

Abstract
Background:
Wilson's disease is one of the most common hereditary causes of unclear hepatopathy.
Patient & Method:
A 34-year old male patient with a history of hyperlipidemia was admitted with symptoms of abdominal pain and slight hematuria. Abnormal liver function tests, ultrasound reports and liver biopsy were suggestive of nonalcoholic steatohepatitis (NASH). The patient received preliminary treatment for NASH. However, on subsequent follow-up, NASH remained unresolved and liver histology showed fibrosis progression from fibrosis stage 1 to stage 3.
Results:
Biochemical tests revealed that the levels of serum ceruloplasmin were decreased (7mg/dl) while the urinary excretion of copper was found to be increased (174.2 μg/day). Wilson's disease was confirmed by diagnostic mutation analysis involving Direct Sequencing. Heterogeneity in the patient's ATP7B gene confirmed Wilson's disease. Administration of D-penicillamine resulted in a decrease in fat deposition in the liver and no further progression in fibrosis after 10 months.
Conclusion:
Adult patient presenting NASH as first symptoms need to be examined for Wilson's disease and other metabolic conditions affecting the liver, prior to initiation of treatment.
Keywords: Wilson's disease, serum ceruloplasmin, hyperlipidemia, liver, non alcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH)
Introduction
Wilson's disease is a rare metabolic disorder characterized by copper accumulation in the body, mainly in the liver and brain. In Japan, one in 40,000 individuals is believed to have Wilson's disease[1]. Usually, onset of symptoms occur between the ages of 6 and 40, caused by mutations of the ATP7B gene on chromosome 13, leading to dysfunction of the copper transporting liver enzyme P-type ATPase. This enzyme is responsible for transporting copper into bile and incorporating it into ceruloplasmin[2–5]. Ceruloplasmin is a protein synthesized in the liver and is the major copper carrying protein in blood, responsible for transporting about 90% of copper in plasma. Liver disease leads to reduction in ceruloplasmin synthesis and a decrease in plasma level. Low plasma level of ceruloplasmin is an indication for Wilson's disease[6–10].
Wilson's disease affecting the liver may be asymptomatic or may present in the form of hepatomegaly, fatty liver, jaundice, acute hepatitis, fulminant hepatic failure, portal hypertension with bleeding varices, cirrhosis and even liver cancer[11–14]. NASH is fatty inflammation of the liver in the absence of excess alcohol consumption. Most cases of NASH are related to insulin resistance and the metabolic syndrome (obesity, hyperlipidemia, type 2 diabetes mellitus and hypertension) and respond well to treatments for type 2 diabetes mellitus, that is, diet restriction, weight loss and other life-style related changes. Usually, NASH is diagnosed following abnormal liver function tests during routine examinations or screening tests for some other disease. Diagnosis of NASH is made in the presence of elevated liver enzymes and hepatic steatosis on abdominal ultrasound and confirmed by liver biopsy, which determines the severity of liver inflammation and fibrosis progression[15–18].
Case Report
A 34-year old man with a history of hyperlipidemia was admitted to a clinic with symptoms of abdominal pain and mild hematuria. Based on the patient's symptoms, urinary tract infection was initially suspected, but no evidence of renal stone was found. Hyperlipidemia and elevated liver enzymes were shown in the biochemical tests (Table 1).
Table 1.
Biochemical data of patient at the time of each biopsy
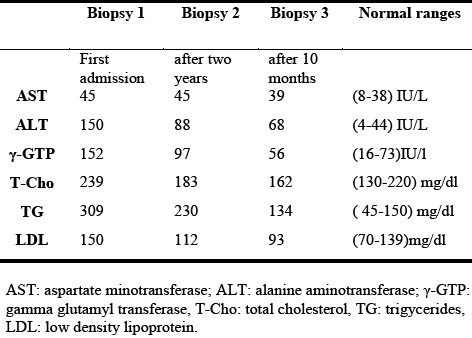
The patient had no history of prior similar illness nor was there a family history of liver disease. The patient's alcohol consumption was around 350 ml of beer 3 times a week. After being diagnosed as fatty liver, the patient was referred to Kawasaki Hospital. Initial laboratory tests revealed the elevated liver enzymes (ALT: 150 IU/L, AST: 45 IU/L, ALP: 466 IU/L, and γ-GTP: 152 Iu/L) and an abnormal lipid profile (T-Cho: 239 mg/dl, TG: 309 mg/dl, and LDL-C: 150 mg/dl), but the markers of viral or autoimmune hepatitis were negative. Subsequent abdominal ultrasound and CT-scan were indicative of fatty liver. Liver biopsy showed diffuse fat deposition (70-80%) in all lobules of hepatocytes with large to medium fat droplets, localized in the portal area. Presence of mild to moderate portal inflammation with the limiting plate intact was also observed in the liver parenchyma. The biopsy assessment was fibrosis stage 1 and activity grade 2 (Fig. 1).
Fig. 1.
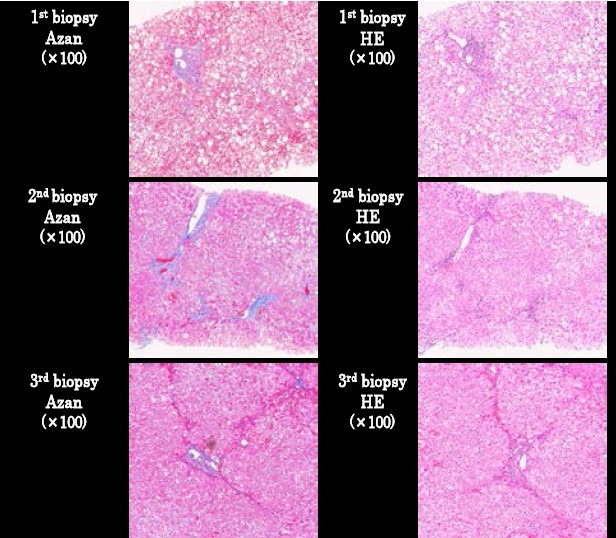
Three sets of biopsy specimens of the patient in HE & Azan staining showing fibrosis stage on first admission (Biopsy 1), 2 years later, (Biopsy 2) and 1 year after the 2nd biopsy (Biopsy 3).
Accordingly, the patient received treatment for NASH in the outpatient clinic, along with advice on making lifestyle changes to improve health. However, on subsequent follow-up, NASH remained unresolved, liver enzymes remained elevated (ALT: 106 IU/L; ALP: 413 IU/L; γ-GTP: 105 IU/L) and liver biopsy showed a continuing inflammation in the portal area with the limiting plate partly broken. Interphase hepatitis with bridging fibrosis was observed in some areas with moderate inflammation. Fat deposition had moderately decreased to about 50%, with the presence of small to moderate sized fat droplets. The biopsy assessment was fibrosis stage 3 and activity grade 2 (Fig. 1).
Further laboratory investigation showed a decrease in the levels of ceruloplasmin (7mg/dl) in sera and the urinary excretion of copper to be 74.2 μg/ in 24 hours, though only traces of copper were detected by liver biopsy. Suspicion of Wilson's disease led to diagnostic mutation analysis via Direct Sequencing. Presence of a heterogeneity in the patient's ATP7B gene confirmed Wilson's disease (Fig. 2).
Fig. 2.
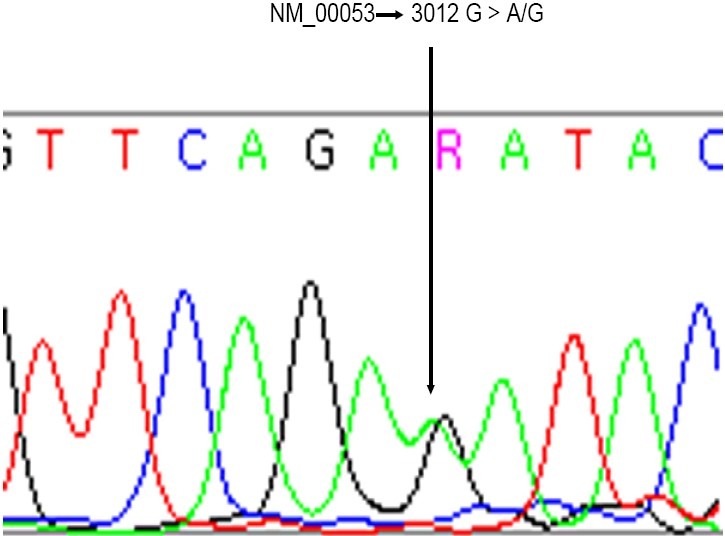
Direct mutation analysis showing the presence of heterogeneity in the patients ATP7B gene.
However, the patient had no apparent neurological sign or symptoms of Wilson's disease. Administration of D-penicillamine at a dose of 795 μg/day resulted in an increase in the urinary excretion of copper at 666.7 μg/day. After 10 months of treatment, liver function tests improved (ALT: 48 IU/L; AST: 28 IU/L; ALP: 332IU/L; γ-GTP: 61 IU/L), and with the help of nutritional counseling, an excess body weight of about 5 kg was lost. Liver biopsy showed overall marked decrease in fat deposition (< 5 %) in the liver, though within the liver parenchyma, bridging fibrosis remained unchanged and fibrotic bands appeared fine but still connected to the portal area (Fig. 1).
Discussion
A progressive case of Wilson's disease showed good response to D-penicillamine treatment. Initially, the patient's presenting symptoms were indicative of NASH[15]. However, treatments for NASH were unable to prevent fibrosis progression of liver tissue[16].
Differential diagnosis of NASH is important, and led to the confirmation of Wilson's disease. Patients with NASH often present with few or no symptoms, though imaging techniques and liver biopsy show fat accumulation in the liver, mostly accompanied by hyperlipidemia. However, evaluation of patients based on fatty liver, hyperlipidemia, and abnormal liver function tests may not be sufficient in detecting the severity of the underlying cause.
Wilson's disease is transmitted through a recessive gene believed to be located on chromosome 13, which disrupts copper metabolism. Since the main sites of copper accumulation in the body are the liver and the brain, hepatic and neuropsychiatric symptoms often predominate in early childhood. Therefore, a great awareness of clinical features of inherited metabolic liver diseases such as Wilson's disease may promote proactive diagnostic evaluation, preemptive treatment and help in preventing the development of phenotypic complications.
Therefore, for the adult and even elderly patients with unexplained histologic findings of steatohepatitis, it is reasonable to consider the possibility of Wilson's disease, before starting any treatment regime.
References
- 1.Horike N, et al. Wilson disease, hemochromatosis. Kantansui. 2007;54:371–376. [Google Scholar]
- 2.Gow PJ, Smallwood RA, Angus PW. Diagnosis of Wilson's disease. An experience over three decades. GUT. 2000;46:415–419. doi: 10.1136/gut.46.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ala A, Walker Ap, Ashkan K, Dooley JS, Schilsky ML. Wilsons Disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 4.Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study. Gut. 2007;56:115–120. doi: 10.1136/gut.2005.087262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology. 2003;37:1475–1492. doi: 10.1053/jhep.2003.50252. [DOI] [PubMed] [Google Scholar]
- 6.Motonishi S, Hayashi H, Fujita Y, Okada H, Kusakabe A, Ito M, Miyamoto K, Ueno T. Copper and iron rich matrices in hepatocellular lipofuscin particles of a young male patient: diagnostic ultrastructures for Wilson's disease. Ultrastruct Pathol. 2006;30:409–414. doi: 10.1080/01913120600854327. [DOI] [PubMed] [Google Scholar]
- 7.Roberts EA, Sarkar B. Liver as a key organ in the supply, storage and excretion of copper. Am J Clin Nutr. 2008;88:851S–854S. doi: 10.1093/ajcn/88.3.851S. [DOI] [PubMed] [Google Scholar]
- 8.Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22:439–458. doi: 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- 9.Yang FM, Freidrichs WE, Cipples RL. Human ceruloplasmin.Tissue-specific expression of transcripts produced by alternative splicing. J Biol. 1990;265:10780–10785. [PubMed] [Google Scholar]
- 10.Kumagi T, Horiike N, Michitaka K, Hasebe A, Kawai K, Tokumoto Y, Nakanishi S. Recent clinical features of Wilson's disease with hepatic presentation. J Gastroenterol. 2004;39:1165–1169. doi: 10.1007/s00535-004-1466-y. [DOI] [PubMed] [Google Scholar]
- 11.Dib N, Valsesia E, Malinge M, Mauras Y, Misrahi M, Cales P. Late onset of Wilson's disease in a family with genetic haemochromtosis. Eur J Gastroenterol Heptol. 2006;18:43–47. doi: 10.1097/00042737-200601000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Seo JK. Wilson disease: an update. Korean J Hepatol. 2006;12:333–363. [PubMed] [Google Scholar]
- 13.Alpinar E, Akhan O. Liver imaging findings of Wilson's disease. Eur J Radiol. 2007;61:25–32. doi: 10.1016/j.ejrad.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Kuloglu Z, Kansu A, Demireken F, Erden E, Fitoz S, Gigin N. An unusual presentation of Wilson's disease in childhood: nodular fatty infiltration in liver. Turk J Pediatr. 2008;50:167–170. [PubMed] [Google Scholar]
- 15.Alffire ME, Treem WR. Nonalcoholic fatty liver disease. (297-299).Pediatr Ann. 2006;35:290–4. doi: 10.3928/0090-4481-20060401-14. [DOI] [PubMed] [Google Scholar]
- 16.Adams LA, Angulo P. Treatment of non-alcoholic fatty liver disease. Postgrad Med J. 2006;82:315–322. doi: 10.1136/pgmj.2005.042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark JM, Diehl AM. Nonalcoholic fatty liver disease: an under recognized cause of cryptogenic cirrhosis. JAMA. 2003;289:3000–3004. doi: 10.1001/jama.289.22.3000. [DOI] [PubMed] [Google Scholar]
- 18.Huang MA, Greenson JK, Chao C. One year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: a pilot study. Am J Gastroenterol. 2005;100:1072–1081. doi: 10.1111/j.1572-0241.2005.41334.x. [DOI] [PubMed] [Google Scholar]