

Abstract
Peroxisome proliferator-activated receptors (PPARs) and retinoic acid receptors (RARs), members of the nuclear receptor superfamily, are transcription factors that regulate a variety of important cellular functions. PPARs form heterodimers retinoid X receptor (RXR), an obligate heterodimeric partner for other nuclear receptors. Several novel links between retinoid metabolism and PPAR responses have been identified, and activation of PPAR/RXR expression has been shown to increase response to retinoids. PPARγ has emerged as a key regulator of cell growth and survival, whose activity is modulated by a number of synthetic and natural ligands. While clinical trials in cancer patients with thiazolidinediones (TZD) have been disappointing, novel structurally different PPARγ ligands, including triterpenoids, have entered clinical arena as therapeutic agents for epithelial and hematopoietic malignancies. Here we shall review the antitumor advances of PPARγ, alone and in combination with RARα ligands in control of cell proliferation, differentiation, and apoptosis and their potential therapeutic applications in hematological malignancies.
1. Introduction
Acute myelogenous leukemia (AML) remains incurable in most patients because of the likelihood of relapse and the development of resistant disease [1]. Many novel agents do not improve survival of patients once relapse occurs, which enforces the need for more effective treatment strategies for AML exploiting apoptosis and/or differentiation induction.
Ligands of nuclear hormone receptors (NHRs) have been shown to induce apoptosis and/or inhibiting proliferation in a variety of preclinical models. The most striking improvement in AML therapy was achieved by the treatment of acute promyelocytic leukemia (APL) using the retinoic acid (RA) receptor- (RAR-) specific ligand, all-trans RA (ATRA) [2, 3]. ATRA, combined with chemotherapy, results in complete remission (CR) rates ranging from 72% to 90% in APL patients with the oncogenic transcriptional repressor PML-RARα [4–8]. However, approximately 10% to 30% of patients relapse [8] and frequently develop resistance to ATRA [9, 10]. Acquisition of specific mutations in the ligand binding site, which leads to altered interactions with transcriptional coregulators, is a well-documented mechanism of acquired ATRA resistance [11, 12]. In addition, several alternative mechanisms such as DNA methylation [13] or impaired telomerase regulation [14] have been proposed to cause ATRA-resistant disease.
Considering the potential of using PPARγ ligands in APL “transcriptional” therapy, this paper summarizes the effects of endogenous and synthetic PPARγ ligands in AML and focuses on elucidating the mechanisms underlying the anti-tumor effects of novel synthetic PPARγ ligand 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) in APL.
2. PPARγ and PPARγ Ligands
PPARs belong to the NHR superfamily of ligand-dependent transcription factors, which includes RAR and RXR among others. Three PPAR isotypes have been identified: PPARγ, PPARα, and PPARβ/δ. PPARγ exists as a heterodimer with RXR, and upon activation by endogenous or synthetic ligands, PPARγ/RXR binds to the specific response elements PPRE in the promoter regions of target genes, respectively, which in turn functions as a transcription factor [15–17].
PPARγ modulates gene networks involved in controlling growth, cellular differentiation, and apoptosis [18]. PPARγ receptor can be activated by endogenous ligands (e.g., prostaglandin D2 (PGD2), 15-deoxy prostaglandin J2 (15dPGJ2), or 15-hydroxyeicosatetraenoic acid (15-HETE)) [19, 20], and synthetic ligands that include insulin sensitizing antidiabetic thiazolidinediones (TZD); troglitazone (TGZ), rosiglitazone (RGZ), ciglitazone (CGZ), or pioglitazone (PGZ) [21–23]; nonsteroidal anti-inflammatory compounds indomethacin, ibuprofen, flufenamic acid, or fenoprofen [24]; triterpenoids 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) [25] are a semisynthetic triterpenoid derived from oleanolic acid, whose structure contains two α, β-unsaturated carbonyl moieties. CDDO was shown to release nuclear receptor corepressor (NCoR) and recruit CCAAT/enhancer-binding protein (CBP/p300) to PPARγ [25] (Figure 1).
Figure 1.
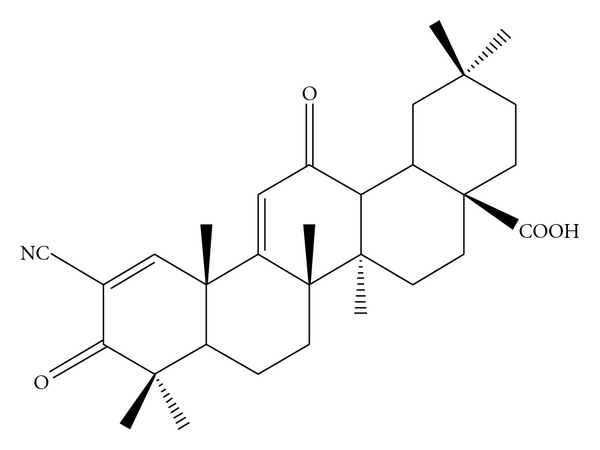
Molecular structure of CDDO 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO).
PPARγ ligands induce differentiation and inhibit proliferation in several tumor models [26–34]. The regulation of gene transcription by ligand-bound PPARγ involves cofactor proteins, which bridge transcription factors to the basal transcriptional machinery or modify chromatin structure. These include release of small accessory molecules known as corepressors (e.g., NCoR or silencing mediator for retinoid receptor and thyroid hormone receptors (SMRT)) and recruitment of coactivators (e.g., CBP/p300, cyclic adenosine monophosphate response-element binding protein (CREB), steroid receptor coactivator-1 (SRC-1), receptor interacting protein 140(RIP140), or PPARγ interacting protein (PRIP/RAP250) [35–40]. The multiprotein complex induces transcription by chromatin remodeling and interaction with the basal transcriptional machinery [41, 42], and the relative levels of cofactor expression (e.g., availability of cofactors CBP/p300 versus SRC-1) also control the specificity of the physiological response to target gene transcription [43].
3. Antitumor Effects of PPARγ in AML
High PPARγ expression was observed in normal bone marrow and peripheral blood CD34+ progenitor cells [44]. Furthermore, significantly higher PPARγ mRNA expression was observed in primary AML cases compared to normal peripheral blood or bone marrow mononuclear cells [45, 46].
The mechanisms of cell differentiation and cell cycle arrest by activated PPARγ depend heavily on the specificity of PPARγ ligands. The induction of differentiation by activation of PPARγ may represent a promising novel therapeutic approach for cancer as already demonstrated for liposarcoma [27] and in xenograft models of prostate [47] and colon cancer [30]. Differentiation therapy may well play a role in acute myeloid leukemias, analogous to ATRA-induced differentiation in APL. PPARγ is known to be induced and/or expressed in cells of the myeloid/monocytic lineage [48, 49].
In PPARγ expressing AML cell lines, PPARγ ligand TGZ suppressed their clonal growth with G1 cell cycle phase arrest, induced differentiation into monocytes, and increased apoptosis at higher concentrations [50, 51]. Troglitazone-induced G0/G1 cell cycle arrest with upregulation of p21 mRNA in myeloid leukemia cell lines [52]. In concert with these findings, PPARγ ligand PGZ and 15dPGJ2 suppressed proliferation, and the combined treatment with ATRA synergistically induced myeloid differentiation in promyelocytic leukemia NB4 cells [53]. Furthermore, simultaneous treatment with TGZ and RXR or RAR ligands resulted in additive suppression of growth indicating that PPARγ ligand combined with a retinoid is a potent inhibitor of clonogenic growth of AML [50]. CDDO has been reported to induce monocytic differentiation of human myeloid leukemia cells and adipogenic differentiation of mouse fibroblasts [54].
CDDO-Me also induced granulo-monocytic differentiation in primary AML cells and cell lines. Combinations with ATRA or the RXR-specific ligand LG100268 enhanced the effects of CDDO-Me on cell viability and/or terminal differentiation of myeloid leukemic cell lines [54]. CDDO-Me-induced enhanced apoptosis when combined with ara-C and retinoids indicating potential activity in the future therapy for AML [55].
With respect to the mechanisms of PPARγ-ligand-induced differentiation, CCAAT enhancer-binding protein alpha (CEBPA) translational upregulation has been reported to be required for CDDO-induced granulocytic differentiation of AML patients samples and cell lines [56]. CDDO increases the ratio of transcriptionally active p42 and the inactive p30 CEBPA isoform, which in turn leads to transcriptional activation of CEBPA-regulated genes and associates with dephosphorylation of eIF2alpha and phosphorylation of eIF4E [56].
PPARγ ligands are additionally known to induce apoptosis. The mechanisms of apoptosis induction by activated PPARγ depend heavily on the specificity of PPARγ ligands. PPARγ activation by natural ligand 15dPGJ2 and synthetic ligand TGZ induce apoptosis accompanied by caspase-3 activation and downregulated c-myc gene expression in myeloid leukemic cells [57]. 15dPGJ2 and TGZ have been also reported to induce upregulation of bax and downregulation of antiapoptotic proteins survivin and bcl-2 in AML and CML [58]. Furthermore, downregulation of cyclooxygenase-2 expression, disruption of mitochondrial membrane potential, activation of caspase-3, downregulation of Bcl-2, Bcl-Xl, and Mcl-1, and upregulation of Bax by these PPARγ agonists 15dPGJ2 and TGZ has been reported in human monocytic leukemia cells [59]. Semisynthetic oleanane triterpenoid CDDO has potent differentiating, antiproliferative, anti-inflammatory, and apoptosis-inducing properties [54]. CDDO has been reported to activate caspase-8 and -3 and to induce mitochondrial cytochrome c release in leukemic cells and in osteosarcoma cells [60–62]. CDDO has been further shown to activate the intrinsic pathway of apoptosis that involves the release of cytochrome c and AIF and initiates caspase-dependent and independent cell death in AML [63]. The C-28 methyl ester of CDDO, CDDO-Me [55], and C-28 imidazolide imide of CDDO (CDDO-Im) [64] has been shown to be more potent than CDDO in inducing apoptosis and differentiation of acute myeloid leukemia (AML) cells. CDDO-Me is 3- to 5-fold more active than CDDO in inhibiting the viability of AML cells in an MDR-1- and p53-independent manner, inducing apoptosis through a loss of mitochondrial membrane potential, and increasing caspase-3 cleavage and proapoptotic Bax protein. It has significantly less cytotoxicity against normal CD34+ progenitor cells, assuring therapeutic window [55].
In addition, CDDO was shown to inhibit NF-κB-mediated gene expression in leukemic cells [62]. CDDO/tumor-necrosis-factor- (TNF-) induced apoptosis occurs through selective inhibition of NF-κB-dependent antiapoptotic proteins, bypassing potential mitochondrial resistance mechanisms [62]. CDDO-Me also inhibits both constitutive and inducible NF-κB through inhibition of IκB α kinase, leading to the suppression of expression of NF-κB-regulated gene products and enhancement of apoptosis induced by TNFα [65].
Notably, certain PPARγ ligands execute anti-tumor activities without requiring interaction with the PPAR ligand binding domain [66]. For example, CDDO, CDDO-Me, and CDDO-Im activate PPARγ-dependent and -independent pathways that inhibit cancer-cell growth [67]. They activate PPARγ in transactivation assays, and CDDO-induced apoptosis was diminished by dominant-negative PPARγ in myeloid HL-60 cells and by T007 in myeloid U937 cells [68], but CDDO-Im-induced differentiation in leukemia cells was not inhibited by the PPARγ antagonist GW9662 [61], and T007 did not affect inhibition of SKOV3 ovarian cancer cell growth by CDDO [69]. In these scenarios, interaction with the PPARγ receptor is irrelevant to the anti-cancer effects, which may depend on cell type, presence/activity of the receptor(s), and cellular abundance of coactivators/corepressors. PPAR-independent effects of PPARγ ligands are due in part to their electrophilic nature, proteasomal degradation of cell cycle-, and apoptosis-regulatory proteins, transcriptional repression, and other mechanisms [70–72]. Both, PPARγ-dependent and -independent pathways that contribute to inhibition of cancer cell growth may be beneficial for cancer chemotherapy [67].
4. Antitumor Effects of PPARγ-Active Triterpenoid CDDO on APL
RARs bind with high affinity to the RA-responsive element (RARE) as a heterodimer with RXR, which also heterodimerizes with other nuclear receptors, such as PPARγ.
In APL cells, the oncogenic transcription factor PML-RARα, a dominant negative transcriptional repressor, targets consist of two copies of an AGGTCA, a highly conserved consensus for RARα. PML-induced dimerization allows the two RARα moieties of PML-RARα to bind very distant monomeric DNA sites. The spectrum of response elements for PML-RARα and PML-RARα-RXR (DR1-DR16 response elements) is much broader than one for the wild-type RAR-RXR (DR1, DR2, and DR5), and PML-RARα-RXR oligomers silence a wide range of nuclear receptor target genes [73].
X-RARα fusion proteins in APL have been demonstrated to negatively affect transactivation of PPARγ [74], indicating that inhibition of PPARγ activity may contribute to the pathophysiology of the differentiation block in APL, and that PPARγ ligands could sensitize APL cells to the differentiating effects of ATRA including ATRA-resistant cells [45].
PML-RARα recruits the nuclear corepressors and histone deacetylase (HDAC), which leads to histone condensation and transcriptional repression [75–77]. ATRA acts by causing the PML-RARα/HDAC complex to dissociate, thereby converting PML-RARα into a transcriptional activator [76]. Reactivation of ATRA target genes by inducing an appropriate level of histone acetylation in their promoters is a potential strategy for restoring anticancer effects of ATRA in refractory APL [77]. Differentiating agents including ATRA, arsenic, cAMP, HDAC inhibitors, and rexinoids relieve this repression through various molecular mechanisms, allowing spontaneous differentiation of leukemic blasts [73].
In fact, it has been demonstrated that HDAC inhibitors (HDACI) such as trichostatin A (TSA), sodium phenylbutyrate (PB), and suberoylanilide hydroxamic acid (SAHA) can augment the cell growth inhibition induced by ATRA, and that ATRA combined with SAHA increased survival and induced remissions in APL transgenic mice harboring the PLZF-RARα translocation [78]. In addition, the PML-RARα fusion protein was observed to induce hypermethylation on RAR promoter, and the DNA methyltransferase inhibitor 5-asa-2′-deoxycytidine (5-Aza-dC) enhanced ATRA-induced RAR promoter transactivation in APL cells [13].
Induction of APL cell differentiation by ATRA is associated with modulation of several critical genes, including RARβ2 [78], C/EBPβ [79], p21 [80], PU.1 [81], or a dominant repressor of RAR signaling PRAME [82]. Notably, PML-RARα has a significant affinity for DR1 [83], a binding site for RXR/PPARγ heterodimers, and negatively contributes to transactivation by ligand-activated PPRE.
The RA-target gene RARβ plays a crucial role in mediating the growth-inhibitory and tumor suppressive effects of retinoids in various cancer cells [84–87], and RARβ is silenced in many tumors [84, 87, 88] and myeloid leukemias [89, 90] including APL [13]. Its upregulation has been proposed as a general mechanism of retinoid-induced growth inhibition and differentiation induction [72]. RARβ2 induction has been implicated in several tumor cell models in which retinoids inhibit growth and induce differentiation [91]. In HeLa cells, the transfected RARβ2 transgene inhibits proliferation, while exogenous RA further increases the ability of the transgene to inhibit proliferation [92]. Disruption of RARβ2 expression in RARβ2 positive cancer cells abolishes RA effects of growth arrest [72], and the presence of RARβ2 antisense predisposed the murine lung tissue to tumor formation [91].
Semisynthetic PPARγ ligand triterpenoid CDDO augmented the ATRA-induced reactivation of RARβ2 in APL via histone acetylation [93]. In combination with ATRA, CDDO may activate the transcription of PPARγ target genes, which in turn increase the affinity of RARβ for βRARE. CDDO caused a prominent increase in RARβ2 binding to the response element in the gel shift assay, and ATRA/CDDO combination increased H3-Lys9 acetylation in RARβP2 and RARβ2 transcription [93]. These findings support the concept that ligation of the PPARγ and RAR nuclear receptors is capable of inducing cell maturation and enhances proapoptotic effects of ATRA in APL cells. PPARγ and RXR form a complex with βRARE in the RARβ promoter, and the combination of ligands of PPARγ and RXR was reported to induce RARβ in ATRA-resistant breast cancer cells in the presence of histone deacetylase inhibitor [94]. Based on these findings, CDDO may induce recruitment of PPARγ/RXR to the RARE, which promotes affinity of RARβ for βRARE.
Ligand-bound RAR/RXR heterodimer has been shown to recruit the histone acetylase PCAF and the coactivator CBP/p300, which accumulates the HAT activity on the heterodimer/DNA complex and finally leads to enhanced retinoid-responsive transcription [95]. Likewise, the regulation of gene transcription by ligand-bound PPARγ involves the recruitment of coactivator proteins, including CBP/p300 and SRC-1 [17, 25, 39, 40]. CDDO has been shown to induce transactivation and PPARγ interaction with multiple coactivators including SRC-1, SRC-2, SRC-3, TRAP 220, CARM-1, and PGC-1 in colon cancer cells [67]. While CDDO alone did not recruit CBP to the RARβ2 promoter, the CDDO/ATRA combination increased ATRA-induced CBP recruitment. Altogether, the ability of ATRA/CDDO to restore RAR signaling and to cause cell maturation might be in part dependent on the PPARγ-mediated induction of histone acetylation and reactivation of ATRA target genes (Figure 2).
Figure 2.
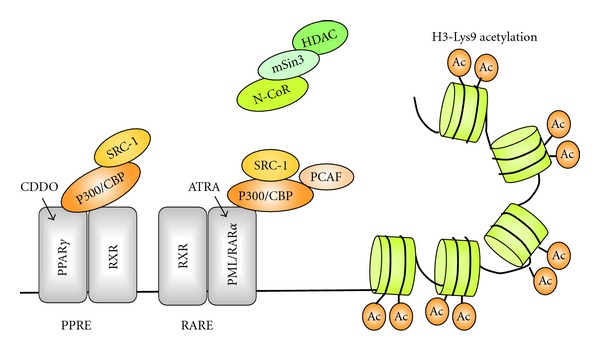
CDDO augments ATRA-induced reactivation of RARβ2 in APL via histone acetylation. Combination of all-trans RA (ATRA) and 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) increases H3-Lys9 acetylation in RARβP2 and RARβ2 transcription. CDDO-bound PPARγ may recruit coactivator proteins, including CBP-p300 and SRC-1 to PPARγ/RXR, which in turn induce histone acetylation and reactivation of ATRA target genes. Ac: acetylated histone H3-Lys9, HDAC: histone deacetylase, mSin3: mammalian homolog of the S. cerevisiae corepressor, Sin 3, NCoR: nuclear receptor corepressor, SRC-1: steroid receptor coactivator-1, CBP/p300: CCAAT/enhancer-binding protein, PCAF: P300/CBP-associated factor.
ATRA is a nonselective retinoid capable of transactivating both, RARα and RXR receptors [96, 97]. Although PPARγ/RXR heterodimers promote transcriptional activity of PPARγ [16], RXR-selective ligand LG100268 and CDDO combination was not sufficient for RARβ2 induction, suggesting that RARβ2 gene induction is not due to ligand-induced RXR activation in APL cells [93].
Whereas CDDO alone failed to induce maturation of APL cells, the combination of CDDO with ATRA induced ATRA sensitive- and resistant-APL cells to differentiate into mature granulocytes with striking increase in Nitro Blue Tetrazolium (NBT) reduction positive and CD11b-positive cells above effects elicited by single agent ATRA [93]. Furthermore, the combined use of CDDO derivative CDDO-Me and ATRA in the murine model of APL resulted in the significant increase of mature granulocytic cells in peripheral blood and prolongation of survival compared to the single compound treatment of ATRA or CDDO. Ikeda et al. [64] also demonstrated that CDDO-Im selectively downregulated expression of PML-RARα fusion protein with an activation of caspase 8, which might contribute to enhanced ATRA-induced differentiation in APL cells, and arsenic-trioxide- (ATO-) induced apoptosis in both ATRA-sensitive NB4 and resistant R2 cell lines and primary APL cells.
RA signaling is a common mechanism in AML other than APL, and HDAC inhibitors have been shown to restore RA-dependent transcriptional activation and trigger terminal differentiation of primary blasts from AML patients [89]. Recent reports of in vivo differentiation of the leukemic clone following HDAC inhibitor valproic acid/ATRA treatment in AML patients [98] further suggest the possibility that the ATRA/CDDO or its more potent derivatives combination may be useful transcriptional/differentiation therapy in non-APL AML. Randomized trial AML HD98B showed that administration of ATRA in addition to intensive chemotherapy improved the outcomes of the patients with genotype of “mutant (mt-) NPM1 without FLT3-ITD” [99]. NPM1 has been reported to be a possible transcriptional corepressor [100]. Inhibition of NPM1 oligomerization or knockdown of NPM1-induced apoptosis and sensitized to ATRA in mt-NPM1-bearing AML cells [101]. These findings suggest new avenues of exploration for ATRA and CDDO derivatives combination therapy targeting “mt-NPM1 wt-FLT3” genotype AML.
Acknowledgment
The authors thank an anonymous for the paper review. There is no conflict of interests between the paper's authors and the companies involved in the paper.
References
- 1.Estey EH. Treatment of relapsed and refractory acute myelogenous leukemia. Leukemia. 2000;14(3):476–479. doi: 10.1038/sj.leu.2401568. [DOI] [PubMed] [Google Scholar]
- 2.Degos L, Chomienne C, Daniel MT, et al. Treatment of first relapse in acute promyelocytic leukaemia with all-trans retinoic acid. The Lancet. 1990;336(8728):1440–1441. doi: 10.1016/0140-6736(90)93135-c. [DOI] [PubMed] [Google Scholar]
- 3.Chen ZX, Xue YQ, Zhang R, et al. A clinical and experimental study on all-trans retinoic acid-treated acute promyelocytic leukemia patients. Blood. 1991;78(6):1413–1419. [PubMed] [Google Scholar]
- 4.Meng-er H, Yu-chen Y, Shu-rong C, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72(2):567–572. [Google Scholar]
- 5.Warrell RP, Frankel SR, Miller WH, et al. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid) New England Journal of Medicine. 1991;324(20):1385–1393. doi: 10.1056/NEJM199105163242002. [DOI] [PubMed] [Google Scholar]
- 6.Mandelli F, Diverio D, Avvisati G, et al. Molecular remission in PML/RARα-positive acute promyelocytic leukemia by combined all-trans retinoic acid and Idarubicin (AIDA) therapy. Blood. 1997;90(3):1014–1021. [PubMed] [Google Scholar]
- 7.Castaigne S, Chomienne C, Daniel MT, et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76(9):1704–1709. [PubMed] [Google Scholar]
- 8.Tallman MS. Therapy of acute promyelocytic leukemia: all-trans retinoic acid and beyond. Leukemia. 1998;12(1):S37–S40. [PubMed] [Google Scholar]
- 9.Warrell RP. Retinoid resistance in acute promyelocytic leukemia: new mechanisms, strategies, and implications. Blood. 1993;82(7):1949–1953. [PubMed] [Google Scholar]
- 10.Cornic M, Chomienne C. Induction of retinoid resistance by all-trans retinoic acid in acute promyelocytic leukemia after remission. Leukemia and Lymphoma. 1995;18(3-4):249–257. doi: 10.3109/10428199509059615. [DOI] [PubMed] [Google Scholar]
- 11.Shao W, Benedetti L, Lamph WW, Nervi C, Miller WH. A retinoid-resistant acute promyelocytic leukemia subclone expresses a dominant negative PML-RARα mutation. Blood. 1997;89(12):4282–4289. [PubMed] [Google Scholar]
- 12.Cote S, Zhou D, Bianchini A, Nervi C, Gallagher RE, Miller WH. Altered ligand binding and transcriptional regulation by mutations in the PML/RARα ligand-binding domain arising in retinoic acid-resistant patients with acute promyelocytic leukemia. Blood. 2000;96(9):3200–3208. [PubMed] [Google Scholar]
- 13.Di Croce L, Raker VA, Corsaro M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295(5557):1079–1082. doi: 10.1126/science.1065173. [DOI] [PubMed] [Google Scholar]
- 14.Pendino F, Sahraoui T, Lanotte M, Ségal-Bendirdijian E. A novel mechanism of retinoic acid resistance in acute promyelocytic leukemia cells through a defective pathway in telomerase regulation. Leukemia. 2002;16(5):826–832. doi: 10.1038/sj.leu.2402470. [DOI] [PubMed] [Google Scholar]
- 15.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor super-family: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulman IG, Shao G, Heyman RA. Transactivation by retinoid X receptor-peroxisome proliferator-activated receptor γ (PPARγ) heterodimers: intermolecular synergy requires only the PPRAγ hormone-dependent activation function. Molecular and Cellular Biology. 1998;18(6):3483–3494. doi: 10.1128/mcb.18.6.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosen ED, Spiegelman BM. PPARγ: a nuclear regulator of metabolism, differentiation, and cell growth. Journal of Biological Chemistry. 2001;276(41):37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 18.Pfahl M, Apfel R, Bendik I, et al. Nuclear retinoid receptors and their mechanism of action. Vitamins and Hormones. 1994;49(C):327–382. doi: 10.1016/s0083-6729(08)61150-4. [DOI] [PubMed] [Google Scholar]
- 19.Huang JT, Welch JS, Ricote M, et al. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400(6742):378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 20.Nagy L, Tontonoz P, Alvarez JGA, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ . Cell. 1998;93(2):229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 21.Berger J, Bailey P, Biswas C, et al. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-γ: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology. 1996;137(10):4189–4195. doi: 10.1210/endo.137.10.8828476. [DOI] [PubMed] [Google Scholar]
- 22.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ) Journal of Biological Chemistry. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 23.Lambe KG, Tugwood JD. A human peroxisome-proliferator-activated receptor-γ is activated by inducers of adipogenesis, including thiazalidinedione drugs. European Journal of Biochemistry. 1996;239(1):1–7. doi: 10.1111/j.1432-1033.1996.0001u.x. [DOI] [PubMed] [Google Scholar]
- 24.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. Journal of Biological Chemistry. 1997;272(6):3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Porter WW, Suh N, et al. A synthetic triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), is a ligand for the peroxisome proliferator-activated receptor γ . Molecular Endocrinology. 2000;14(10):1550–1556. doi: 10.1210/mend.14.10.0545. [DOI] [PubMed] [Google Scholar]
- 26.Tontonoz P, Singer S, Forman BM, et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor γ and the retinoid X receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(1):237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demetri GD, Fletcher CDM, Mueller E, et al. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-γ ligand troglitazone in patients with liposarcoma. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(7):3951–3956. doi: 10.1073/pnas.96.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elstner E, Müller C, Koshizuka K, et al. Ligands for peroxisome proliferator-activated receptory and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(15):8806–8811. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller E, Sarraf P, Tontonoz P, et al. Terminal differentiation of human breast cancer through PPARγ . Molecular Cell. 1998;1(3):465–470. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- 30.Sarraf P, Mueller E, Jones D, et al. Differentiation and reversal of malignant changes in colon cancer through PPARγ . Nature Medicine. 1998;4(9):1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- 31.Sarraf P, Mueller E, Smith WM, et al. Loss-of-function mutations in PPARγ associated with human colon cancer. Molecular Cell. 1999;3(6):799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 32.Mueller E, Smith M, Sarraf P, et al. Effects of ligand activation of peroxisome proliferator-activated receptor γ in human prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(20):10990–10995. doi: 10.1073/pnas.180329197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang TH, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor γ in non-small cell lung cancer. Cancer Research. 2000;60(4):1129–1138. [PubMed] [Google Scholar]
- 34.Marra F, Efsen E, Romanelli RG, et al. Ligands of peroxisome proliferator-activated receptor γ modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119(2):466–478. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- 35.Westin S, Kurokawa R, Nolte RT, et al. Interactions controlling the assembly of nuclear-receptor heterodimers and co-activators. Nature. 1998;395(6698):199–202. doi: 10.1038/26040. [DOI] [PubMed] [Google Scholar]
- 36.Puigserver P, Adelmant G, Wu Z, et al. Activation of PPARγ coactivator-1 through transcription factor docking. Science. 1999;286(5443):1368–1371. doi: 10.1126/science.286.5443.1368. [DOI] [PubMed] [Google Scholar]
- 37.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 38.Nolte RT, Wisely GB, Westin S, et al. Ligand binding and co-activator assembly of the peroxisome proliferator- activated receptor-γ . Nature. 1998;395(6698):137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 39.Yang W, Rachez C, Freedman LP. Discrete roles for peroxisome proliferator-activated receptor γ and retinoid X receptor in recruiting nuclear receptor coactivators. Molecular and Cellular Biology. 2000;20(21):8008–8017. doi: 10.1128/mcb.20.21.8008-8017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kodera Y, Takeyama KI, Murayama A, Suzawa M, Masuhiro Y, Kato S. Ligand type-specific interactions of peroxisome proliferator-activated receptor γ with transcriptional coactivators. Journal of Biological Chemistry. 2000;275(43):33201–33204. doi: 10.1074/jbc.C000517200. [DOI] [PubMed] [Google Scholar]
- 41.Chen J, Kinyamu HK, Archer TK. Changes in attitude, changes in latitude: nuclear receptors remodeling chromatin to regulate transcription. Molecular Endocrinology. 2006;20(1):1–13. doi: 10.1210/me.2005-0192. [DOI] [PubMed] [Google Scholar]
- 42.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes and Development. 2006;20(11):1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 43.Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W. From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Progress in Lipid Research. 2006;45(2):120–159. doi: 10.1016/j.plipres.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Ikezoe T, Miller CW, Kawano S, et al. Mutational analysis of the peroxisome proliferator-activated receptor γ in human malignancies. Cancer Research. 2001;61(13):5307–5310. [PubMed] [Google Scholar]
- 45.Konopleva M, Andreeff M. Role of peroxisome proliferator-activated receptor-γ in hematologic malignancies. Current Opinion in Hematology. 2002;9(4):294–302. doi: 10.1097/00062752-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 46.Tsao T, Kornblau S, Safe S, et al. Role of peroxisome proliferator-activated receptor-γ and its coactivator DRIP205 in cellular responses to CDDO (RTA-401) in acute myelogenous leukemia. Cancer Research. 2010;70(12):4949–4960. doi: 10.1158/0008-5472.CAN-09-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kubota T, Koshizuka K, Williamson EA, et al. Ligand for peroxisome proliferator-activated receptor γ (Troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Research. 1998;58(15):3344–3352. [PubMed] [Google Scholar]
- 48.Tontonoz P, Nagy L, Alvarez JGA, Thomazy VA, Evans RM. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93(2):241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 49.Moore KJ, Rosen ED, Fitzgerald ML, et al. The role of PPAR-γ in macrophage differentiation and cholesterol uptake. Nature Medicine. 2001;7(1):41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 50.Asou H, Verbeek W, Williamson E, et al. Growth inhibition of myeloid leukemia cells by troglitazone, a ligand for peroxisome proliferator activated receptor gamma, and retinoids. International Journal of Oncology. 1999;15(5):1027–1031. doi: 10.3892/ijo.15.5.1027. [DOI] [PubMed] [Google Scholar]
- 51.Fujimura S, Suzumiya J, Nakamura K, Ono J. Effects of troglitazone on the growth and differentiation of hematopoietic cell lines. International Journal of Oncology. 1998;13(6):1263–1267. doi: 10.3892/ijo.13.6.1263. [DOI] [PubMed] [Google Scholar]
- 52.Sugimura A, Kiriyama Y, Nochi H, et al. Troglitazone suppresses cell growth of myeloid leukemia cell lines by induction of p21WAF1/CIP1 cyclin-dependent kinase inhibitor. Biochemical and Biophysical Research Communications. 1999;261(3):833–837. doi: 10.1006/bbrc.1999.1049. [DOI] [PubMed] [Google Scholar]
- 53.Yasugi E, Horiuchi A, Uemura I, et al. Peroxisome proliferator-activated receptor γ ligands stimulate myeloid differentiation and lipogenensis in human leukemia NB4 cells. Development Growth and Differentiation. 2006;48(3):177–188. doi: 10.1111/j.1440-169X.2006.00855.x. [DOI] [PubMed] [Google Scholar]
- 54.Suh N, Wang Y, Honda T, et al. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9- dien-28-oic acid, with potent differentiating, antiproliferative, and anti- inflammatory activity. Cancer Research. 1999;59(2):336–341. [PubMed] [Google Scholar]
- 55.Konopleva M, Tsao T, Ruvolo P, et al. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002;99(1):326–335. doi: 10.1182/blood.v99.1.326. [DOI] [PubMed] [Google Scholar]
- 56.Koschmieder S, D’Alò F, Radomska H, et al. CDDO induces granulocytic differentiation of myeloid leukemic blasts through translational up-regulation of p42 CCAAT enhancer-binding protein alpha. Blood. 2007;110(10):3695–3705. doi: 10.1182/blood-2006-11-058941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamakawa-Karakida N, Sugita K, Inukai T, et al. Ligand activation of peroxisome proliferator-activated receptor γ induces apoptosis of leukemia cells by down-regulating the c-myc gene expression via blockade of the Tcf-4 activity. Cell Death and Differentiation. 2002;9(5):513–526. doi: 10.1038/sj.cdd.4401000. [DOI] [PubMed] [Google Scholar]
- 58.Liu JJ, Huang RW, Lin DJ, et al. Expression of survivin and bax/bcl-2 in peroxisome proliferator activated receptor-γ ligands induces apoptosis on human myeloid leukemia cells in vitro. Annals of Oncology. 2005;16(3):455–459. doi: 10.1093/annonc/mdi077. [DOI] [PubMed] [Google Scholar]
- 59.Liu JJ, Liu PQ, Lin DJ, et al. Downregulation of cyclooxygenase-2 expression and activation of caspase-3 are involved in peroxisome proliferator-activated receptor-γ agonists induced apoptosis in human monocyte leukemia cells in vitro. Annals of Hematology. 2007;86(3):173–183. doi: 10.1007/s00277-006-0205-2. [DOI] [PubMed] [Google Scholar]
- 60.Ito Y, Pandey P, Place A, et al. The novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid induces apoptosis of human myeloid leukemia cells by a caspase-8-dependent mechanism. Cell Growth and Differentiation. 2000;11(5):261–267. [PubMed] [Google Scholar]
- 61.Ito Y, Pandey P, Sporn MB, Datta R, Kharbanda S, Kufe D. The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Molecular Pharmacology. 2001;59(5):1094–1099. doi: 10.1124/mol.59.5.1094. [DOI] [PubMed] [Google Scholar]
- 62.Stadheim TA, Suh N, Ganju N, Sporn MB, Eastman A. The novel triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) potently enhances apoptosis induced by tumor necrosis factor in human leukemia cells. Journal of Biological Chemistry. 2002;277(19):16448–16455. doi: 10.1074/jbc.M108974200. [DOI] [PubMed] [Google Scholar]
- 63.Konopleva M, Tsao T, Estrov Z, et al. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Research. 2004;64(21):7927–7935. doi: 10.1158/0008-5472.CAN-03-2402. [DOI] [PubMed] [Google Scholar]
- 64.Ikeda T, Kimura F, Nakata Y, et al. Triterpenoid CDDO-Im downregulates PML/RARα expression in acute promyelocytic leukemia cells. Cell Death and Differentiation. 2005;12(5):523–531. doi: 10.1038/sj.cdd.4401574. [DOI] [PubMed] [Google Scholar]
- 65.Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IκBα kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor κB-regulated gene products in human leukemic cells. Clinical Cancer Research. 2006;12(6):1828–1838. doi: 10.1158/1078-0432.CCR-05-2044. [DOI] [PubMed] [Google Scholar]
- 66.Simpson-Haidaris PJ, Pollock SJ, Ramon S, et al. Anticancer role of PPARgamma agonists in hematological malignancies found in the vasculature, marrow, and eyes. PPAR Research. 2010;2010, article 814609 doi: 10.1155/2010/814609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chintharlapalli S, Papineni S, Konopleva M, Andreef M, Samudio I, Safe S. 2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid and related compounds inhibit growth of colon cancer cells through peroxisome proliferator-activated receptor γ-dependent and -independent pathways. Molecular Pharmacology. 2005;68(1):119–128. doi: 10.1124/mol.105.011437. [DOI] [PubMed] [Google Scholar]
- 68.Konopleva M, Elstner E, McQueen TJ, et al. Peroxisome proliferator-activated receptor and retinoid X receptor ligands are potent inducers of differentiation and apoptosis in leukemias. Molecular Cancer Therapeutics. 2004;3(10):1249–1262. [PubMed] [Google Scholar]
- 69.Melichar B, Konopleva M, Hu W, Melicharova K, Andreeff M, Freedman RS. Growth-inhibitory effect of a novel synthetic triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, on ovarian carcinoma cell lines not dependent on peroxisome proliferator-activated receptor-γ expression. Gynecologic Oncology. 2004;93(1):149–154. doi: 10.1016/j.ygyno.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 70.Ray DM, Morse KM, Hilchey SP, et al. The novel triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) induces apoptosis of human diffuse large B-cell lymphoma cells through a peroxisome proliferator-activated receptor gamma-independent pathway. Experimental Hematology. 2006;34(9):1202–1211. doi: 10.1016/j.exphem.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 71.Ray DM, Akbiyik F, Phipps RP. The peroxisome proliferator-activated receptor γ (PPARγ) ligands 15-deoxy-Δ12,14-prostaglandin J2 and ciglitazone induce human B lymphocyte and B cell lymphoma apoptosis by PPARγ-independent mechanisms. Journal of Immunology. 2006;177(8):5068–5076. doi: 10.4049/jimmunol.177.8.5068. [DOI] [PubMed] [Google Scholar]
- 72.Wei S, Yang J, Lee SL, Kulp SK, Chen CS. PPARγ-independent antitumor effects of thiazolidinediones. Cancer Letters. 2009;276(2):119–124. doi: 10.1016/j.canlet.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kamashev D, Vitoux D, De Thé H. PML-RARA-RXR oligomers mediate retinoid and rexinoid/cAMP cross-talk in acute promyelocytic leukemia cell differentiation. Journal of Experimental Medicine. 2004;199(8):1163–1174. doi: 10.1084/jem.20032226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamadani SA, Zhang T, Dorrell C, et al. X-retinoic acid receptor a fusion genes in acute promyelocytic leukemia interfere with retinoid and peroxisome-proliferator signaling pathways. Blood. 2001;98:p. 88a. [Google Scholar]
- 75.He LZ, Guidez F, Tribioli C, et al. Distinct interactions of PML-RARα and PLZF-RARα with co-repressors determine differential responses to RA in APL. Nature Genetics. 1998;18(2):126–135. doi: 10.1038/ng0298-126. [DOI] [PubMed] [Google Scholar]
- 76.Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391(6669):815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 77.He LZ, Tolentino T, Grayson P, et al. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. Journal of Clinical Investigation. 2001;108(9):1321–1330. doi: 10.1172/JCI11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Faria TN, Mendelsohn C, Chambon P, Gudas LJ. The targeted disruption of both alleles of RARβ2 in F9 cells results in the loss of retinoic acid-associated growth arrest. Journal of Biological Chemistry. 1999;274(38):26783–26788. doi: 10.1074/jbc.274.38.26783. [DOI] [PubMed] [Google Scholar]
- 79.Duprez E, Wagner K, Koch H, Tenen DG. C/EBPβ: a major PML-RARA-responsive gene in retinoic acid-induced differentiation of APL cells. EMBO Journal. 2003;22(21):5806–5816. doi: 10.1093/emboj/cdg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu M, Iavarone A, Freedman LP. Transcriptional activation of the human p21(WAF1/CIP1) gene by retinoic acid receptor. Correlation with retinoid induction of U937 cell differentiation. Journal of Biological Chemistry. 1996;271(49):31723–31728. doi: 10.1074/jbc.271.49.31723. [DOI] [PubMed] [Google Scholar]
- 81.Mueller BU, Pabst T, Fos J, et al. ATRA resolves the differentiation block in t(15;17) acute myeloid leukemia by restoring PU.1 expression. Blood. 2006;107(8):3330–3338. doi: 10.1182/blood-2005-07-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Epping MT, Wang L, Edel MJ, Carlée L, Hernandez M, Bernards R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell. 2005;122(6):835–847. doi: 10.1016/j.cell.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 83.Perez A, Kastner P, Sethi S, Lutz Y, Reibel C, Chambon P. PMLRAR homodimers: distinct DNA binding properties and heteromeric interactions with RXR. EMBO Journal. 1993;12(8):3171–3182. doi: 10.1002/j.1460-2075.1993.tb05986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Y, Dawson MI, Agadir A, et al. Regulation of RARβ expression by RAR- and RXR-selective retinoids in human lung cancer cell lines: effect on growth inhibition and apoptosis induction. International Journal of Cancer. 1998;75:88–95. doi: 10.1002/(sici)1097-0215(19980105)75:1<88::aid-ijc14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 85.Houle B, Rochette-Egly C, Bradley WEC. Tumor-suppressive effect of the retinoic acid receptor β in human epidermoid lung cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(3):985–989. doi: 10.1073/pnas.90.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu Y, Lee MO, Wang HG, et al. Retinoic acid receptor β mediates the growth-inhibitory effect of retinoic acid by promoting apoptosis in human breast cancer cells. Molecular and Cellular Biology. 1996;16(3):1138–1149. doi: 10.1128/mcb.16.3.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li C, Wan YJY. Differentiation and antiproliferation effects of retinoic acid receptor β in hepatoma cells. Cancer Letters. 1998;124(2):205–211. doi: 10.1016/s0304-3835(97)00475-8. [DOI] [PubMed] [Google Scholar]
- 88.Haugen BR, Larson LL, Pugazhenthi U, et al. Retinoic acid and retinoid X receptors are differentially expressed in thyroid cancer and thyroid carcinoma cell lines and predict response to treatment with retinoids. Journal of Clinical Endocrinology and Metabolism. 2004;89(1):272–280. doi: 10.1210/jc.2003-030770. [DOI] [PubMed] [Google Scholar]
- 89.Ferrara FF, Fazi F, Bianchini A, et al. Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Research. 2001;61(1):2–7. [PubMed] [Google Scholar]
- 90.Lehmann S, Paul C, Törmä H. The expression of cellular retinoid binding proteins in non-APL leukemic cells and its association with retinoid sensitivity. Leukemia and Lymphoma. 2002;43(4):851–858. doi: 10.1080/10428190290016999. [DOI] [PubMed] [Google Scholar]
- 91.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB Journal. 1996;10(9):940–954. [PubMed] [Google Scholar]
- 92.Si SP, Lee X, Tsou HC, et al. ARγ2-mediated growth inhibition in HeLa cells. Experimental Cell Research. 1996;223:102–111. doi: 10.1006/excr.1996.0062. [DOI] [PubMed] [Google Scholar]
- 93.Tabe Y, Konopleva M, Kondo Y, et al. PPARγ-active triterpenoid CDDO enhances ATRA-induced differentiation in APL. Cancer Biology and Therapy. 2007;6(12):1967–1977. doi: 10.4161/cbt.6.12.4982. [DOI] [PubMed] [Google Scholar]
- 94.James SY, Lin F, Kolluri SK, Dawson MI, Zhang XK. Regulation of retinoic acid receptor β expression by peroxisome proliferator-activated receptor γ ligands in cancer cells. Cancer Research. 2003;63(13):3531–3538. [PubMed] [Google Scholar]
- 95.Blanco JCG, Minucci S, Lu J, et al. The histone acetylase PCAF is a nuclear receptor coactivator. Genes and Development. 1998;12(11):1638–1651. doi: 10.1101/gad.12.11.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mangelsdorf DJ, Ong ES, Dyck JA, Evans RM. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature. 1990;345(6272):224–229. doi: 10.1038/345224a0. [DOI] [PubMed] [Google Scholar]
- 97.Heyman RA, Mangelsdorf DJ, Dyck JA, et al. 9-Cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell. 1992;68(2):397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- 98.Cimino G, Lo-Coco F, Fenu S, et al. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia. Cancer Research. 2006;66(17):8903–8911. doi: 10.1158/0008-5472.CAN-05-2726. [DOI] [PubMed] [Google Scholar]
- 99.Schlenk RF, Döhner K, Kneba M, et al. German-Austrian AML Study Group (AMLSG). Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia: results from the AMLSG Trial AML HD98B. Haematologica. 2009;94:54–60. doi: 10.3324/haematol.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu H, Tan BCM, Tseng KH, et al. Nucleophosmin acts as a novel AP2α-binding transcriptional corepressor during cell differentiation. EMBO Reports. 2007;8(4):394–400. doi: 10.1038/sj.embor.7400909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Balusu R, Fiskus W, Rao R, et al. Targeting levels or oligomerization of nucleophosmin 1 induces differentiation and loss of survival of human AML cells with mutant NPM1. Blood. 2011;118:3096–3106. doi: 10.1182/blood-2010-09-309674. [DOI] [PMC free article] [PubMed] [Google Scholar]