, epidemiology, etiology, pathogenesis, evaluation and prognosis. Am J Dermatopathol. 2005;27:504–528. doi: 10.1097/01.dad.0000181109.54532.c5. [DOI] [PubMed] [Google Scholar]
- 3.Reumaux D, Duthilleul P, Roos D. Pathogenesis of diseases associated with antineutrophil cytoplasm autoantibodies. Hum Immunol. 2004;65:1–12. doi: 10.1016/j.humimm.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Kain R, Matsui K, Exner M, Binder S, Schaffner G, Sommer EM, Kerjaschki D. A novel class of autoantigens of anti-neutrophil cytoplasmic antibodies in necrotizing and crescentic glomerulonephritis: the lysosomal membrane glycoprotein h-lamp-2 in neutrophil granulocytes and a related membrane protein in glomerular endothelial cells. J Exp Med. 1995;181:585–597. doi: 10.1084/jem.181.2.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henry JB. 19th ed. Vol. 1556. Philadelphia: W.B. Saunders; 1996. Clinical Diagnosis and Management by Laboratory Methods; pp. 992–993. [Google Scholar]
- 6.Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med. 1988;318:1651–1657. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- 7.Seo P, Stone J. The Antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med. 2004;117:39–50. doi: 10.1016/j.amjmed.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 8.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci U S A. 1990;87:4115–4119. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337:1512–1523. doi: 10.1056/NEJM199711203372106. [DOI] [PubMed] [Google Scholar]
- 10.Lucena-Fernandes MF, Duhaime N, Richard C, Boire G, Ménard HA. Antineutrophil cytoplasmic antibodies in human pathologies. Union Med Can. 1993;2:103–108. [PubMed] [Google Scholar]
- 11.Hogan SL, Falk RJ, Chin H, et al. Predictors of relapse and treatment resistance in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Ann Intern Med. 2005;143:621–631. doi: 10.7326/0003-4819-143-9-200511010-00005. [DOI] [PubMed] [Google Scholar]
- 12.Chen KR, Carlson JA. Clinical approach to cutaneous vasculitis. Am J Clin Dermatol. 2008;2:71–92. doi: 10.2165/00128071-200809020-00001. [DOI] [PubMed] [Google Scholar]
- 13.Akar H, Ozbasli-Levi C, Senturk T, Kadikoylu G, Levi E, Bolaman Z. MPO-ANCA-associated small vessel vasculitis presenting as fever of unknown origin. Report of one case. Nephron. 2002;3:73–675. doi: 10.1159/000064068. [DOI] [PubMed] [Google Scholar]
- 14.Cormane RH, Giannetti A. IgD in various dermatoses: immunofluorescence studies. Br J Derm. 1971;84:523. doi: 10.1111/j.1365-2133.1971.tb02541.x. [DOI] [PubMed] [Google Scholar]
- 15.Kai Kessenbrock K, Krumbholz M, Schönermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nature Medicine. 2009;15:623–625. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]