

Abstract
The development of experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis, has been studied in mice that were (i) vitamin D-deficient, (ii) minus the vitamin D receptor, (iii) minus a vitamin D 25-hydroxylase, and (iv) minus the vitamin D 25-hydroxyvitamin D-1α-hydroxylase. EAE development was markedly suppressed in mice lacking the vitamin D receptor and partially suppressed in vitamin D-insufficient mice. However, the absence of either of the two key hydroxylases (i.e., 25-hydroxylase and 1α-hydroxylase) neither inhibits nor enhances the development of EAE. These results indicate that vitamin D and the vitamin D receptor are required for the development of EAE. The results also suggest that 1,25-dihydroxyvitamin D3 may not play a role in this autoimmune response.
Keywords: autoimmune disease, serum calcium, serum 25-hydroxyvitamin D3, CYP27B1 knockout mice, CYP2R1 knockout mice
As early as 1974, the work by Agranoff and Goldberg (1) suggested that the incidence of multiple sclerosis is inversely related to the degree of sunlight exposure based on geographical distribution of multiple sclerosis. The prevalence of multiple sclerosis dramatically increases with latitude in both hemispheres (2). Sunlight was also believed to play a key role in the lower incidence of multiple sclerosis in the tropical areas. It was natural to assume that the effect of sunlight might be through production of vitamin D in skin. Therefore, vitamin D supplements have been considered as a potential therapeutic strategy for multiple sclerosis patients. So far, convincing evidence that vitamin D3 supplementation can prevent or reduce multiple sclerosis is lacking (3).
For vitamin D to function in its classical actions, it must be converted first to 25-hydroxyvitamin D (25-OH-D) and further, to 1α,25-dihydroxyvitamin D (1,25-(OH)2D; the hormonal form) (4). Administration of higher doses of 1,25-(OH)2D3 can clearly reduce or prevent experimental autoimmune encephalomyelitis (EAE), an experimental model of multiple sclerosis, but this suppression occurs only when hypercalcemia occurs (5). Furthermore, 1,25-(OH)2D3 loses its effect when low calcium diets are fed (6). Hypercalcemia itself suppresses EAE (7); thus, it is not at all clear whether any vitamin D compound can suppress this disease without hypercalcemia.
The question has recently been raised by two independent groups as to whether vitamin D deficiency increases susceptibility of mice to EAE (8, 9). To their surprise, the incidence of disease, severity of disease, and onset of EAE were all markedly reduced by vitamin D deficiency. Although it is clear that vitamin D deficiency does affect the autoimmunity process, the result was exactly the opposite of what was expected. This result forced a reconsideration of the idea that high sunlight exposure suppresses multiple sclerosis by increasing the amount of vitamin D. On examining this hypothesis, the work by Becklund et al. (10) found that the effect of UV light in suppressing EAE was not mediated by vitamin D production. These experiments and the experiments on vitamin D-deficient mice described above led us to examine further whether the vitamin D system is in any way related to EAE and its treatment.
In the present study, we used vitamin D receptor (VDR) KO mice, 1α-hydroxylase KO mice, 25-hydroxylase KO mice, and vitamin D deficiency to determine how the vitamin D system relates to the development of EAE.
Results
There was no change in EAE severity in both minus vitamin D 25-hydroxylase (CYP2R1) KO mice and minus the vitamin D 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1) KO mice (Figs. 1 and 2). In the previous studies, vitamin D-deficient mice had been shown to have reduced EAE severity and delayed onset (8). The CYP2R1 KO mice had a serum 25-OH-D level reduced to one-half of the level of WT. The results were similar in male and female mice and were combined for the results shown in Fig. 1. The KO of CYP2R1 had no effect on either severity or day of onset of EAE (Fig. 1). Serum calcium was also unchanged by this mutation. Serum 25-OH-D level was increased in CYP27B1 KO mice, and as expected, the serum calcium level was also unchanged because of the 1.2% dietary calcium level (Fig. 2 C and D). Severity and day of onset of EAE remained unchanged in the CYP27B1 KO mice (Fig. 2A). The decreased body weight in week 4 of EAE also suggested that EAE progressed similarly in both WT and CYP27B1 KO mice (Fig. 2B).
Fig. 1.

EAE severity is unchanged in CYP2R1 KO and WT mice. All mice were immunized with MOG35–55/CFA emulsion. Clinical score was measured daily for 30 d after immunization. (A) Time course of the development of EAE. (B) Body weights. (C) Serum calcium levels determined at termination of the experiment. (D) Serum 25-OH-D measured at termination of the experiment. *P < 0.05 compared with WT. The results obtained were similar in male and female mice.
Fig. 2.

EAE development is not affected by ablation of CYP27B1. (A) Clinical score after immunization. (B) Body weight. (C) Serum calcium and (D) serum 25-OH-D levels at termination of the experiment. Serum 25-OH-D in CYP27B1 KO mice was significantly elevated. *P < 0.05.
Dramatic Suppression of EAE in VDR KO Mice.
The clinical score was dramatically decreased in VDR KO mice when fed the purified diet both with and without vitamin D supplementation (Fig. 3 A and B). The body weight and serum calcium level were not changed in VDR KO mice, because they were fed the 1.2% calcium and 20% lactose diet (g/kg) (Fig. 3 C–F). The incidence, mean severity, and cumulative disease index (CDI) were all significantly decreased in VDR KO mice (Table 1).
Fig. 3.

EAE is markedly suppressed in VDR KO mice. Clinical score as a function of time after immunization in (A) +D and (B) −D mice. (C and D) Weight record of +D and −D mice. (E and F) Terminal serum calcium values. Serum calcium levels in all animals were kept in the normal range by feeding 1.2% calcium and 20% lactose. *P < 0.05 compared with WT in Fig. 3 A and B; *P < 0.05 compared with WT in week 1 and 2 in Fig. 3C and week 1 in WT in Fig. 3D (n = 9–11).
Table 1.
Suppressed EAE progression in VDR KO mice
| Treatment | Incidence | Day of onset | Mean severity | CDI |
| VDR-WT | 92% (11/12) | 13 ± 4 | 2.4 ± 0.4 | 43 ± 12 |
| VDR-KO | 40% (4/10)* | 14 ± 1 | 1.1 ± 0.4* | 9 ± 11* |
*P < 0.05, compared with WT group.
Suppression of EAE in Vitamin D-Deficient Mice.
We also tested EAE development in vitamin D-deficient mice. Vitamin D deficiency was confirmed by measuring both serum calcium and serum 25-OH-D levels. The −D mice were fed a purified diet with different levels of calcium (0.47% and 1.2% calcium) plus vitamins A, E, and K. The +D mice were fed the same purified diets and vitamins A, D, E, and K. The score of EAE was decreased in −D mice with both 0.47% and 1.2% calcium diets compared with the +D mice, confirming previous reports (Fig. 4A). At the end of the experiment, the serum calcium level was in the normal range in the −D mice given 1.2% calcium and lower in the −D mice fed the 0.47% calcium diet (Fig. 4C). Compared with the +D mice, −D mice with 0.47% calcium had lower CDI (Fig. 4). Interestingly, both +D and −D mice fed the 1.2% calcium diet had decreased CDI compared with those mice fed the 0.47% calcium diet, suggesting that high dietary calcium plays a suppressive role in the development of EAE (Fig. 4).
Fig. 4.
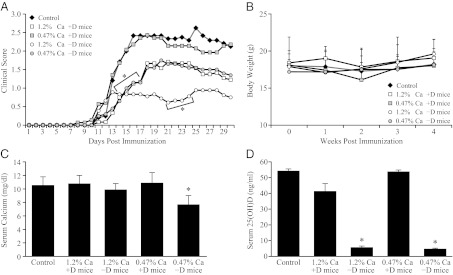
Vitamin D deficiency suppresses EAE severity. (A) Time course of EAE development. (B) Body weights. (C) Serum calcium and (D) serum 25-OH-D levels measured at termination (day 30 postimmunization). Vitamin D deficiency reduced serum 25-OH-D (P < 0.05) and EAE scores *P < 0.05 compared with control in Fig. 4C; *P < 0.05 compared with 0.47% calcium +D in Fig. 4C; P < 0.05 compared with control and +D mice in Fig. 4D (n = 9–12).
Discussion
The hormonal form of vitamin D has been clearly used to suppress models of autoimmune diseases (for example, multiple sclerosis, type 1 diabetes, and inflammatory bowel disease) (11–13). The protective role of 1,25-(OH)2D3 against EAE is related to high dietary calcium levels (5), and hypercalcemia accompanies the suppressive action of 1,25-(OH)2D3. Furthermore, hypercalcemia produced by parathyroid hormone suppresses EAE in female mice (7). Many proposed mechanisms where 1,25-(OH)2D can suppress autoimmune diseases have emerged (14–16), but a clear explanation remains to be realized given the presence of the VDR in cells of the immune system.
The incidence of multiple sclerosis is inversely related to latitude (17). This relationship is thought to be based on vitamin D production in skin in response to degree of sunlight exposure. Vitamin D supplementation should provide protection if the assumption that the geographic distribution of multiple sclerosis is related to vitamin D production is correct. Thus far, clinical studies on this point have yet to surface (18, 19). As expected, human multiple sclerosis incidence does correlate with low serum 25-OH-D, the most reliable index of vitamin D status (18). However, this finding does not establish a cause and effect relationship. At least two studies have shown that an analog (paricalcitol) or vitamin D itself in the absence of hypercalcemia does not reduce multiple sclerosis lesions (3).
Although EAE in mice has limitations as a model of multiple sclerosis, it is still the most widely used. Two independent studies have clearly revealed that severe vitamin D deficiency itself impairs the development of EAE (8, 9). Furthermore, severe vitamin D deficiency impairs delayed hypersensitivity in mice. Both of these findings suggest a significant role of vitamin D in mounting an immune response (20). However, this result would be at variance with a protective role of vitamin D against autoimmune diseases.
In examining the role of UV light in protecting against EAE, an effect independent of vitamin D production was discovered (10). Important questions distill from these accumulated results. Does vitamin D have any protective role against multiple sclerosis, and even further, is vitamin D signaling required for the development of EAE? Because both vitamin D deficiency and an absence of VDR largely prevent the development of EAE, it is clear that a lack of vitamin D signaling is not a risk factor in EAE.
A short-term (1 wk) lack of 1,25-(OH)2D3 does not seem to prevent EAE, whereas a lack of vitamin D itself does interfere with EAE development. We were unable to eliminate production of 25-OH-D3 completely, and a partial reduction did not affect EAE development. Because VDR and vitamin D are required for the development of EAE, it seems that some form of vitamin D is needed for development of elements of the immune system that are required for the autoimmune disease EAE. Because 1,25-(OH)2D3 was absent for only 1 wk before immunization, it is possible that elements of the immune system that require 1,25-(OH)2D3 had developed, making them responsive to EAE induction. For this reason, a possible role of 1,25-(OH)2D3 cannot be completely eliminated. Because 1 wk is in excess of 10 half-lives of 1,25-(OH)2D3 in vivo, it is nevertheless clear that 1,25-(OH)2D does not play a direct role in the postimmunization events resulting in EAE.
These studies do not address the important question of whether any form of vitamin D treatment independent of hypercalcemia is effective against EAE. All results from this laboratory are consistent with the idea that hypercalcemia mediates the protective effect of 1,25-(OH)2D3, 25-OH-D3, or their analogs (5, 6, 10). Certainly, VDR is required for vitamin D compounds to suppress EAE (21). However, the lack of VDR also prevents the vitamin D-induced rise in serum calcium.
This study provides an unexpected result that strongly supports the role of a form of vitamin D in mounting an immune response independent of calcium. It also intensifies a critical examination of whether vitamin D therapy is useful in the treatment of EAE and multiple sclerosis.
Materials and Methods
Animals and Diets.
Three different gene KO mice and vitamin D-deficient mice were used in this study.
i) CYP2R1 KO mice. The WT and KO mice were generated from heterozygote pairs and confirmed by PCR analysis. The mice were maintained on chow diet (Purina Chow 5015 containing a 3.3 IU vitamin D/g diet) during the entire experiment.
ii) CYP27B1 KO mice. CYP27B1 WT and CYP27B1 KO mice were generated from the heterozygote pairs as described previously (6). The genotype was confirmed by PCR analysis. Breeders were maintained on Purina Chow 5015. Confirmed homozygotes were then grown for 3 wk on purified diet 11 (0.47% calcium and 0.3% phosphorus) supplemented with 4 ng 1,25-(OH)2D3 per mouse per day dissolved in Wesson Oil until 1 wk before the induction of EAE (7).
iii) VDR KO mice. VDR heterozygote pairs of C57BL/6J background were purchased from Jackson Laboratory. The VDR WT and VDR KO mice were generated from heterozygote breeders. The genotype was determined by PCR. Mice were fed a rescue diet (1.2% calcium and 20% lactose) from weaning and during the experiment;
iv) Vitamin D-deficient mice (−D). To generate vitamin D-deficient mice, the breeders were fed a vitamin D-deficient diet (1.2% calcium) supplemented with oil containing the fat-soluble vitamins A, E, and K (22) and housed under incandescent lighting to prevent vitamin D synthesis in skin. Weanling pups were then fed the purified diet containing 0.47% calcium for 3 wk and the same diet essentially devoid of calcium for 1 wk. Serum calcium and serum 25-OH-D3 levels were measured to confirm vitamin D deficiency. At 8 wk, the −D mice were randomly divided into two groups: one group was fed a diet containing 0.47% calcium, and the other group was fed a diet containing 1.2% calcium and 20% lactose. The animals on both diets were supplemented with vitamins A, E, and K. As controls, the C57BL/6J mice were randomly divided into two groups: one group was fed the diet containing 0.47% calcium, and the other group was fed the diet containing 1.2% calcium and 20% lactose supplemented with 25 units vitamin D per day in the diet. Each group had 6–12 mice (2–3 males). All of the procedures were done in accordance with the Research Animal Resources Committee of the College of Agricultural & Life Sciences, University of Wisconsin–Madison. Animal protocols were approved by the University of Wisconsin–Madison Institutional Animal Care and Use Committee.
EAE Induction.
EAE was induced by subcutaneous injection of myelin oligodendrocyte glycoprotein peptide (MOG35–55; MEVGWYRSPFSRVVHLYRNGK) injection (9). MOG35–55 was synthesized at the University of Wisconsin–Madison Biotechnology Center and purified to ≥95% by reverse-phase HPLC. To make MOG35–55 emulsion, 4 mg/mL MOG35-55 solution was mixed by 1:1 with Complete Freund's adjuvant (CFA) containing 5 mg/mL inactivated Mycobacterium tuberculosis H37Ra (DIFCO Laboratories). Each mouse was injected s.c. with 100 μL MOG35–55 emulsion and injected i.p. with 100 μL pertussis toxin (200 ng; List Biological Laboratories). At 48 h, the mice received a second pertussis toxin injection. Mice were scored daily by a technician blinded to treatments for clinical signs of EAE using the following scale: 0, no clinical disease; 1, loss of tail tone; 2, unsteady gait; 3, hindlimb paralysis; 4, forelimb paralysis; 5, death.
Serum Calcium Measurement.
Blood was collected by retro-orbital bleeding at termination of the experiments. Blood samples were spun at 3,000 × g for 15 min followed by a second spin at 17,000 × g for 1 min. Serum calcium levels were determined from two measurements per sample in 0.1% LaCl3 by atomic absorption spectroscopy (Perkin-Elmer) (13).
Serum 25-OH-D Measurement.
Blood was collected at the initial and/or final time points throughout the experiment. Serum was collected through two successive centrifugation steps as described above. Serum 25-OH-D levels were determined using a 125I-radioimmunological assay using the manufacturer’s instructions (DiaSorin). Samples above the range of standard curve were diluted before reanalysis. Radioactivity was quantified using a Cobra 5002 γ-scintillation counter (PerkinElmer).
Statistical Analysis.
Data were expressed as mean ± SD. Statistical analyses were performed using the two-tailed Fisher exact probability test for incidence, the Mann–Whitney nonparametric u test for clinical scores, and the unpaired Student t test for all other measurements. A value of P < 0.05 was considered statistically significant.
Acknowledgments
We thank Billy Blaser, Erin Gudmundson, and Wendy Hellwig for their technical support; Jean Prahl for supplying the mice; and Pat Mings for her assistance in the preparation of the manuscript. This work was supported by a fund from the Wisconsin Alumni Research Foundation.
Footnotes
The authors declare no conflict of interest.
*Fleming JO, et al. (2000) Vitamin D treatment of relapsing-remitting multiple sclerosis (RRMS): A MRI-based pilot study. Neurology 54(Suppl 3):A338. Abstract.
References
- 1.Agranoff BW, Goldberg D. Diet and the geographical distribution of multiple sclerosis. Lancet. 1974;2:1061–1066. doi: 10.1016/s0140-6736(74)92163-1. [DOI] [PubMed] [Google Scholar]
- 2.Simpson S, Jr, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: A meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82:1132–1141. doi: 10.1136/jnnp.2011.240432. [DOI] [PubMed] [Google Scholar]
- 3.Kriegel MA, Manson JE, Costenbader KH. Does vitamin D affect risk of developing autoimmune disease?: A systematic review. Semin Arthritis Rheum. 2011;40:512–531. doi: 10.1016/j.semarthrit.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holick MF, Schnoes HK, DeLuca HF. Identification of 1,25-dihydroxycholecalciferol, a form of vitamin D3 metabolically active in the intestine. Proc Natl Acad Sci USA. 1971;68:803–804. doi: 10.1073/pnas.68.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci USA. 1996;93:7861–7864. doi: 10.1073/pnas.93.15.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantorna MT, Humpal-Winter J, DeLuca HF. Dietary calcium is a major factor in 1,25-dihydroxycholecalciferol suppression of experimental autoimmune encephalomyelitis in mice. J Nutr. 1999;129:1966–1971. doi: 10.1093/jn/129.11.1966. [DOI] [PubMed] [Google Scholar]
- 7.Meehan TF, Vanhooke J, Prahl J, Deluca HF. Hypercalcemia produced by parathyroid hormone suppresses experimental autoimmune encephalomyelitis in female but not male mice. Arch Biochem Biophys. 2005;442:214–221. doi: 10.1016/j.abb.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 8.DeLuca HF, Plum LA. Vitamin D deficiency diminishes the severity and delays onset of experimental autoimmune encephalomyelitis. Arch Biochem Biophys. 2011;513:140–143. doi: 10.1016/j.abb.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Fernandes de Abreu DA, Ibrahim EC, Boucraut J, Khrestchatisky M, Féron F. Severity of experimental autoimmune encephalomyelitis is unexpectedly reduced in mice born to vitamin D-deficient mothers. J Steroid Biochem Mol Biol. 2010;121:250–253. doi: 10.1016/j.jsbmb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Becklund BR, Severson KS, Vang SV, DeLuca HF. UV radiation suppresses experimental autoimmune encephalomyelitis independent of vitamin D production. Proc Natl Acad Sci USA. 2010;107:6418–6423. doi: 10.1073/pnas.1001119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemire JM, Archer DC. 1,25-Dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Invest. 1991;87:1103–1107. doi: 10.1172/JCI115072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zella JB, McCary LC, DeLuca HF. Oral administration of 1,25-dihydroxyvitamin D3 completely protects NOD mice from insulin-dependent diabetes mellitus. Arch Biochem Biophys. 2003;417:77–80. doi: 10.1016/s0003-9861(03)00338-2. [DOI] [PubMed] [Google Scholar]
- 13.Cantorna MT. Vitamin D, multiple sclerosis and inflammatory bowel disease. Arch Biochem Biophys. 2011 doi: 10.1016/j.abb.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niino M. Vitamin D and its immunoregulatory role in multiple sclerosis. Drugs Today (Barc) 2010;46:279–290. doi: 10.1358/dot.2010.46.4.1476498. [DOI] [PubMed] [Google Scholar]
- 15.von Essen MR, et al. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol. 2010;11:344–349. doi: 10.1038/ni.1851. [DOI] [PubMed] [Google Scholar]
- 16.Joshi S, et al. 1,25-Dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol. 2011;31:3653–3669. doi: 10.1128/MCB.05020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramagopalan SV, et al. Relationship of UV exposure to prevalence of multiple sclerosis in England. Neurology. 2011;76:1410–1414. doi: 10.1212/WNL.0b013e318216715e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson S, Jr, et al. Higher 25-hydroxyvitamin D is associated with lower relapse risk in multiple sclerosis. Ann Neurol. 2010;68:193–203. doi: 10.1002/ana.22043. [DOI] [PubMed] [Google Scholar]
- 19.Lonergan R, et al. Multiple sclerosis prevalence in Ireland: Relationship to vitamin D status and HLA genotype. J Neurol Neurosurg Psychiatry. 2011;82:317–322. doi: 10.1136/jnnp.2010.220988. [DOI] [PubMed] [Google Scholar]
- 20.Yang S, Smith C, Prahl JM, Luo X, DeLuca HF. Vitamin D deficiency suppresses cell-mediated immunity in vivo. Arch Biochem Biophys. 1993;303:98–106. doi: 10.1006/abbi.1993.1260. [DOI] [PubMed] [Google Scholar]
- 21.Meehan TF, DeLuca HF. The vitamin D receptor is necessary for 1alpha,25-dihydroxyvitamin D(3) to suppress experimental autoimmune encephalomyelitis in mice. Arch Biochem Biophys. 2002;408:200–204. doi: 10.1016/s0003-9861(02)00580-5. [DOI] [PubMed] [Google Scholar]
- 22.Suda T, DeLuca HF, Tanaka Y. Biological activity of 25-hydroxyergocalciferol in rats. J Nutr. 1970;100:1049–1052. doi: 10.1093/jn/100.9.1049. [DOI] [PubMed] [Google Scholar]