

Abstract
Flavonoids possess several biological and pharmacological activities. Quercetin (Q), a naturally occurring flavonoid, has been shown to downregulate inflammatory responses and provide cardioprotection. However, the mechanisms behind the anti-inflammatory properties of Q in cardiac cells are poorly understood. In inflammation, nitric oxide (NO) acts as a proinflammatory mediator and is synthesized by inducible nitric oxide synthase (iNOS) in response to pro-inflammatory agents such as lipopolysaccharide (LPS), a causative agent in myocardial depression during sepsis. In the present study, we evaluated the protective effect of Q on rat cardiac dysfunction during sepsis induced by LPS. Pretreatment of H9c2 cardiomyoblasts with Q inhibited LPS-induced iNOS expression and NO production and counteracted oxidative stress caused by the unregulated NO production that leads to the generation of peroxynitrite and other reactive nitrogen species. In addition, Q pretreatment significantly counteracted apoptosis cell death as measured by immunoblotting of the cleaved caspase 3 and caspase 3 activity. Q also inhibited the LPS-induced phosphorylation of the stress-activated protein kinases (JNK/SAPK) and p38 MAP kinase that are involved in the inhibition of cell growth as well as the induction of apoptosis. In conclusion, these results suggest that Q might serve as a valuable protective agent in cardiovascular inflammatory diseases.
1. Introduction
Systemic bacterial infection is associated with multiorgan dysfunctions including heart failure, which is the main cause of morbidity and mortality in septic patients [1, 2]. Lipopolysaccharides (LPSs), a major constituent of bacterial outer membrane, have been demonstrated to play a critical role in the initiation of the pathophysiological cascades [3–5]. Under septic conditions, excessive LPS activates numerous types of cells upon recognition by toll-like receptor 4 (TLR-4) resulting in enhanced production of proinflammatory cytokines that contribute to myocardial dysfunction [6–8]. Recent studies have shown that cardiomyoblasts express TLR-4 through which LPS has direct adverse effect on cardiomyocyte physiology [9, 10]. Activation of TLR-4 by LPS triggers NF-κB signaling and results in decreased cardiomyocyte contractility and substantial expressions of proinflammatory cytokines such as intercellular adhesion molecule-1 (ICAM-1) [11, 12], tumor necrosis factor-α (TNF-α) [13, 14], and inducible nitric oxide synthase (iNOS) [13]. The increased expression of iNOS, especially in noninflammatory cells, may have deleterious effects [15–17]. Potential subcellular mechanisms involved in these harmful consequences include excessive direct reactions of NO with a wide variety of proteins and enzymes including reactions with amino, thiol (SH), diazo, and tyrosyl groups, and with heme and Fe2+ or sulfur centers [18]. Moreover, unregulated NO production, also associated with oxidative stress, can result in the generation of peroxynitrite and other reactive nitrogen species (RNS) that alter protein function via nitration and oxidation reactions [16, 19, 20]. It has been demonstrated that reducing systemic inflammatory response could improve myocardial function [21]. Activation of multiple stress signaling processes such as oxidative stress and mitogen-activated protein kinases (MAPKs) plays pivotal roles in the pathogenesis of septic cardiac dysfunction [22]. While activation of ERK1/2 MAP-kinase has been identified to enhance cell growth and migration, the stress-activated protein kinases JNK/SAPK and p38 MAP-kinase are involved in the inhibition of cell growth as well as the induction of apoptosis [23].
Quercetin (Q) is a flavonoid and more specifically a flavonol that possesses a broad range of pharmacological properties, including anti-inflammatory effects [24], antiproliferative effects [25], and protective effects against oxidative stress [26]. Foods rich in quercetin include apples, black and green tea, onions, red wine, red grapes, citrus fruit, broccoli and other leafy green vegetables, cherries, and a number of berries including raspberries and cranberries [27]. Normally, human quercetin plasma concentrations are in the nanomolar range, but after quercetin intake, they may reach the micromolar range [28, 29]. A recent study demonstrated that low to moderate oral dose of quercetin for two weeks increased plasma quercetin concentrations dose-dependently in healthy individuals [30], confirming its bioavailability. In a previous study, we have demonstrated that Q is uptaken by the cardiomyoblast H9c2 cell line and counteracts cardiac oxidative stress via both its well-known antioxidant activity and through the modulation of two key fundamental protein kinases involved in prosurvival signaling pathways, Akt and ERK1/2 [31]. Moreover, we have shown that Q is able to strongly upregulate different antioxidant and phase 2 enzymes in neonatal rat cardiomyocytes demonstrating its indirect antioxidant activity [32].
In this paper, we evaluated the anti-inflammatory effects of Q in cardiac cells, focusing on the possible mechanisms by which this polyphenol counteracts LPS-induced inflammatory responses.
2. Materials and Methods
2.1. Materials
PhosSTOP was purchased from Roche Diagnostics (Mannheim, Germany). CelLytic M, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), mammalian protease inhibitor mixture, DMEM, fetal bovine serum (FBS), penicillin, streptomycin, L-glutamine, acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (Ac-DEVD-AMC), quercetin, and all other chemicals of the highest analytical grade were purchased from Sigma Chemical (St. Louis, MO).
2.2. Cell Cultures
Rat embryonic heart-derived myogenic cell line H9c2 (European Collection of Cell Cultures Salisbury, UK) was grown in DMEM supplemented with 10% heat-inactivated FBS, penicillin (100 U/mL), streptomycin (100 mg/mL), and L-glutamine (2 mM) in a humidified incubator with 5% CO2 at 37°C. Cells were split 1 to 4 at subconfluence (80%). H9c2 cells were seeded at a density of 5 × 104 cells/cm2 72 hours prior to treatments.
2.3. Measurement of Nitric Oxide Production
Released nitrite, a stable product of NO in aqueous medium, was measured using the Griess Reagent System (Promega, Madison, WI, USA). Briefly, H9c2 cells were treated with 30 μM Q for 2 hours prior to stimulation with 10 μg/mL LPS for 24 hours. At the end of this time period, the culture medium was mixed with an equal volume of sulfanilamide solution (1% in 5% phosphoric acid) and of N-1-naphtylethylenediamine dihydrochloride solution (0.1% in water). The absorbance was measured at 540 nm. Nitrite concentrations were determined from a calibration curve of standard 0.1 M sodium nitrite concentrations 0.5–25 μM against absorbance.
2.4. Detection of Intracellular Reactive Oxygen Species
The formation of ROS was evaluated using a fluorescent probe, DCFH-DA, as described by Wang and Joseph [33]. Briefly, H9c2 cells were pretreated with 30 μM Q for 24 h and then incubated with 5 μM DCFH-DA in PBS for 30 min. After DCFH-DA removal, the cells were stimulated with 10 μg/mL LPS for different periods (0.5–24 hours). Cell fluorescence was measured using 485 nm excitation and 535 nm emission with a microplate spectrofluorometer (VICTOR3 V Multilabel Counter, Perkin-Elmer, Wellesley, MA). Intracellular antioxidant activity was expressed as percentage of control cells.
2.5. Caspase-3 Activity Assay
The activity of caspase-3 was measured by hydrolysis of Ac-DEVD-AMC by caspase-3, resulting in the release of the fluorescent 7-amino-4-methylcoumarin moiety (AMC) [34]. H9c2 cells were treated with 30 μM Q for 2 hours prior to stimulation with 10 μg/mL LPS for different periods (0.5–24 hours). Cells were lysated in lysis buffer (50 mM Tris, 0.1% Triton X-100, 150 mM NaCl, 2 mM EGTA/EDTA, 1 mM sodium pyrophosphate, 10 mg/mL phenylmethylsulfonyl fluoride, 1 mM sodium vanadate, 50 mM sodium fluoride, and 1 mg/mL aprotinin) and then centrifuged 5 min at 5,000 g and the supernatant was added to the assay buffer (100 mM HEPES pH 7.0, 5 mM dithiothreitol, 0.1% CHAPS, 10% sucrose, and 0.15 mM Ac-DEVD-AMC). The specific cleavage of the fluorogenic peptide Ac-DEVDA-MC was monitored following AMC cleavage at 370 nm excitation and 455 nm emission. One unit is defined as the amount of enzyme activity cleaving 1.0 nmol of substrate per minute at 25°C under the conditions described.
2.6. Western Immunoblotting
H9c2 cells were treated with 30 μM Q and after 2 hours were stimulated with 10 μg/mL LPS for different periods (0.5–24 hours). Cells were washed with ice-cold PBS and proteins extracted with CelLytic M cell lysis reagent with mammalian protease inhibitor mixture (1 : 100 dilution) and PhosSTOP. Proteins were boiled at 98°C for 3 min in loading buffer (62.5 mM Tris, pH 6.8 containing 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, and 0.0025% bromophenol blue). Protein extracts were separated by SDS-PAGE (20 μg/lane) and then transferred onto nitrocellulose membrane (Hybond-C; GE Healthcare, Buckinghamshire, UK) at 110 V for 90 min using Tris-glycine buffer. Membranes were then incubated in a blocking buffer containing 5% (w/v) skimmed milk and incubated with anti-NOS2, anti-phospho SAPK/JNK (Thr183/Tyr185), anti-SAPK/JNK, anti-phospho p38 MAPK, anti-p38 MAPK (Cell Signaling Technology, Beverly, MA), and anti-β-actin (Sigma), overnight at 4°C on a three-dimensional rocking table. The blots were then incubated with goat anti-rabbit IgG conjugated to horseradish peroxidase for 60 min at room temperature. The results were visualized by chemiluminescence using ECL advance reagent according to the manufacturer's protocol (GE Healthcare). Semiquantitative analysis of specific immunolabeled bands was performed using a Fluor S image analyzer (Bio-Rad, Hercules, CA, USA).
2.7. Protein Concentration
The protein concentration of the cell lysates was determined by the Bio-Rad Bradford protein assay (Bio-Rad Laboratories).
2.8. Statistics
Each experiment was performed at least three times, and all values are represented as means ± SD. One-way analysis of variance (ANOVA) was used to compare differences among groups followed by Bonferroni's test (Prism 5, GraphPad Software Inc., San Diego, CA, USA). Values of P < 0.05 were considered as statistically significant.
3. Results
3.1. Effect of Q on iNOS Expression and NO Production
The first aim of the study was to identify the effect of LPS treatment on iNOS expression (Figure 1). LPS treatment (10 μg/mL) of H9c2 cells for 0.5 hours did not influence iNOS protein expression. On the contrary, LPS stimulation for 2 and 24 hours was able to significantly induce iNOS. In particular, LPS increased the enzyme protein expression in a time-dependent manner (r = 0.9888, P < 0.05). Treatment with 30 μM Q alone for 2 hours had no effect on iNOS protein expression, while pretreatment with Q, prior to LPS exposure, was able to significantly reduce iNOS protein expression at each LPS stimulation time.
Figure 1.
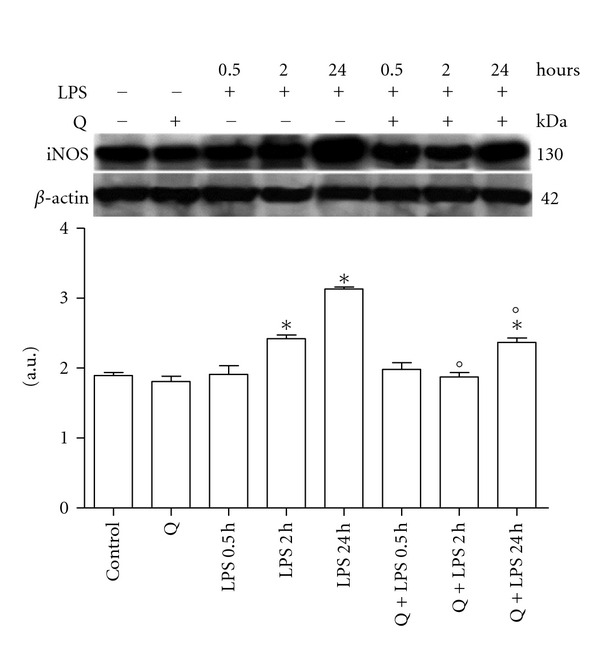
Effect of Q on iNOS expression in LPS-stimulated H9c2 cells. Cells were pretreated with 30 μM Q for 2 hours before stimulation with 10 μg/mL LPS. After the indicated time points, cells were harvested and lysed. Crude homogenates (20 μg) were immunoblotted with an antibody that detects endogenous levels of iNOS (NOS2). Equal loading was verified with an anti-β-actin antibody. Densitometric analysis of the protein bands was performed using Bio-Rad Quantity One 1-D Analysis software. Each column represents the mean ± SD of three independent experiments. Data were analyzed by one-way ANOVA followed by Bonferroni's test. *P < 0.05 compared to Control; °P < 0.05 compared to the corresponding LPS exposure times.
We next evaluated total nitrite release as an indicator of NO production (Figure 2). As at 24 hours LPS stimulation had the strongest effect on iNOS protein expression, we used this time to stimulate H9c2. In agreement with the previous results, LPS significantly increased the release of NO in the culture medium, 30 μM Q alone did not influence NO production, and pretreatment with 30 μM Q before LPS stimulation significantly reduced NO production to level comparable to control cells.
Figure 2.
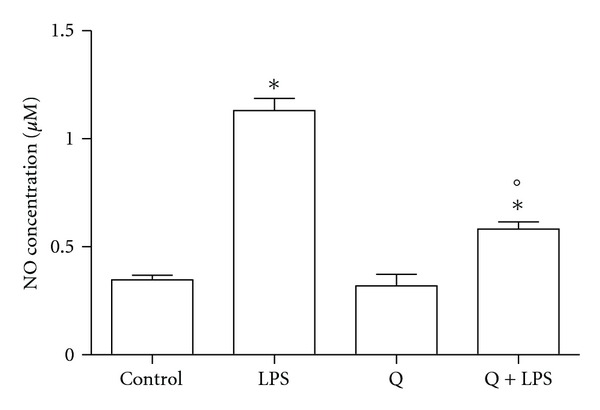
Effect of Q on NO production in LPS-stimulated H9c2 cells. Cells were pretreated with 30 μM Q for 2 hours before stimulation with 10 μg/mL LPS for 24 hours. After the treatment, the culture medium was collected for NO assay. The concentration of NO was determined by the Griess reagent using NaNO2 as standard. Each column represents the mean ± SD of three independent experiments. Data were analyzed by one-way ANOVA followed by Bonferroni's test. *P < 0.05 compared to Control; °P < 0.05 compared to LPS.
3.2. Effect of Q on Intracellular ROS Production
To verify if the increase in RNS was accompanied by a concomitant increase in ROS production, we evaluated intracellular ROS accumulation in H9c2 cells stimulated with LPS for different periods in the presence or the absence of 30 μM Q (Figure 3). Intracellular ROS levels were significantly increased in cells stimulated with LPS for 24 hours compared to control cells, as indicated by the increase in DCF fluorescence. However, the increase in DCF fluorescence was significantly decreased by Q pretreatment suggesting that Q was capable of reducing intracellular ROS accumulation following LPS stimulation.
Figure 3.
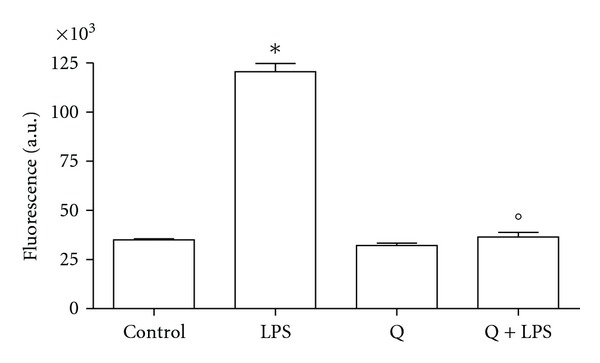
Effect of Q on intracellular ROS production in LPS-stimulated H9c2 cells. Cells were pretreated with 30 μM Q for 2 hours before stimulation with 10 μg/mL LPS. After 24 h intracellular ROS were measured using the peroxide-sensitive fluorescent probe DCHF-DA. Each column represents the mean ± SD of three independent experiments. Data were analyzed by one-way ANOVA followed by Bonferroni's test. *P < 0.05 compared to Control; °P < 0.05 compared to LPS.
3.3. Effect of Q on LPS-Induced Apoptosis in H9c2 Cells
Q demonstrated antiapoptotic effects in different cell types, therefore, we next investigated whether Q protects cardiomyoblasts from LPS-induced apoptosis. H9c2 cells were pretreated with 30 μM Q before exposure to 10 μg/mL LPS for different exposure times and cleaved caspase 3 protein expression (Figure 4) and caspase 3 activity (Figure 5) were evaluated. Immunoblot analyses showed that stimulation with LPS for 24 hours induced a significant cleavage of caspase 3 in respect to control cells, while 0.5 and 2 hours LPS exposure times had no effect on cleaved caspase 3. Pretreatment with Q before LPS stimulation was able to strongly reduce cleavage of caspase 3 to level comparable to control cells. Q alone did not modify caspase 3 cleavage. These data were confirmed by caspase 3 activity assay. Only 24 hours LPS stimulation was able to significantly increase caspase 3 activity and pretreatment with Q significantly reduced caspase 3 activity to level comparable to control cells, demonstrating the antiapoptotic effect of Q against LPS-induced damage.
Figure 4.
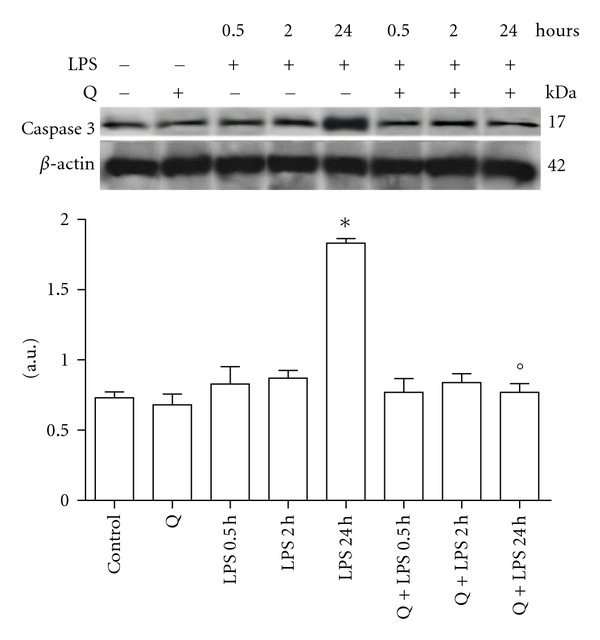
Effect of Q on cleaved caspase 3 protein expression in LPS-stimulated H9c2 cells. Cells were pretreated with 30 μM Q for 2 hours before stimulation with 10 μg/mL LPS. At the indicated time points, cells were harvested and lysed. Crude homogenates (20 μg) were immunoblotted with an antibody that detects endogenous levels of cleaved caspase 3. Equal loading was verified with an anti-β-actin antibody. Densitometric analysis of the protein bands was performed using Bio-Rad Quantity One 1-D Analysis software. Each column represents the mean ± SD of three independent experiments. Data were analyzed by one-way ANOVA followed by Bonferroni's test. *P < 0.05 compared to Control; °P < 0.05 compared to the corresponding LPS exposure times.
Figure 5.
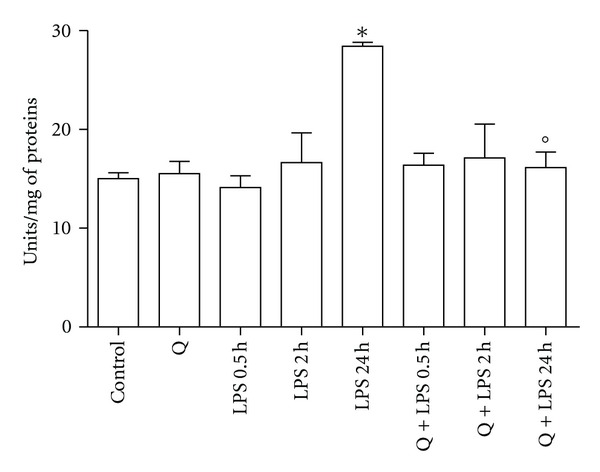
Effect of Q on caspase 3 activity in LPS-stimulated H9c2 cells. Cells were pretreated with 30 μM Q for 2 hours before stimulation with 10 μg/mL LPS. At the indicated time points, caspase 3 activity was measured spectrofluorimetrically in cell lysates as reported in Section 2. Each column represents the mean ± SD of three independent experiments. Data were analyzed by one-way ANOVA followed by Bonferroni's test. *P < 0.05 compared to Control; °P < 0.05 compared to the corresponding LPS exposure times.
3.4. Effect of Q on LPS Activation of MAPK in H9c2 Cells
To better clarify the mechanisms behind Q protection against LPS induced apoptosis, we investigated the role of Q in the modulation of two fundamental proapoptotic signaling pathways in H9c2 cells: JNK and p38 MAPK. H9c2 cells were pretreated with 30 μM Q before exposure to 10 μg/mL LPS for different exposure times and cell lysates were analyzed by immunoblotting with anti-phospho-p38 and anti-p38 antibodies (Figure 6) or anti-phospho-JNK and anti-JNK antibodies (Figure 7). LPS treatment for 0.5 and 2 hours significantly activated p38 MAPK as measured by phospho/total p38 ratio, with the highest increase at 0.5 hours. At the longest exposure time p38, MAPK was no longer activated as phospho/total p38 ratio was comparable to control cells. Q was able to significantly inhibit p38 MAPK activation. LPS treatment for 0.5 hours significantly activated JNK as measured by phospho/total JNK ratio, while after 2 and 24 hours LPS stimulation did not influence JNK phosphorylation. Q was able to significantly reduce JNK activation after 0.5 hours LPS exposure time at level comparable to control cells.
Figure 6.
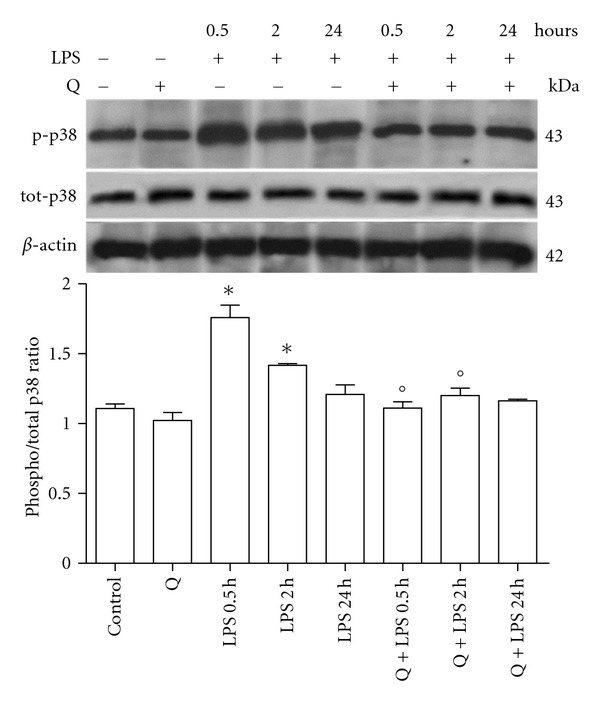
Effect of Q on p38 MAPK activation in LPS-stimulated H9c2 cells. Cells were pretreated with 30 μM Q for 2 hours before stimulation with 10 μg/mL LPS. At the indicated time points, cells were harvested and lysed. Crude homogenates (20 μg) were immunoblotted with anti-phospho-p38 and anti-p38 antibodies. Equal loading was verified with an anti-β-actin antibody. Densitometric analysis of the protein bands was performed using Bio-Rad Quantity One 1-D Analysis software. Each column represents the mean ± SD of three independent experiments. Data were analyzed by one-way ANOVA followed by Bonferroni's test. *P < 0.05 compared to Control; °P < 0.05 compared to the corresponding LPS exposure time.
Figure 7.
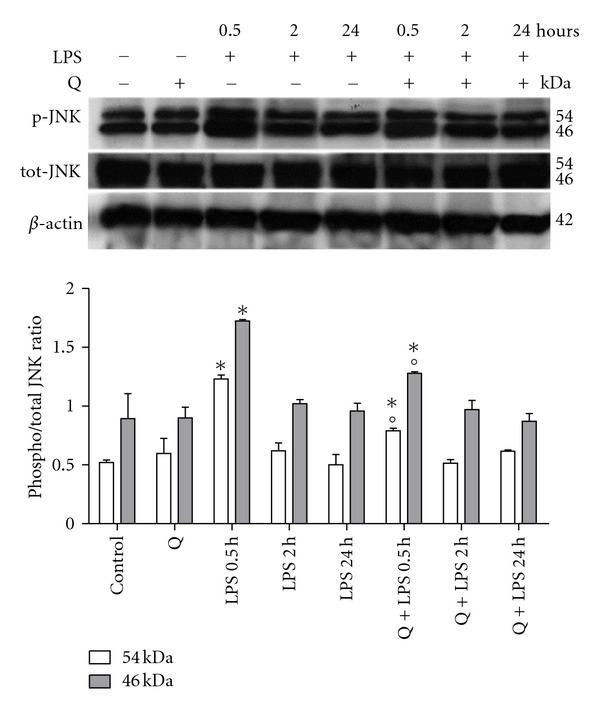
Effect of Q on JNK activation in LPS-stimulated H9c2 cells. Cells were pretreated with 30 μM Q for 2 hours before stimulation with 10 μg/mL LPS. At the indicated time points, cells were harvested and lysed. Crude homogenates (20 μg) were immunoblotted with anti-phospho-JNK and anti-JNK antibodies. Equal loading was verified with an anti-β-actin antibody. Densitometric analysis of the protein bands was performed using Bio-Rad Quantity One 1-D Analysis software. Each column represents the mean ± SD of three independent experiments. Data were analyzed by one-way ANOVA followed by Bonferroni's test. *P < 0.05 compared to Control; °P < 0.05 compared to the corresponding LPS exposure time.
4. Discussion
In this study, we have evaluated the anti-inflammatory role of Q in H9c2 cells demonstrating that this polyphenol attenuated LPS-induced inflammatory events inhibiting iNOS induction, reducing NO production and oxidative stress, and counteracting apoptosis through the modulation of two key protein kinases p38 MAPK and JNK.
A range of clinical conditions are associated with a dysregulation of inflammatory responses. Although the most common of these is sepsis, high concentrations of cytokines are also generated by ischemia-reperfusion, trauma, acute rejection, antigen-specific immune responses, and different acute inflammatory states [35]. Several studies suggest a beneficial effect for cardiac dysfunction through inhibition of cardiac inflammatory processes in sepsis [13].
LPS of Gram-negative bacteria has been recognized as a causative agent in myocardial depression during sepsis [36]. It has been shown that the inflammatory responses induced by LPS in cardiomyocytes are characterized by an increased production of ROS which leads to the activation of transcription factors and intracellular signaling pathways and to the induction of inflammatory mediators, including TNF-α, ICAM-1, and iNOS. All of these mediators may be involved in the depression of cardiac function [13, 37, 38]. In the cardiovascular system, NO produced by iNOS is a major pathophysiologic mediator of septic shock [39] and has been shown to mediate the negative inotropic effects of cytokines [40]. In agreement with previous studies, performed using cardiomyocytes stimulated with LPS for at least 20 h [41, 42], we demonstrated that iNOS protein expression and NO production were significantly increased in LPS-stimulated H9c2. On the contrary, a study of Chen et al. [43] demonstrated an increase in iNOS mRNA level while NO production was not influenced by a 4 h LPS stimulation in rat cardiomyoblasts. This discrepancy could be ascribed to the lower LPS exposure time that is probably too short to significantly increase NO in the culture medium. In our study, Q treatment was able to significantly reduce both iNOS expression and NO production.
As NO may react with ROS such as the superoxide radical to yield the highly reactive oxidant species peroxynitrite, leading to more aggressive oxidative and nitrosative stress [44], we evaluated the intracellular ROS production in LPS-stimulated H9c2 cells. As expected, LPS significantly increased intracellular ROS level while Q pretreatment was able to reduce this production to level comparable to control cells. We previously demonstrated the ability of Q to reduce oxidative stress in H9c2 cells [31] and in rat cardiomyocytes [32] through both a direct and indirect antioxidant mechanisms.
The action of NO has been related to the induction of programmed cell death, or apoptosis, in various cells [45]. The capacity of NO to induce apoptosis was first appreciated by Albina et al. [46], who showed that NO caused apoptosis in macrophages. Many reports suggest that during the ischemia-reperfusion event, NO mediates tissue injury [47, 48]. Moreover, induction of iNOS by cytokines in primary rat cardiac myocytes was associated with an increased myocytes apoptosis, which was ameliorated by the administration of an inhibitor of NO synthase, which blocked the expression of iNOS in response to cytokines [45]. These results suggest that NO is markedly involved in cardiomyopathy, which leads to the contribution of impaired cardiac function. Our results demonstrated that Q was able to counteract LPS-induced apoptosis by inhibiting caspase-3 activation and significantly reducing caspase-3 activity. Different studies have demonstrated that the mechanism underlying the cytoprotection of Q may be mediated by inhibition of NF-κB activation [44, 49, 50], which controls the expression of inflammatory mediators [51]. A crucial step in the activation of NF-κB is the degradation of IκBa [52]. LPS stimulates the canonical NF-κB activation pathway through degradation of IκBa and phosphorylation of NFκB p65 subunit [53, 54], NF-κB p65 subsequently translocates from the cytoplasm to the nucleus and, in turn, triggers a large amount of genes encoded for inflammatory mediators [54].
The mitogen-activated protein kinase (MAPK) family, including extracellular signal-regulated c-Jun N-terminal kinases 1 and 2 (JNK) and p38 MAP kinases, plays an important role in cell fate decisions and has been implicated in death/survival signaling in cardiac myocytes [55]. Evidence also indicates that ROS contribute to LPS-stimulated MAPKs signaling pathways in myocardial cells [13]. These findings are in line with the crucial role of MAPKs signaling and oxidative stress in sepsis-induced myocardial contractile dysfunction [13, 37, 38]. Data reported in this paper demonstrated that LPS stimulation activates both p38 MAPK and JNK. In particular, JNK activation was quick and transient and after 2 h LPS stimulation JNK phosphorylation was comparable to control cells, while p38 MAPK activation lasted for at least 2 h. Our data are in agreement with the results of Peng et al. [56] that demonstrated an immediate and transient increase in p38 MAPK activation after LPS stimulation, which was followed by TNF-α production in the myocardium and the inhibition of p38 MAPK activation improved cardiac function and survival during endotoxemia in mice. Activation of JNK signal transduction cascades has been implicated in the regulation of hypertrophic and apoptotic responses in the myocardium [57]. Peng et al. [58] showed that LPS increases JNK activation in cultured cardiomyocytes. It has been demonstrated that the activation of JNK/SAPK is crucial for NO toxicity in H9c2 cardiac muscle cells [59], moreover, Liu et al. [60] observed that incubation of LPS-treated myocardial cells with the JNK1/2 inhibitor SP600125 resulted in marked inhibition of LPS-induced phosphorylation of IκB, degradation of IκB and upregulation of TNF-α, leading to myocardial apoptotic responses.
Q pretreatment was able to reduce the activation of the two kinases to values comparable to control cells, demonstrating that the antiapoptotic effect of Q could be also mediated by the modulation of p38 MAPK and JNK whose detrimental role in sepsis has been largely demonstrated.
In conclusion, Q inhibited iNOS induction in LPS-stimulated H9c2 cells in a time-dependent manner, reduced NO production, and counteracted LPS-induced apoptosis. This protective effect might be mediated by the suppression of the activation of p38 MAPK and JNK. Therefore, these results suggest that Q might serve as a valuable protective agent in cardiovascular and inflammatory diseases.
Acknowledgments
This work has been supported by grants from Ministero dello Sviluppo Economico (Italy), Project Made in Italy-over 50, and Fondazione del Monte di Bologna e Ravenna (Italy).
References
- 1.Parrillo JE, Burch C, Shelhamer JH, Parker MM, Natanson C, Schuette W. A circulating myocardial depressant substance in humans with septic shock. Septic shock patients with a reduced ejection fraction have a circulating factor that depresses in vitro myocardial cell performance. Journal of Clinical Investigation. 1985;76(4):1539–1553. doi: 10.1172/JCI112135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma AC. Sepsis-induced myocardial dysfunction. Shock. 2007;28(3):265–269. doi: 10.1097/01.shk.0000235090.30550.fb. [DOI] [PubMed] [Google Scholar]
- 3.Mann DL, Topkara VK, Evans S, Barger PM. Innate immunity in the adult mammalian heart: for whom the cell tolls. Transactions of the American Clinical and Climatological Association. 2010;121:34–51. [PMC free article] [PubMed] [Google Scholar]
- 4.von Haehling S, Schefold JC, Lainscak M, Doehner W, Anker SD. Inflammatory biomarkers in heart failure revisited: much more than innocent bystanders. Heart Failure Clinics. 2009;5(4):549–560. doi: 10.1016/j.hfc.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Charalambous BM, Stephens RCM, Feavers IM, Montgomery HE. Role of bacterial endotoxin in chronic heart failure: the gut of the matter. Shock. 2007;28(1):15–23. doi: 10.1097/shk.0b013e318033ebc5. [DOI] [PubMed] [Google Scholar]
- 6.Hickson-Bick DLM, Jones C, Buja LM. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. Journal of Molecular and Cellular Cardiology. 2008;44(2):411–418. doi: 10.1016/j.yjmcc.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Knapp S, Branger J, Van der Poll T. Advances in research of the inflammatory response: the importance of Toll-like receptors. Wiener Medizinische Wochenschrift. 2002;152(21-22):552–554. doi: 10.1046/j.1563-258x.2002.02098.x. [DOI] [PubMed] [Google Scholar]
- 8.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Nemoto S, Vallejo JG, Knuefermann P, et al. Escherichia coli LPS-induced LV dysfunction: role of toll-like receptor-4 in the adult heart. American Journal of Physiology. 2002;282(6):H2316–H2323. doi: 10.1152/ajpheart.00763.2001. [DOI] [PubMed] [Google Scholar]
- 10.Knuefermann P, Nemoto S, Misra A, et al. CD14-deficient mice are protected against lipopolysaccharide-induced cardiac inflammation and left ventricular dysfunction. Circulation. 2002;106(20):2608–2615. doi: 10.1161/01.cir.0000038110.69369.4c. [DOI] [PubMed] [Google Scholar]
- 11.Davani EY, Boyd JH, Dorscheid DR, et al. Cardiac ICAM-1 mediates leukocyte-dependent decreased ventricular contractility in endotoxemic mice. Cardiovascular Research. 2006;72(1):134–142. doi: 10.1016/j.cardiores.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 12.Boyd JH, Chau EH, Tokunanga C, et al. Fibrinogen decreases cardiomyocyte contractility through an ICAM-1-dependent mechanism. Critical Care. 2008;12(1, article R2) doi: 10.1186/cc6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ceylan-Isik AF, Zhao P, Zhang B, Xiao X, Su G, Ren J. Cardiac overexpression of metallothionein rescues cardiac contractile dysfunction and endoplasmic reticulum stress but not autophagy in sepsis. Journal of Molecular and Cellular Cardiology. 2010;48(2):367–378. doi: 10.1016/j.yjmcc.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu H, Shan L, Schiller PW, Mai A, Peng T. Histone deacetylase-3 activation promotes Tumor Necrosis Factor-α (TNF-α) expression in cardiomyocytes during lipopolysaccharide stimulation. Journal of Biological Chemistry. 2010;285(13):9429–9436. doi: 10.1074/jbc.M109.071274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funakoshi H, Kubota T, Kawamura N, et al. Disruption of inducible nitric oxide synthase improves β-adrenergic inotropic responsiveness but not the survival of mice with cytokine-induced cardiomyopathy. Circulation Research. 2002;90(9):959–965. doi: 10.1161/01.res.0000017632.83720.68. [DOI] [PubMed] [Google Scholar]
- 16.Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circulation Research. 2000;87(3):241–247. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 17.Shindo T, Ikeda U, Ohkawa F, Kawahara Y, Yokoyama M, Shimada K. Nitric oxide synthesis in cardiac myocytes and fibroblasts by inflammatory cytokines. Cardiovascular Research. 1995;29(6):813–819. [PubMed] [Google Scholar]
- 18.Champion HC, Skaf MW, Hare JM. Role of nitric oxide in the pathophysiology of heart failure. Heart Failure Reviews. 2003;8(1):35–46. doi: 10.1023/a:1022142904202. [DOI] [PubMed] [Google Scholar]
- 19.Oyama JI, Shimokawa H, Momii H, et al. Role of nitric oxide and peroxynitrite in the cytokine-induced sustained myocardial dysfunction in dogs in vivo. Journal of Clinical Investigation. 1998;101(10):2207–2214. doi: 10.1172/JCI986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez BL, Liu GL, Christopher TA, Ma XL. Peroxynitrite, the product of nitric oxide and superoxide, causes myocardial injury in the isolated perfused rat heart. Coronary Artery Disease. 1997;8(3-4):149–153. doi: 10.1097/00019501-199703000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi K, Del Nido PJ, Poutias DN, Cowan DB, Munakata M, McGowan FX. Vesnarinone restores contractility and calcium handling in early endotoxemia. Circulation. 2000;102(19, supplement 3):III365–III369. doi: 10.1161/01.cir.102.suppl_3.iii-365. [DOI] [PubMed] [Google Scholar]
- 22.Yuan H, Perry CN, Huang C, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. American Journal of Physiology. 2009;296(2):H470–H479. doi: 10.1152/ajpheart.01051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 24.Boots AW, Haenen GRMM, Bast A. Health effects of quercetin: from antioxidant to nutraceutical. European Journal of Pharmacology. 2008;585(2-3):325–337. doi: 10.1016/j.ejphar.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Borska S, Gebarowska E, Wysocka T, Drag-Zalesińska M, Zabel M. The effects of quercetin vs cisplatin on proliferation and the apoptotic process in A549 and SW1271 cell lines in in vitro conditions. Folia Morphologica. 2004;63(1):103–105. [PubMed] [Google Scholar]
- 26.Saito A, Sugisawa A, Umegaki K, Sunagawa H. Protective effects of quercetin and its metabolites on H2O2-induced chromosomal damage to WIL2-NS cells. Bioscience, Biotechnology and Biochemistry. 2004;68(2):271–276. doi: 10.1271/bbb.68.271. [DOI] [PubMed] [Google Scholar]
- 27.Hakkinen SH, Karenlampi SO, Heinonen IM, Mykkanen HM, Torronen AR. Content of the flavonols quercetin, myricetin, and kaempferol in 25 edible berries. Journal of Agricultural and Food Chemistry. 1999;47(6):2274–2279. doi: 10.1021/jf9811065. [DOI] [PubMed] [Google Scholar]
- 28.Conquer JA, Maiani G, Azzini E, Raguzzini A, Holub BJ. Supplementation with quercetin markedly increases plasma quercetin concentration without effect on selected risk factors for heart disease in healthy subjects. Journal of Nutrition. 1998;128(3):593–597. doi: 10.1093/jn/128.3.593. [DOI] [PubMed] [Google Scholar]
- 29.Moon JH, Nakata R, Oshima S, Inakuma T, Terao J. Accumulation of quercetin conjugates in blood plasma after the short-term ingestion of onion by women. American Journal of Physiology. 2000;279(2):R461–R467. doi: 10.1152/ajpregu.2000.279.2.R461. [DOI] [PubMed] [Google Scholar]
- 30.Egert S, Wolffram S, Bosy-Westphal A, et al. Daily quercetin supplementation dose-dependently increases plasma quercetin concentrations in healthy humans. Journal of Nutrition. 2008;138(9):1615–1621. doi: 10.1093/jn/138.9.1615. [DOI] [PubMed] [Google Scholar]
- 31.Angeloni C, Spencer JPE, Leoncini E, Biagi PL, Hrelia S. Role of quercetin and its in vivo metabolites in protecting H9c2 cells against oxidative stress. Biochimie. 2007;89(1):73–82. doi: 10.1016/j.biochi.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Angeloni C, Leoncini E, Malaguti M, Angelini S, Hrelia P, Hrelia S. Role of quercetin in modulating rat cardiomyocyte gene expression profile. American Journal of Physiology. 2008;294(3):H1233–H1243. doi: 10.1152/ajpheart.01091.2007. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radical Biology and Medicine. 1999;27(5-6):612–616. doi: 10.1016/s0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- 34.Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376(6535):37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 35.Landry DW, Oliver JA. Mechanisms of disease: the pathogenesis of vasodilatory shock. New England Journal of Medicine. 2001;345(8):588–595. doi: 10.1056/NEJMra002709. [DOI] [PubMed] [Google Scholar]
- 36.Peng T, Lu X, Lei M, Feng Q. Endothelial nitric-oxide synthase enhances lipopolysaccharide-stimulated tumor necrosis factor-α expression via cAMP-mediated p38 MAPK pathway in cardiomyocytes. Journal of Biological Chemistry. 2003;278(10):8099–8105. doi: 10.1074/jbc.M207288200. [DOI] [PubMed] [Google Scholar]
- 37.Haudek SB, Spencer E, Bryant DD, et al. Overexpression of cardiac I-κBα prevents endotoxin-induced myocardial dysfunction. American Journal of Physiology. 2001;280(3):H962–H968. doi: 10.1152/ajpheart.2001.280.3.H962. [DOI] [PubMed] [Google Scholar]
- 38.Davani EY, Dorscheid DR, Lee CH, Van Breemen C, Walley KR. Novel regulatory mechanism of cardiomyocyte contractility involving ICAM-1 and the cytoskeleton. American Journal of Physiology. 2004;287(3):H1013–H1022. doi: 10.1152/ajpheart.01177.2003. [DOI] [PubMed] [Google Scholar]
- 39.Maragos CM, Ji Ming Wang, Hrabie JA, Oppenheim JJ, Keefer LK. Nitric oxide/nucleophile complexes inhibit the in vitro proliferation of A375 melanoma cells via nitric oxide release. Cancer Research. 1993;53(3):564–568. [PubMed] [Google Scholar]
- 40.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257(5068):387–389. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 41.Comini L, Boraso A, Bachetti T, et al. Effects of endotoxic shock on neuronal NOS and calcium transients in rat cardiac myocytes. Pharmacological Research. 2005;51(5):409–417. doi: 10.1016/j.phrs.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Schwartz IF, Hershkovitz R, Iaina A, et al. Garlic attenuates nitric oxide production in rat cardiac myocytes through inhibition of inducible nitric oxide synthase and the arginine transporter CAT-2 (cationic amino acid transporter-2) Clinical Science. 2002;102(5):487–493. [PubMed] [Google Scholar]
- 43.Chen TH, Wo HT, Wu CC, et al. Exendin-4 attenuates lipopolysaccharides induced inflammatory response but does not protects H9c2 cells from apoptosis. Immunopharmacology and Immunotoxicology. 2012;34(3):484–490. doi: 10.3109/08923973.2011.630398. [DOI] [PubMed] [Google Scholar]
- 44.Dias AS, Porawski M, Alonso M, Marroni N, Collado PS, González-Gallego J. Quercetin decreases oxidative stress, NF-κB activation, and iNOS overexpression in liver of streptozotocin-induced diabetic rats. Journal of Nutrition. 2005;135(10):2299–2304. doi: 10.1093/jn/135.10.2299. [DOI] [PubMed] [Google Scholar]
- 45.Chae HJ, Chae SW, Kim HR. Cyclic adenosine monophosphate inhibits nitric oxide-induced apoptosis of cardiac muscle cells in a c-Jun N-terminal kinase-dependent manner. Immunopharmacology and Immunotoxicology. 2004;26(2):249–263. doi: 10.1081/iph-120037722. [DOI] [PubMed] [Google Scholar]
- 46.Albina JE, Cui S, Mateo RB, Reichner JS. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. Journal of Immunology. 1993;150(11):5080–5085. [PubMed] [Google Scholar]
- 47.Matheis G, Sherman MP, Buckberg GD, Haybron DM, Young HH, Ignarro LJ. Role of L-arginine-nitric oxide pathway in myocardial reoxygenation injury. American Journal of Physiology. 1992;262(2):H616–H620. doi: 10.1152/ajpheart.1992.262.2.H616. [DOI] [PubMed] [Google Scholar]
- 48.Galea E, Feinstein DL, Reis DJ. Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10945–10949. doi: 10.1073/pnas.89.22.10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho SY, Park SJ, Kwon MJ, et al. Quercetin suppresses proinflammatory cytokines production through MAP kinases and NF-κB pathway in lipopolysaccharide-stimulated macrophage. Molecular and Cellular Biochemistry. 2003;243(1-2):153–160. doi: 10.1023/a:1021624520740. [DOI] [PubMed] [Google Scholar]
- 50.Mu MM, Chakravortty D, Sugiyama T, et al. The inhibitory action of quercetin on lipopolysaccharide-induced nitric oxide production in RAW 264.7 macrophage cells. Journal of Endotoxin Research. 2001;7(6):431–438. doi: 10.1179/096805101101533034. [DOI] [PubMed] [Google Scholar]
- 51.Valen G, Yan ZQ, Hansson GK. Nuclear factor kappa-B and the heart. Journal of the American College of Cardiology. 2001;38(2):307–314. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 52.Schmitz ML, Mattioli I, Buss H, Kracht M. NF-κB: a multifaceted transcription factor regulated at several levels. ChemBioChem. 2004;5(10):1348–1358. doi: 10.1002/cbic.200400144. [DOI] [PubMed] [Google Scholar]
- 53.Wright G, Singh IS, Hasday JD, et al. Endotoxin stress-response in cardiomyocytes: NF-κB activation and tumor necrosis factor-α expression. American Journal of Physiology. 2002;282(3):H872–H879. doi: 10.1152/ajpheart.00256.2001. [DOI] [PubMed] [Google Scholar]
- 54.Hall G, Singh IS, Hester L, Hasday JD, Rogers TB. Inhibitor-κB kinase-β regulates LPS-induced TNF-α production in cardiac myocytes through modulation of NF-κB p65 subunit phosphorylation. American Journal of Physiology. 2005;289(5):H2103–H2111. doi: 10.1152/ajpheart.00393.2005. [DOI] [PubMed] [Google Scholar]
- 55.Andréka P, Tran T, Webster KA, Bishopric NH. Nitric oxide and promotion of cardiac myocyte apoptosis. Molecular and Cellular Biochemistry. 2004;263(1):35–53. doi: 10.1023/B:MCBI.0000041847.63338.b8. [DOI] [PubMed] [Google Scholar]
- 56.Peng T, Lu X, Lei M, Moe GW, Feng Q. Inhibition of p38 MAPK decreases myocardial TNF-alpha expression and improves myocardial function and survival in endotoxemia. Cardiovascular Research. 2003;59(4):893–900. doi: 10.1016/s0008-6363(03)00509-1. [DOI] [PubMed] [Google Scholar]
- 57.Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. Journal of Molecular and Cellular Cardiology. 2003;35(12):1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 58.Peng T, Lu X, Feng Q. NADH oxidase signaling induces cyclooxygenase-2 expression during lipopolysaccharide stimulation in cardiomyocytes. The FASEB Journal. 2005;19(2):293–295. doi: 10.1096/fj.04-2289fje. [DOI] [PubMed] [Google Scholar]
- 59.Chae HJ, So HS, Chae SW, et al. Sodium nitroprusside induces apoptosis of H9C2 cardiac muscle cells in a c-Jun N-terminal kinase-dependent manner. International Immunopharmacology. 2001;1(5):967–978. doi: 10.1016/s1567-5769(01)00033-9. [DOI] [PubMed] [Google Scholar]
- 60.Liu CJ, Lo JF, Kuo CH, et al. Akt mediates 17β-estradiol and/or estrogen receptor-α inhibition of LPS-induced tumor necresis factor-α expression and myocardial cell apoptosis by suppressing the JNK1/2-NFκB pathway. Journal of Cellular and Molecular Medicine. 2009;13(9 B):3655–3667. doi: 10.1111/j.1582-4934.2009.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]