

Abstract
Background:
We reported a new variant of endemic pemphigus foliaceus in El Bagre, Colombia.
Aims:
Our study performed Complex Segregation Analysis (CSA) and short tandem repeats to discriminate between environmental and/or genetic factors in this disorder.
Materials and Methods:
The CSA analysis was carried out according to the unified model, implemented using the transmission probabilities implemented in the computer program POINTER, and evaluated by using a software package for population genetic data analysis (GDA), Arlequin. We performed pedigree analyses by using Cyrillic 2.1 software, with a total of 30 families with 50 probands (47 males and 3 females) tested. In parallel to the CSA, we tested for the presence of short tandem repeats from HLA class II, DQ alpha 1, involving the gene locus D6S291 by using the Hardy-Weinberg- Castle law.
Results
Our results indicate that the best model of inheritance in this disease is a mixed model, with multifactorial effects within a recessive genotype. Two types of possible segregation patterns were found; one with strong recessive penetrance in families whose phenotype is more Amerindian-like, and another of possible somatic mutations.
Conclusion:
The penetrance of 10% or less in female patients 60 years of age or older indicates that hormones could protect younger females. The greatest risk factor for men being affected by the disorder was the NN genotype. These findings are only possible due to somatic mutations, and/or strong environmental effects. We also found a protective role for two genetic loci (D6S1019 AND D6S439) in the control group.
Keywords: Endemic pemphigus foliaceus, short tandem repeats (STRs), complex segregation analysis (CSA)
Introduction
Endemic pemphigus foliaceus (EPF) represents a geographically restricted, autoimmune disease occurring in South, in Central America, and in Tunisia, Africa[1–5]. The Brazilian form, known as fogo selvagem (FS), predominantly affects young people; both sexes are equally affected. In this disorder, many cases present a genotype clustered within families; and autoantibodies are found not only in patients, but also often in relatives of the patients[1–4]. The autoantibodies are less commonly observed in healthy people living within the endemic regions[1–6]. Few genetic studies have been performed in the patients affected by endemic pemphigus foliaceus.
The major histocompatibility complex, class II, DQ alpha 1 (HLA-II), also known as HLA-DQA1, is a human gene present on the short arm of chromosome 6 (6p21.3) and also denotes the genetic locus which contains this gene[7]. The protein encoded by this gene is one of two proteins that are required to form the DQ heterodimer, a cell surface receptor essential to the function of the immune system[7]. HLA-DQA1 belongs to the HLA class II alpha chain paralogues. The class II molecule is a heterodimer consisting of an alpha (DQA) and a beta chain (DQB), both anchored in the membrane[7]. It plays a central role in the immune system by presenting peptides derived from extracellular proteins[7]. Class II molecules are expressed in antigen-presenting cells (APCs), B lymphocytes, dendritic cells and macrophages[7]. The alpha chain contains 5 exons. Exon one encodes the leader peptide, exons 2 and 3 encode the two extracellular protein domains, exon 4 encodes the transmembrane domain and the cytoplasmic tail[7]. Within the DQ molecule, both the alpha chain and the beta chain contain polymorphisms specifying the peptide binding specificities, resulting in up to 4 different molecules[7].
Selected previous studies had addressed the role of HLA class II alleles in fogo selvagem, suggesting a locus of major predisposition genes; although contradictory reports also exist[8–13]. Other previous studies in endemic pemphigus foliaceus addressed the possible polymorphism of one of the most common auto antigens in EPF, namely the desmoglein 1 (Dsg1) gene, and its interaction in an epistatic manner with the major histocompatibility complex class II genes[14,15]. Regrettably, these studies have also demonstrated contradictory findings[14,15]. We described a new variant of endemic pemphigus occurring in a mining town in northeastern Colombia in the El Bagre area (El Bagre-EPF). We had performed an 11-year prospective, controlled epidemiologic, humanitarian, and immunologic fieldwork case-control survey[16–17]. The disease appeared in 4.7% of middle-aged and older men and postmenopausal women from these rural areas[16–17]. The disease differs from previously described forms of endemic pemphigus foliaceus. El Bagre-EPF shares some heterogeneous immunoreactivity with paraneoplastic pemphigus, but is not associated with malignant tumors[16–17]. The disease resembles Senear-Usher syndrome (i.e., pemphigus and lupus), but occurs endemically. El Bagre-EPF manifests either as 1) a localized form (clinically and immunologically stable), or 2) as a more florid form, where systemic compromise seems to occur. The systemic form may affect organs other than skin, and is characterized by episodic relapses and poor prognosis in comparison with the localized form[16–17]. Heterogeneous antigenic reactivity is observed as in paraneoplastic pemphigus, but with no evidence of association with neoplasia. In addition, constant exogenous antigenic stimulation and a genetic predisposition may be required in the pathogenesis of this disease[16–17]. Thus, the objective of this study was to perform complex segregation analysis (CSA) and associated short tandems repeat (STR), studies to discriminate between genetic and/or environmental factors in this disorder. We performed pedigrees, and utilized other genetic tools to further ascertain a possible model(s) of inheritance for the El Bagre-EPF. We took into consideration variables such as age, sex, and that the population of Colombia is an admixture of Caucasoid, Negroid and Mongoloid peoples in our analysis[18]. Thus, we: performed the pedigrees, created a DNA repository, searched for a possible major gene, and applied a unified model of complex segregation analysis and using the Genetic Data Analysis (GDA) and Arlequin analysis programs[18–26]. In parallel, based on the documented findings that down-regulated human leukocyte antigen (HLA) class I expression is 1) frequently correlated with allelic loss at 6p21.3 (the location of the HLA coding sequence) and 2) is further associated with several microsatellite rich regions spanning the 4 megabase HLA region, we tested for selected short tandems repeat loci. Our purpose was to find either linkage disequilibrium, and/or possibly a direct contributory gene (s) relevant to the development of clinical El Bagre-EPF[18–26].
Materials and Methods
Samples and controls for the genetic study: An active search was performed of El Bagre population data collected during the 1992-2001 period. Patients that fulfilled the clinical and immunologic criteria for a diagnosis of El Bagre-EPF were included in this study[16,17]. We randomly selected 33 cases from this pool, according to a sequential sampling strategy. In addition, we selected 33 healthy controls from the endemic area (CEAs) including some relatives of the affected patients, matched to the affected patients by age, sex, working activity and living area[16,17].
Subjects of study and obtaining the original clinical and laboratory data: We studied 50 patients who fulfilled the diagnosis of El Bagre-EPF as described by us[16,17], and 50 controls from the endemic area matched by age, sex living and working conditions. All patient consents were obtained, as well as Institutional Review Board (IRB) permission. We tested a total of 30 families with 50 probands (50 El Bagre-EPF patients; 47 males and 3 females) and tested for the presence of positive intercellular staining between the keratinocytes by direct immunofluorescence and by indirect immunofluorescence[16,17]. The El Bagre-EPF cases included those who fulfilled the clinical phenotype, epidemiological and immunological criteria. To qualify as a case of El Bagre-EPF, a patient's sera was required to immunoprecipitate a Con-A affinity purified bovine tryptic fragment of pemphigus foliaceus antigen, as previously described[16,17]. In addition to the above-mentioned criteria, the sera of the subjects of the study were also tested by immunoblotting (IB) for reactivity against Dsg1, as well as other antigens including plakins by ELISA testing. The El Bagre-EPF cases were required to recognize desmoglein 1 and or other plakins molecules by immunoblotting[16,17]. The sera of some patients affected by fogo selvagem from Brazil, two sera from cases of sporadic pemphigus foliaceus and healthy subject sera from the USA were used as controls.
Notations, assumptions, ascertainment, and parameters the Hardy–Weinberg-Castle law: Mendelian genetics were rediscovered in 1900. However, it remained somewhat controversial for several years as it was not then known how the model could account for continuous characteristics. Udny Yule argued against Mendelism because he thought that dominant alleles would increase in the population[27]. William E. Castle,[28] showed that without selection, the genotype frequencies would remain stable. Karl Pearson,[29] found one equilibrium position with values of p = q = 0.5. The Hardy–Weinberg-Castle law greatly advanced the genetics field[30,31]. The basis of the principle follows: Suppose that Aa is a pair of mendelian characters, A being dominant, and that in any given generation the number of pure dominants (AA), heterozygotes (Aa), and pure recessives (aa) are as p:2q:r. Also, suppose that the numbers of individuals are large, so that mating may be regarded as random, that the sexes are evenly distributed among the three varieties, and that all are equally fertile. Thus, in the next generation, the numbers will be as (p+q)2 :2(p+q) (q+r) :( q+r)2, or as p1:2q1:r1. The interesting question is —in what circumstances will this distribution be the same as that in the generation before? It is easy to see that the condition for this is q2 = pr. And since q12 = p1r1, whatever the values of p, q, and r may be, the distribution would continue unchanged after the second generation. No natural population can meet all the requirements for a Hardy–Weinberg-Castle law[29–31]. The Hardy–Weinberg-Castle law states that both allele and genotype frequencies in a population remain constant—that is, they are in equilibrium—from generation to generation, unless specific disturbing influences are introduced. Those disturbing influences include non-random mating, mutations, selection, limited population size, random genetic drift and gene flow[29–31]. The Hardy–Weinberg-Castle law equilibrium is impossible in nature. Genetic equilibrium is an ideal state that provides a baseline to measure genetic change. Static allele frequencies in a population across generations assume: 1) random mating, 2) no mutation (the alleles don’t change), 3) no migration or emigration (no exchange of alleles between populations), 4) infinitely large population size, and 5) no selective pressure for or against any traits[29–31].
In the simplest case of a single locus with two alleles: the dominant allele is denoted A and the recessive a and their frequencies are denoted by p and q; freq (A) = p; freq (a) = q; p + q = 1. If the population is in equilibrium, then we will have freq (AA) = p2 for the AA homozygotes in the population, freq (aa) = q2 for the aa homozygotes, and freq (Aa) = 2pq for the heterozygotes. Based on these equations, we can determine useful but difficult-to-measure facts about a population. The Hardy–Weinberg-Castle law could be used to work backward from disease occurrence to the frequency of heterozygous recessive individuals.[30,31]. Thus, we took into consideration the population's effective size, heterozygosity levels, and inbreeding coefficients for particular individuals[25–28]. We utilized a “pointer” individual (a disease proband, defined as a relative of extreme phenotype). The pedigrees and data were analyzed using the genetics data analysis Arlequin and Cyrillic software systems [18–26, 27–31]. The unified model allowed the underlying liability for El Bagre-EPF to be separated into three independent components: a diallelic single major gene locus component, a polygenic background component, and a random environmental component.
Complex segregation analysis (CSA): A total of 30 families with 50 probands (50 El Bagre-EPF patients) (47 males and 3 females) were ascertained and the complex segregation analysis was carried out, with the aim to determine the real value of the genetic component versus the environmental component (s). This was performed according to the unified model of Lalouel et al, 1981[18–26]. The ascertainment probability was calculated according to Simpson's method for each set of pedigrees in each syndromic group according to the equation a (a-1)/a(r-1) where a, number of probands and r, total number of affected. The model partitions the total variation in the underlying liability to El Bagre-EPF into three independent components: a diallelic single major locus component, a polygenic background, and a random environmental component[18–26].
Genetic linkage, linkage mapping and linkage disequilibrium: The amount of crossing over between different linked genes led to the concept that crossover frequency might indicate the distance separating genes on the chromosome[32,33]. Therefore a genetic map, also called a linkage map, was created based on the fact that the greater the distance between linked genes, the greater the chance that non-sister chromatids would cross over in the region between the genes[32,33]. By working out the number of recombinants it is possible to obtain a measure for the distance between the genes[32,33]. This distance is called a genetic map unit (m.u.) or a centimorgan and is defined as the distance between genes for which one product of meiosis in 100 is recombinant[32,33]. A recombinant frequency (RF) of 1 % is equivalent to 1 m.u. A linkage map is created by finding the map distances between a number of traits that are present on the same chromosome, ideally avoiding having significant gaps between traits (to avoid the inaccuracies that will occur due to the possibility of multiple recombination events)[32,33]. Linkage mapping is critical for identifying the location of genes that cause genetic diseases. In an ideal population, genetic traits and markers will occur in all possible combinations, with the frequencies of combinations determined by the frequencies of the individual genes. For example, if alleles A and a occur with respective frequencies of 90% and 10%, and alleles B and b at a different genetic locus occur with respective frequencies 70% and 30%, the frequency of individuals having the combination AB would be 63%, the product of the frequencies of A and B, regardless of how close together the genes are. However, if a mutation in gene B that causes a disease happened recently in a particular subpopulation, it almost always occurs within a particular allele of the gene,[32,33]. assuming, that the individual in which the mutation occurred in fact had that variant of gene B, and there have not been sufficient generations for recombination to happen between the alleles (presumably due to tight linkage on the genetic map)[32,33]. In this case, called linkage disequilibrium, it is possible to search potential markers in the subpopulation, and identify which marker the mutation is close to, thus determining the mutation's location on the map, and identifying the gene at which the mutation occurred[32,33].
Family haplotype analysis: Haplotypes were constructed through the application of the Cyrillic pedigree data program and by visual inspection, utilizing the minimum number of recombination events observed in each family. At each marker locus, an ancestral allele was defined as that allele occurring in the greatest frequency in the affected cohort compared with the unaffected cohort. The assumed ancestral haplotype, presumed to carry the disease mutation, was then constructed using these alleles[18,26].
Deoxyribonucleic acid (DNA) extraction: The blood samples were collected in ethylenediaminetetraacetic acid (EDTA) tubes; the DNA extraction was performed using the Gentra Puregene Blood Kit (QIAGEN, Germantown, MD, USA) and following the manufacturer's recommendations. The precipitated DNA was stored at –20°C for further amplification.
Molecular Genotyping: The analysis is performed by extracting nuclear DNA from the cells of the subjects of the study, and then amplifying specific polymorphic regions of the extracted DNA by means of the polymerase chain reaction (PCR). Once these sequences had been amplified, they were resolved either through gel electrophoresis and/or capillary electrophoresis, which allowed determination of how many repeats of the short tandems repeat sequence were present. We specifically study the microsatellite loci neighboring the major histocompatibility complex region on the 6p chromosome, namely D6S276, D6S265, D6S273 and D6S291. These regions were amplified by PCR using fluorescently labeled primers specific for each locus. The PCR procedures were carried out in 10 µl reactions, containing 30 ng of genomic DNA, 1 X PCR reaction and 5 picomol of each primer. An initial denaturation was performed at 95 °C for 2 min, and then the reaction mix was amplified for 30 cycles using the following conditions: 1 min 94 °C denaturation, 1 min 56-58 °C annealing, 1 min 72 °C extension and a final 5 min 72 °C extension. The PCR products were denatured at 95°C for 5 min and placed on ice. Then, they were electrophoresed according to the manufacturer's protocols on an ABI-PRISM 310 Genetic Analyzer (Perkin-Elmer) system. The PCR products were identified by their molecular weights relative to standardized markers. Detection of fluorescent products was performed automatically using GeneScan 2.1 software and the data analyzed and exported as a text file for subsequent analyses. The PCR products for each allele were further characterized using a 6% SDS polyacrylamide denaturing gel and silver staining techniques.
Statistical evaluation: Medical software for Windows, as well as the POINTER and PRISM GRAPH PAD programs was utilized for statistical analysis.
Results
We performed our study by first examining 50 El Bagre-EPF patients and 30 extended members of their families, and building multigenerational clinical pedigrees. We then tested 50 controls matched by age, sex and work occupation from the endemic area, and 50 disease probands (El Bagre-EPF patients; 47 males and 3 females) (π=0.76). We also utilized genealogical data from several kindreds, displaying a strong aggregation in families affected by El Bagre-EPF; the average siblingship size in these families was 5.3.
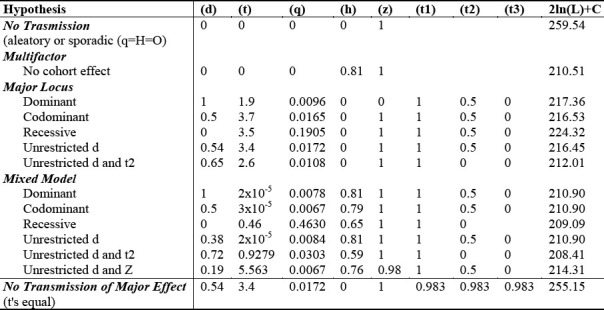
In Table 1, the complex segregation analysis of our data is shown. Fifteen hypothetical models were evaluated using the likelihood ratio test, and likelihood values for each comparison were examined using the Chi squared (χ2) test. Parameter estimates corresponding to maximum likelihood models under each set of constraints are shown for each model. In the process, 50 probands (47 males and 3 females), and 31 extended and multigenerational pedigrees were analyzed; they were manifested as 262 nuclear components and 1642 records.
Table 1.
Models of Complex Segregation Analysis in multiple, hypothetical disease modes of inheritance (left column) are displayed.
Based on the fact that transmission probabilities are not correctly implemented in the computer program POINTER, we then chose another 17 affected individuals pointed out by the probands (16 males and 1 female) (π=0.6531) for correction.
Our results indicate that the best model for this disease is that of a mixed model (Table 1) with strong multifactorial effects. Our study revealed more than 81% phenotypic variance, and a low major gene effect (approximately 19%) (Table 1). Although the effect of a major gene epistatic influence in the predisposition to this disease can not be rejected, its contribution to the phenotypic variance is weak. The major gene effect is demonstrated in the approximately 50% of the men from the 2, 3 and 4 susceptibility classes belonging to heterozygote genotypes, with an actual risk close to 50% (Table 1).
The comparison among the hypotheses of 1) multifactor component only and 2) that of the existence of a major gene only (i.e., a comparison between model 2 and model 7) did not show significant differences, although the value of p was close to 0.05 (χ2 2 DF = 5.94, p > 0.05). Among the models postulating a major gene effect only, the iteration of the t2 parameter of the general model (i.e., model 7 vs. model 8) showed significant differences (χ2 1 DF = 4.44, p < 0.05). It is important to observe that each of the mixed major gene models (i.e., models 9, 10 and 11) showed a significant better fit than models assuming the presence of only one major gene. Moreover, it was impossible to determine significant differences among the mixed models (Table 1). Also, the values of t (representing the standard deviations among the homozygotes AA and aa) were lower than 0.5, indicating an admixture of distributions as a consequence of the polygenic component. The model postulating no polygenic component in the mixed model was rejected (χ2 1 DF = 5.55, p <0.05). The model postulating the non-existence of a major gene component in the mixed model could not be rejected (i.e., comparison of models 2 and 12) (χ2 3df = 0.39, p>0.05). There were also no significant differences in the fitting of the general model when t2 (in the major gene component) and Z (in the polygenic component) parameters were not restricted. The model of no transmission of major effect was rejected (χ2 3df = 38.7, p <0.0001) (Table 1).
In the women, the major gene effect is represented exclusively by the heterozygote genotype (with a theoretical risk of 100% representing the probability that new cases will be the fruit of new somatic mutations or environmentally induced), and is close to 0% (Table 2). The presence of female hormones was noted as a possible protective factor in the development of this disease. The hypothesis of no familial transmission of El Bagre-EPF in these families was rejected (χ25 DF = 48.64, p < 0.0001) (Table 1)
Table 2.
Summarizes the classes of disease susceptibility according to sex and age at disease onset, and respective correlation of death probabilities (nqx).
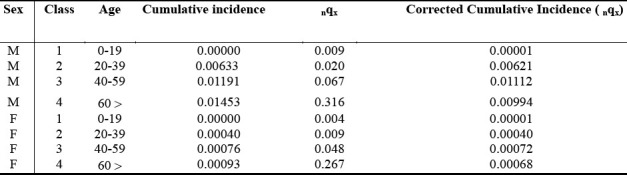
Table 3 Shows that the onset age of the disease does not comprise individuals younger than age 20, and most of the affected individuals (approximately 80%) are between 20 and 60 years of age, with a mean age of 45. In females, the age onset of the disease is within the post-menopausal period (i.e., 45 to 50 years)
Table 3.
Correction of onset by age
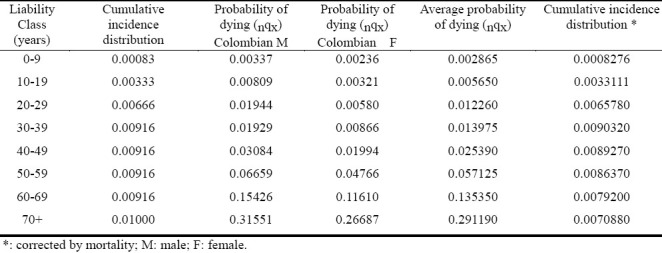
Table 4.
Provides information on Complex Segregation Analysis performed in our study, with total number of subjects, sexes, probands, and endemic area controls documented.
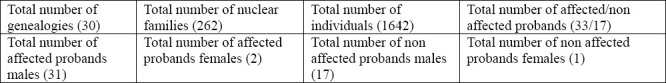
Table 5.
Summarizes both the linkage disequilibrium analyses and expected Hardy–Weinberg-Castle law genotype proportions, for both the El Bagre-EPF patients and the controls.
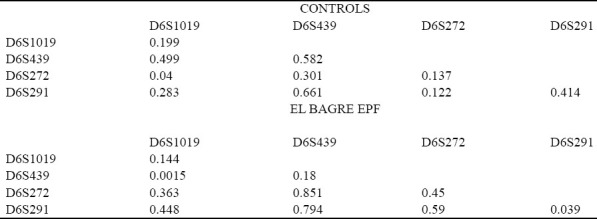
Table 6.
Markers detected in people affected by El Bagre-EPF and by controls from the endemic area matched by age, sex, and living activities.
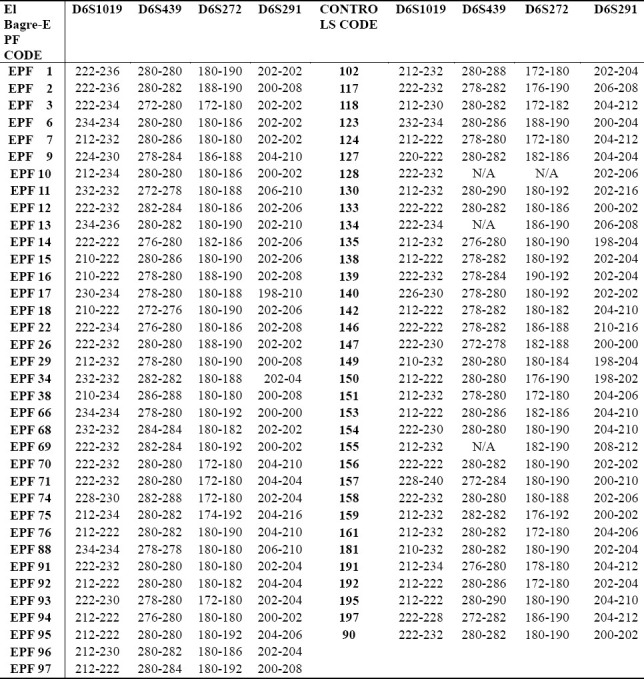
Figure 1.
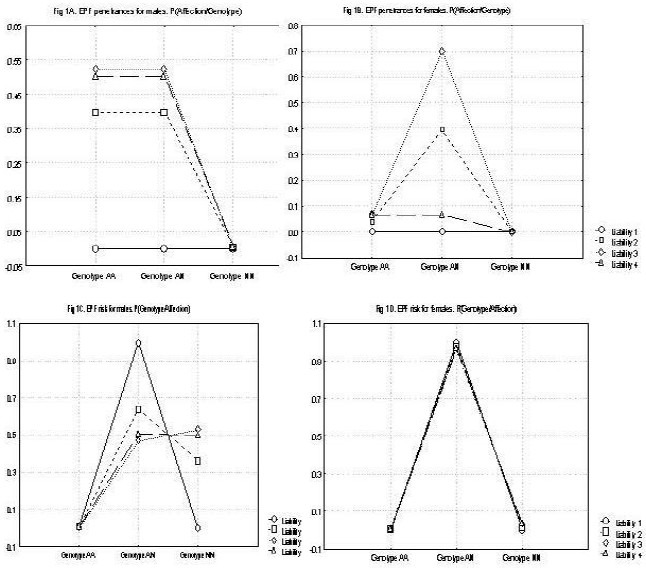
A: shows the penetrance for males (affection/genotype by sex) in people affected by El Bagre-EPF. B. Shows the penetrance for males (affection/genotype by sex) in people affected by El Bagre-EPF. C. Shows the risk for males for developing El Bagre-EPF (affection/genotype by sex). D. Shows the risk for females for developing El Bagre-EPF (affection/genotype by sex).
Discussion
The objective of this study was to perform complex segregation analysis (CSA) and associated short tandems repeat (STR), studies to discriminate between genetic and/or environmental factors in patients affected by a new variant of endemic pemphigus foliaceus in El Bagre, Colombia, South America.
We performed pedigrees and utilized other genetic tools to further ascertain a possible model(s) of inheritance for the El Bagre-EPF.
In this study we also used linkage disequilibrium to try to search potential markers in the subpopulation affected by a new variant of endemic pemphigus foliaceus in El Bagre, and try to identify which marker any such mutation is close to (thus determining the mutation's location on the genetic map and identifying the gene at which the mutation occurred). We were unable to locate a particular gene on a chromosome (gene locus) at this time. However, by using genetic linkage (that occurs when particular genetic loci or alleles for genes are inherited jointly) we were able to determine some degree of genetic linkage.
Our findings constitute a preliminary basis for further genetic molecular studies, based on the fact that genetic loci on the same chromosome are physically connected and tend to stay together during meiosis, and are thus genetically linked, i.e., autosomal linkage[31,32]. Alleles for genes on different chromosomes are usually not linked, due to independent assortment of chromosomes during meiosis. Because there is some crossing over of DNA when the chromosomes segregate, alleles on the same chromosome can be separated and go to different daughter cells[31,32]. In this variant of El Bagre-EPF, there is a greater probability of such separation if the alleles are far apart on the chromosome, and more likely that a cross-over will occur between them. The relative distance between two genes can be calculated using the offspring of an organism showing two linked genetic traits, and finding the percentage of the offspring where the two traits do not run together[31,32]. The higher the percentage of descendants that do not show both traits, the farther apart on the chromosome the two genes must be[31,32].
Among individuals of an experimental population or species, some phenotypes or traits occur randomly with respect to one another in a manner known as independent assortment. Today, scientists understand that independent assortment occurs when the genes affecting the phenotypes are found on different chromosomes or separated by a great enough distance on the same chromosome that recombination occurs at least half of the time[31,32]. An exception to independent assortment develops when genes appear near one another on the same chromosome.
When genes occur on the same chromosome, they are usually inherited as a single unit[31,32]. Genes inherited in this way are said to be linked, and are referred to as “linkage groups.” For example, in fruit flies the genes affecting eye color and wing length are inherited together because they appear on the same chromosome. But in many cases, even genes on the same chromosome that are inherited together produce offspring with unexpected allele combinations[31,32]. These events result from the process of crossing over. Our results point to possible crossing-over in our population. At the beginning of normal meiosis, a chromosome pair (made up of a chromosome from the mother and a chromosome from the father) intertwines and exchange sections or fragments of chromosome[31,32]. The pair then breaks apart to form two chromosomes, with a new combination of genes that differs from the combination supplied by the parents. Through this process of recombining genes, organisms can produce offspring with new combinations of maternal and paternal traits that may contribute to or enhance survival[31,32].
Our complex segregation analyses constitute initial steps to address the disease susceptibility inheritance of people affected by this new variant of El Bagre-EPF. Specifically, the effect of a major gene submitted to epistatic influences in the predisposition to this disease cannot be rejected, and its contribution to the phenotype variance was found to be weak. The major gene effect is represented in the approximately 50% of the men from the 2, 3 and 4 susceptibility classes belonging to heterozygote genotype, and with a risk close to 50%.
Thus far, the most parsimonious model is that of a mixed model with strong multifactorial effects (polygenes, mercury, metals, metalloids, tropical disease agent(s), ultraviolet radiation and other environmental effects, plus a lack of female hormones; more than 81% of the phenotypic variance) and a low major gene effect (approximately 19%). Although the effects of the major gene on the mixed model are very low, its existence is evidenced in rejecting the “non-existence of major gene effect” hypothesis and observing the existence of epistatic effects which were detected when the t2 parameter was iterated. The effect of a major gene submitted to epistatic influence in the predisposition to this disease can not be rejected; however, its contribution to the phenotypic variance is weak. The major gene effect is represented in approximately 50% of the men from the 2, 3 and 4 susceptibility classes belonging to heterozygote genotype and with a risk close to the 50%. In the women, the major effect is represented exclusively by the heterozygote genotype and with a risk of 100%, with the probability that new cases will be the fruit of new somatic mutations or environmentally induced close to 0%.
One socioeconomic issue that should be accounted for in the etiopathogenesis of El Bagre-EPF is the practice of inbreeding in the families of Amerindian origin, due to difficulty of travel and other social aspects of possessing the disease itself[29–32]. Our hypothesis regarding such possible inbreeding practices is evidenced by 1) the malodor that characterize some open blisters of the affected patients; 2) several ancestral Amerindian tribes (such as the Zenues and Embera-Catios) that have historically lived in closed communities, and practiced interracial outbreeding. Of note, in reviewing pedigrees of individual patients whose facial and anthropologic features most closely resembled the Amerindians, we detected 5 affected males in one family of 7 individuals. In contrast, on review of pedigrees from affected individuals who had lived briefly in the endemic area, the only individuals affected were indeed the probands.
Many pedigrees represented an intermediate presentation between these extremes. Given these findings, we believe that additional community-based studies may be more useful if those studies are include the presence of autoantibodies to El Bagre-EPF antigens, in contradistinction to strictly physical phenotypic data. We base our recommendation on the high rate of autoantibodies found by us, and reported by others in relatives of patients affected by endemic pemphigus foliaceus[16,17].
In regards of the short tandems repeats and to the genome per se, and our wide linkage analyses had identified a locus encoding susceptibility to El Bagre-EPF, and placed this gene between in the 12 cM interval between markers D6S426 and D6S276 on chromosome 6p21.3;. This area represents a broad region, encompassing the human leukocyte antigen complex[7]. There are 4 commonly encountered DQA1 alleles: DQA1*0101, *0102, *0103, *0104. These alleles are always found in haplotypes with HLA-DQB1*05 (DQ5) and HLA-DQB1*06 (DQ6). DQ1 is a serotype, rare among serotypes for human class II antigens, in that the antibodies to DQ1 react to the alpha chain of HLA DQ, these DQA1 allele gene products[7]. The other DQA1 alleles have no defined serotype. There are 5 groups, DQA1*02, *03, *04, *05, *06. DQA1 within these groups are either invariant or produce the same α-chain subunit. DQA1*02 and DQA1*06 contain only one allele. DQA1*03 has three alleles which each produce nearly identical α3. For DQA1*05, the DQA1*0501 and DQA1*0505 produce identical α5. Other DQA1*05 exist that produce variant α5var, but these are rare[7]. We have sought to localize the susceptibility gene more precisely by exploiting linkage, haplotype, and linkage disequilibrium information available through genotyping.
Pemphigus vulgaris is reported to be associated with HLA DR4 and/or DR6, and some studies in patients affected by pemphigus vulgaris pointed to a possible common ancestral origin for disease susceptibility[33]. Pemphigus vulgaris is reported to be associated with human leukocyte antigen DR4 and/or DR6 whereas few data are available on pemphigus foliaceus, including fogo selvagem, which is reported to be associated with DR1 and DR4.
In fogo selvagem, which is reported to be associated with HLA DR1 and DR4, family cases are frequent and not everyone living in the endemic region develops this disease, suggesting that host factors play a role in determining whether exposed individuals will be affected. Other studies in fogo selvagem with Brazilian Mestizos and with Xavante and Terena Indians indicate the possibility that some human leukocyte antigen alleles confer increased risk for endemic pemphigus disease. In fogo selvagem, family cases are frequent and not everyone living in endemic region develops this disease, suggesting that host factors play a role in determining whether exposed individuals will be affected. Other authors’ previous work with Brazilian Mestizos and with Xavante and Terena Indians indicate the possibility that some HLA alleles confer increased risk for endemic pemphigus disease. Many studies have been performed in order to try to correlate the HLA system with the pemphigus disease
A short tandem repeat in DNA occurs when a pattern of two or more nucleotides are repeated and the repeated sequences are directly adjacent to each other. The pattern can range in length from 2 to 10 base pairs (bp) (for example (CATG) n in a genomic region) and is typically in the non-coding intron region. By identifying repeats of specific sequences, it is possible to create a genetic profile of an individual. There are currently over 10,000 published short tandem repeat sequences in the human genome, because short tandem repeat analysis has become the prevalent analysis method for determining genetic profiles. In our study of El Bagre-EPF patients, the application of the Hardy-Weinberg-Castle law to short tandem repeats from HLA II favors the locus D6S291 for disease susceptibility, and suggests a protective role for loci D6S1019 and D6S439 in our controls (matched by sex, age, living and working conditions) from the endemic area. Based on the medical literature regarding HLA loci, it is likely that in El Bagre-EPF, the HLA region exerts an epistatic effect over multiple autoimmune control genes.
Conclusions
Our results suggest that other linked genes and/or environmental factors should play a facilitating role in the outbreak of endemic pemphigus foliaceus. Thus, our final question is what social and environmental features might fogo selvagem patients and El Bagre-EPF patients have in common? First, we suggest an increase in population intermarriage (due to religious beliefs, and/or geographic isolation) compared to other human populations. Secondly, it is likely that in both El Bagre-EPF and in fogo selvagem an association may exist with ambient pesticides, selected ambient elements, electrically charged molecules and/or other tropical diseases,, raising the host's susceptibility to somatic mutations.
References
- 1.Brown M. Fogo selvagem (pemphigus foliaceus) A.M.A Arch Derm Syphilo. 1954;69:589. doi: 10.1001/archderm.1954.01540170059008. [DOI] [PubMed] [Google Scholar]
- 2.Castro RM, Proenca NG. Semelhancas e diferencas entre o fogo selvagem e o penfigo foliaceo de Cazanave. Similarities and differences between South American pemphigus foliaceus and Cazanaves pemphigus foliaceus. A Bras Dermatol. 1983;53:137–139. [Google Scholar]
- 3.Diaz LA, Sampaio SAP, Rivitti EA, Martins CR, Cunha PR, Lombardi C. Endemic pemphigus foliaceus (fogo selvagem) Clinical features and immunopathology J Am Acad Dermatol. 1989;20:657–669. doi: 10.1016/s0190-9622(89)70079-7. [DOI] [PubMed] [Google Scholar]
- 4.Morini JP, Jomaa B, Gorgi Y, Saguem MH, Nouira R, Roujeau JC, Revuz J. An Endemic pemphigus foliaceus focus in the Sousse area of Tunisia Arch Dermatol. 1993;129:69–73. doi: 10.1001/archderm.129.1.69. [DOI] [PubMed] [Google Scholar]
- 5.Pavoni DP, Roxo VM, Marquart Filho A, Petzl-Erler ML. Dissecting the associations of endemic pemphigus foliaceus (Fogo Selvagem) with HLA-DRB1 alleles and genotypes. Genes Immun. 2003;4:110–116. doi: 10.1038/sj.gene.6363939. [DOI] [PubMed] [Google Scholar]
- 6.Cerna M, Fernandez-Viña M, Friedman H, Moraes JR, Moraes ME, Diaz L, et al. Genetic markers for susceptibility to endemic Brazilian pemphigus foliaceus (Fogo Selvagem) in Xavante Indians. Tissue Antigens. 1993;42:138–140. doi: 10.1111/j.1399-0039.1993.tb02180.x. [DOI] [PubMed] [Google Scholar]
- 7.Marsh SG, Bodmer JG. HLA class II nucleotide sequences. Tissue Antigens. 1993;40:229–243. doi: 10.1111/j.1399-0039.1992.tb02050.x. [DOI] [PubMed] [Google Scholar]
- 8.Moraes JR, Moraes ME, Fernandez-Viña M. HLA antigens and risk for development of pemphigus foliaceus (fogo selvagem) in endemic areas of Brazil. Immunogenetics. 1991;33:388–391. doi: 10.1007/BF00216698. [DOI] [PubMed] [Google Scholar]
- 9.Petzl-Erber ML, Santamaria J. Are HLA class II genes controlling susceptibility and resistance to Brazilian pemphigus foliaceus (fogo selvagem) Clin Exp Dermatol. 1989;14:51–55. doi: 10.1111/j.1399-0039.1989.tb01684.x. [DOI] [PubMed] [Google Scholar]
- 10.Pavoni DP, Cerqueira LB, Roxo VM, Petzl-Erler ML. Polymorphism of the promoter region and exon 1 of the CTLA4 gene in endemic pemphigus foliaceus (fogo selvagem) Braz J Med Biol Res. 2006;39:1227–1232. doi: 10.1590/s0100-879x2006000900010. [DOI] [PubMed] [Google Scholar]
- 11.Köhler KF, Petzl-Erler ML. No evidence for association of the TP53 12139 and the BAX-248 polymorphisms with endemic pemphigus foliaceus (fogo selvagem) Int J Immunogenet. 2006;33:141–144. doi: 10.1111/j.1744-313X.2006.00585.x. [DOI] [PubMed] [Google Scholar]
- 12.Patrus O. Antigenos de histocompatibilidad immunocomplexos e complemento no penfigo foliaceo. Thesis. Minas Gerais, Brazil, Facultade de Medicina Universidade Federal de Minas Gerais. 1980 [Google Scholar]
- 13.Moraes ME, Fernandez-Vina M, Lazaro A, Diaz LA, Filho GH, Friedman H, et al. An epitope in the third hypervariable region of the DRB1 gene is involved in the susceptibility to endemic pemphigus foliaceus (fogo selvagem) in three different Brazilian populations. Tissue Antigens. 1997;49:35–40. doi: 10.1111/j.1399-0039.1997.tb02707.x. [DOI] [PubMed] [Google Scholar]
- 14.Mouquet H, Farci S, Joly P, Maillere B, Leblond J, Drouot L. A truncated alternative spliced isoform of human desmoglein 1 contains a specific T cell epitope binding to the pemphigus foliaceus-associated HLA class II DRbeta1*0102 molecule. J Immunol. 2006;177:6517–6526. doi: 10.4049/jimmunol.177.9.6517. [DOI] [PubMed] [Google Scholar]
- 15.Petzl-Erler ML, Malheiros D. Pemphigus foliaceus and desmoglein 1 gene polymorphism: is there any relationship? J Autoimmun. 2005;25:121–125. doi: 10.1016/j.jaut.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Abréu-Vélez AM, Beutner E, Montoya F, Bollag WB, Hashimoto T. Analyses of autoantigens in a new form of endemic pemphigus foliaceus in Colombia. J Am Acad Dermatol. 2003;49:609–614. doi: 10.1067/s0190-9622(03)00852-1. [DOI] [PubMed] [Google Scholar]
- 17.Abréu-Vélez AM, Hashimoto T, Tobón S, Londoño ML, Montoya F, Bollag A unique form of endemic pemphigus in Northern Colombia. J Am Acad Dermatol. 2003;4:599–680. doi: 10.1067/s0190-9622(03)00851-x. [DOI] [PubMed] [Google Scholar]
- 18.Builes JJ, Bravo ML, Moreno MA, Gaviria AM, Espinal C. Genetic data analysis of HLA-DQA1 in four Colombian populations: Pereira, Armenia, Manizales, and Chocó. J Forensic Sci. 2003;48:447–448. [PubMed] [Google Scholar]
- 19.Abel L, Garcia A, Demenais F. Complex segregation analysis of familial diseases with variable age of onset: comparison of different methods by a simulation study. Genet Epidemiology. 1995;12:231–249. doi: 10.1002/gepi.1370120302. [DOI] [PubMed] [Google Scholar]
- 20.Briollais L, Demenais F. Regressive threshold model for familial analysis of complex diseases with variable age of onset. Genet Epidemiol. 2002;23:375–397. doi: 10.1002/gepi.10202. [DOI] [PubMed] [Google Scholar]
- 21.Morton NE. Genetic tests under incomplete ascertainment. Am J Hum Genet. 1959;11:1–16. [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider S, Kueffer JM, Roessli D, Excoffier L. Arlequin: A Software for Population Genetic Data Analysis, Version 1.1. Univ Geneva: Genetics and Biometry Lab, Dept. of Anthropology; 1997. [Google Scholar]
- 23.Elandt-Johnson RC. Complex segregation analysis. II. Multiple classifications. Am J Hum Genet. 1971;23:17–32. [PMC free article] [PubMed] [Google Scholar]
- 24.Morton NE, Yee S, Lew R. Complex segregation analysis. Am J Hum Genet. 1971;23:602–611. [PMC free article] [PubMed] [Google Scholar]
- 25.Lalouel JM, Rao DC, Morton NE, Elston RC. A unified model for complex segregation analysis. Am J Hum Genet. 1983;35:816–826. [PMC free article] [PubMed] [Google Scholar]
- 26.Iselius L, Morton NE. Transmission probabilities are not correctly implemented in the computer program POINTER. Am J Hum Genet. 1991;49:459. [PMC free article] [PubMed] [Google Scholar]
- 27.Yule GU. Mendel's laws and their probable relation to intra-racial heredity. New Phytol. 1902;207:222–238. [Google Scholar]
- 28.Castle WE. The laws of Galton and Mendel and some laws governing race improvement by selection. Proc. Amer. Acad. Arts Sci. 1903;35:233–242. [Google Scholar]
- 29.Pearson K. Mathematical contributions to the theory of evolution. XI. On the influence of natural selection on the variability and correlation of organs. Philosophical Transactions of the Royal Society of London, Ser. 1903A;200:1–66. [Google Scholar]
- 30.Emigh TH. A comparison of tests for Hardy–Weinberg equilibrium. Biometrics. 1980;36:627–642. [PubMed] [Google Scholar]
- 31.Hardy GH. Mendelian proportions in a mixed population 1908. Yale J Biol Med. 2003;76(2):79–80. [PMC free article] [PubMed] [Google Scholar]
- 32.Griffiths AJF, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM. “Chapter 5”. An Introduction to Genetic Analysis. 5th ed. New York: W.H. Freeman and Company; 1993. ISBN 0-7167-2285-2. [Google Scholar]
- 33.Poehlman JM, Sleper DA. “Chapter 3”. Breeding Field Crops. 4th ed. Iowa: Iowa State Press; 1995. [Google Scholar]