

Abstract
Survivors of hereditary retinoblastoma have a high risk of second primary malignancies, but it has not been investigated whether specific RB1 germline mutations are associated with greater risk of second primary malignancies in a large cohort. We conducted a retrospective cohort study of 199 survivors of hereditary retinoblastoma with a documented RB1 germline mutation diagnosed between 1905 and 2005. In total, 44 hereditary retinoblastoma survivors developed a second primary malignancy after a median follow-up of 30.2 years (range 1.33–76.0). A significantly increased risk of second primary malignancy was observed among carriers of one of the 11 recurrent CGA>TGA nonsense RB1 mutations (hazard ratio (HR) = 3.53; [95% confidence interval (CI) = 1.82–6.84]; P = .000), and there was a significantly lower risk for subjects with a low penetrance mutation (HR = .19; [95% CI = .05–.81]; P = .025). Our findings suggest a genotype-phenotype correlation for second primary cancers of retinoblastoma survivors and may impact on long-term surveillance protocols of patients with hereditary retinoblastoma, if confirmed by future studies.
Keywords: Retinoblastoma, RB1 gene, Second primary malignancies, Germline mutation, Genotype-phenotype correlation
Introduction
Retinoblastoma is the most common primary intraocular malignancy of childhood [1]. Mutational inactivation of both alleles of the RB1 tumor suppressor gene in the developing retina initiates the formation of retinoblastoma [2, 3]. The RB1 gene consists of 27 exons and is located on chromosome 13q14 (GenBank accession number L11910, MIM#180200). The gene encodes a ubiquitously expressed nuclear protein, which is involved in cell cycle regulation, cellular differentiation and survival [4]. About 40% of retinoblastoma patients have a hereditary predisposition, caused by a heterozygous germline mutation in the RB1 gene and are usually bilaterally affected [5]. Over 600 different pathogenic mutations have been described. Patients with nonhereditary retinoblastoma only have one eye affected, no germline mutation in the RB1 gene and two somatic retinal RB1 mutations.
As is known from long-term follow-up studies [6–10], hereditary retinoblastoma subjects have a strongly increased risk for second primary malignancies, (including osteosarcoma, soft tissue sarcoma, melanoma and epithelial cancers) which is associated with excess mortality [11–13]. So far, it has not been examined in a large cohort of retinoblastoma patients whether specific RB1 mutations might be associated with greater risk of second malignancy.
The objective of the present study was to investigate the RB1 genotype in relation to second malignancy risk in hereditary retinoblastoma subjects.
Materials and methods
Patients
In the Netherlands we have data available of Dutch retinoblastoma subjects diagnosed from 1862 onwards. Detailed information on data collection and follow-up has been described previously [9]. Relevant data collected for the present study were family history of retinoblastoma, tumor laterality, treatment for retinoblastoma, reports on invasive cancers, and date and (underlying) cause of death. Only the first cancer after retinoblastoma was included in this study. Time at risk for a second primary cancer began at diagnosis of retinoblastoma and ended on the date of second malignancy diagnosis, emigration, the date last known to be alive, the date of death, or the closing date of the study, whichever came first.
Patients with bilateral disease, a positive family history of retinoblastoma, or a germline mutation in the RB1 gene detected by chromosomal or DNA analysis were classified as hereditary. The remaining patients, those with unilateral retinoblastoma, no family history of retinoblastoma, and no germline mutation detected in the RB1 gene, were classified as having non-hereditary retinoblastoma.
Eligible subjects for the current study included all hereditary retinoblastoma patients from the Dutch retinoblastoma cohort (1862–2005), in whom a germline RB1 mutation was documented. If a retinoblastoma patient had died before DNA-testing could be performed, but a RB1 mutation was determined in the family, the patient was considered to be a carrier of the familial mutation (n = 26). Every affected family member was handled as a single case in the analysis. Of the 1,028 retinoblastoma patients in the Dutch cohort, we identified a total of 410 (39.9%) hereditary cases. Two-hundred eleven patients were excluded because no DNA analysis could be performed (n = 180) or DNA analysis did not detect a RB1 mutation (n = 31). The remaining 199 patients were included in this study (see Fig. 1), of whom 168 were alive at the time of inclusion.
Fig. 1.
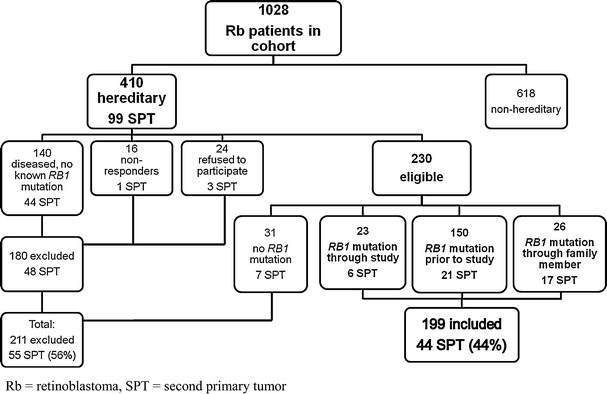
Flow chart showing reasons for inclusion and exclusion of retinoblastoma patients with hereditary retinoblastoma from our cohort. In the total group of 410 hereditary retinoblastoma patients from our cohort, 99 primary tumors (SPT) have been diagnosed. In the flow chart is also depicted in which in- or excluded group these SPT’s have occurred. Percentage is calculated from the total of 99 SPT’s
This study was approved by the Medical Ethics Committees of all participating hospitals, and was conducted in accordance with the principles of the Helsinki declaration.
Mutation screening
Since the beginning of the 1990s all newly diagnosed retinoblastoma patients in the Netherlands undergo germline RB1 mutation analysis. Many patients who were diagnosed before that time underwent DNA-testing between 1990 and 2005, when they were referred to the clinical genetics department (n = 90). Rb patients diagnosed prior to 1990, in whom mutation testing had not yet been performed at the time of the study and who wanted to participate in the study, were invited to undergo DNA-testing and were offered genetic counseling (n = 23).
DNA analysis included direct sequencing of exons 1 and 15, and the RB1 promoter and Denaturing Gradient Gel Electrophoresis (DGGE) analysis of the other exons and flanking intronic sequences. To detect large deletions and duplications Multiplex Ligation-dependent Probe Amplification (MLPA) analysis was performed. If warranted, e.g. when dysmorphic features or mental retardation was noted, karyotyping was performed to detect chromosomal rearrangements. With these techniques we have been able to detect 90% of mutations in familial and/or bilateral cases.
Type of RB1 mutation
In the RB1 gene are several methylated CGA codons known to lead to 11 recurrent nonsense mutations by C>T transitions [14–16]. An important factor in the high recurrence of mutations at these sites was shown to be deamination of 5-methylcytosine [17].
For this study mutations in the promoter, exon 1, missense mutations and deletions of the complete RB1 gene were regarded as low penetrance mutations, based on previous studies [18–21]. Four out of six familial splice mutations were also regarded as low penetrance mutations, based on a diseased eye ratio (DER) of ≤1.5, defined as the total number of affected eyes per family divided by the number of mutation carriers in the family [22, 23]. To exclude possible mosaicism as a cause of milder expression the first mutation carrier in these families was excluded from the analysis.
Statistical methods
We compared the frequency of second primary cancers among hereditary retinoblastoma survivors with specific documented RB1 mutations, and tested for differences using Chi-square tests.
Multivariate Cox regression analysis was performed to quantify the effects of specific RB1 mutations on the risk of second primary malignancies. Therapy, age, laterality, sex, and familial or sporadic occurrence were taken into account as possible confounders (SPSS, Chicago, IL).
Results
Median age of all patients included in the study cohort was 30.0 years (range 1.0–75.0). Of the total of 199 participants, 111 were familial cases and 88 concerned sporadic patients. After a median follow-up time of 30.2 years (range 1.33–76.0), 44 carriers of a RB1 mutation from 31 different families developed a second primary malignancy. Table 1 shows the number of germline RB1 mutations according to type of mutation and lists the number of second primary tumors according to type of mutation. The mutations found in patients who developed a second malignancy were distributed throughout most of the RB1 gene and did not appear to cluster in one region (Fig. 2). There was no correlation between the different types of second malignancies diagnosed in these patients and the type of mutation or the region of the gene where the mutation was located. In the group of retinoblastoma survivors who developed a second malignancy, only nonsense and frameshift mutations, certain splice mutations and large rearrangements were observed.
Table 1.
Number and type of second primary tumor (SPT) by mutation type
| Type of RB1 mutationa | Number of carriers n (%)a |
Number of cases with SPT n (%)b |
Type of SPT | |||
|---|---|---|---|---|---|---|
| Sarcomac | Melanoma | Epithelial cancer | Otherd | |||
| Nonsense/frameshift mutation | 117 (58.8) | 31 (26.5) | 11 | 8 | 10 | 2 |
| Recurrent nonsense mutation | 49 (41.9) | 17 (34.7) | 7 | 7 | 2 | 1 |
| Low penetrance mutation = exon 1 | 7 (6) | 1 (14.3) | ||||
| Splice mutation | 34 (17.1) | 7 (20.6) | 2 | 1 | 4 | 0 |
| Low penetrance mutation | 11 (32.4) | 0 | ||||
| Large rearrangements | 35 (17.6) | 6 (17.1) | 2 | 2 | 2 | 0 |
| Low penetrance mutation | 21 (60) | 1 (4.8) | 1 | |||
| Missense mutation | 11 (5.5) | 0 | 0 | 0 | 0 | 0 |
| Low penetrance mutation | 11 (100) | 0 | ||||
| Promoter mutation | 2 (1) | 0 | 0 | 0 | 0 | 0 |
| Low penetrance mutation | 2 (100) | 0 | ||||
| Total | 199 | 44 (22.1) | 15 | 11 | 16 | 2 |
a Subclassification of the mutation type is shown in italics, percentage as compared to total number of cases with this mutation type
b Percentage of cases with an SPT as compared to total number of cases with this mutation type
c Including soft tissue sarcoma, osteosarcoma, and chondrosarcoma
d Malignant tumor not otherwise specified and brain tumor
Fig. 2.

Graphical representation of RB1 and mutations found among hereditary retinoblastoma subjects diagnosed with a second primary malignancy (n = 44). Exons 1 through 27 are not drawn to scale. Every symbol represents a retinoblastoma subject diagnosed with a second primary malignancy. Black symbols represent sporadic hereditary retinoblastoma subjects. Greyscale coloured symbols represent subjects with familial retinoblastoma. Downward-pointing symbols represent mutations in exons, and upward-pointing symbols represent mutations in introns. The horizontal lines below depict large rearrangements
Table 2 displays the mutations of all patients who developed a second primary malignancy, along with clinical details, listed according to subcategories of germline RB1 mutation type.
Table 2.
Descriptions of retinoblastoma patients with a second primary malignancy according to subcategories of RB1 mutation type
| Subject no.a,b |
Familyc | Number of RB patients in the family without second primary malignancy (age at inclusion) | Site | Mutation descriptiond | Protein | Laterality | First in family | Therapy | Type of second primary malignancye | Second primary malignancy (age at diagnosis) |
|---|---|---|---|---|---|---|---|---|---|---|
| Nonsense or frameshift mutation | ||||||||||
| 1 | F22 | NA | Exon 1 | g.2121dupC | p.Ala22GlyfsX9 | Bil | Yes | RT | Epithelial | B (59) |
| 2 | F11 | 1 (37) | Exon 2 | g.5446G>T | p.Glu54X | Bil | Yes | Enucl | Epithelial | Bl (52) |
| 3 | F12 | 1 (12) | Exon 2 | g.5505_5506dup | p.Ala74GlufsX4 | Bil | Yes | RT + CH | Other | (44) |
| 4 | F16 | 4 (59, 40, 14, 6) | Exon 6 | g.45855delT | p.Leu199TyrfsX2 | Bil | No | RT | Osteosarcoma | (5) |
| 5a | F31 | NA | Exon 8 | g.59695C>T | p.Arg255X | Bil | Yes | RT | Osteosarcoma | (12) |
| 6 | F17 | 2 (4f, 29) | Exon 8 | g.59702_59703dup | p.Asn258ArgfsX7 | Ul | Yes | Enucl | Epithelial | Pa (56) |
| 7 | F17 | Exon8 | g.59702_59703dup | p.Asn258ArgfsX7 | Bil | No | RT | Epithelial | Bl (51) | |
| 8 | F17 | Exon8 | g.59702_59703dup | p.Asn258ArgfsX7 | Bil | No | RT + CH | Epithelial | L (39) | |
| 9a | F4 | 0 | Exon 10 | g.64348C>T | p.Arg320X | Bil | Yes | Enucl | Epithelial | C (49) |
| 10 a | F4 | Exon 10 | g.64348C>T | p.Arg320X | Bil | No | RT | Melanoma | (25) | |
| 11a | F5 | 2 (26, 1f) | Exon 11 | g.65386C>T | p.Arg358X | Bil | Yes | RT + CH | Sarcoma | LMS (41) |
| 12 | F6 | 2 (47, 41) | Exon 12 | g.70261C>T | p.Gln383X | Ul | Yes | Enucl | Epithelial | Bl (62) |
| 13a | F8 | 3 (26, 11, 49) | Exon 14 | g.76430C>T | p.Arg445X | Bil | Yes | Enucl | Epithelial | L (65) |
| 14 a | F8 | Exon 14 | g.76430C>T | p.Arg445X | Bil | No | Enucl | Melanoma | (23) | |
| 15 a | F8 | Exon 14 | g.76430C>T | p.Arg445X | Bil | No | RT | Melanoma | (37) | |
| 16 a | F8 | Exon 14 | g.76430C>T | p.Arg445X | Bil | No | RT | Melanoma | (45) | |
| 17 a | F8 | Exon 14 | g.76430C>T | p.Arg445X | Bil | No | RT | Melanoma | (21) | |
| 18a | F23 | NA | Exon 15 | g.76898C>T | p.Arg467X | Bil | No | RT + CH | Melanoma | (28) |
| 19a | F9 | 1 (54) | Exon 17 | g.78238C>T | p.Arg552X | Bil | Yes | RT | Sarcoma | LMS (33) |
| 20 a | F9 | Exon 17 | g.78238C>T | p.Arg552X | Bil | No | RT + CH | Osteosarcoma | (12) | |
| 21a | F10 | 0 | Exon 18 | g.150037C>T | p.Arg579X | Ul | Yes | Missing | Other | (59) |
| 22 a | F10 | Exon 18 | g.150037C>T | p.Arg579X | Bil | No | RT | Melanoma | (35) | |
| 23a | F24 | NA | Exon 18 | g.150037C>T | p.Arg579X | Bil | No | RT | Osteosarcoma | (10) |
| 24 | F25 | NA | Exon 19 | g.153211T>A | p.Tyr606X | Bil | No | RT | Melanoma | (35) |
| 25 | F13 | 3 (42f, 70, 51) | Exon 20 |
g.156787_156788 delGC |
p.Thr687ProfsX4 | Bil | No | Enucl | Epithelial | O (72) |
| 26 | F13 | Exon 20 |
g.156787_156788 delGC |
p.Thr687ProfsX4 | Ul | No | Enucl | Sarcoma | Unknown location (50) | |
| 27 | F14 | 2 (53, 50) | Exon 21 | g.160832G>T | p.Glu737X | Ul | Yes | Enucl | Epithelial | O (58) |
| 28 | F14 | Exon 21 | g.160832G>T | p.Glu737X | Bil | No | RT | Osteosarcoma | (20) | |
| 29a | F27 | NA | Exon 23 | g.162237C>T | p.Arg787X | Bil | No | RT + CH | Sarcoma | LMS (32) |
| 30a | F28 | NA | Exon 23 | g.162237C>T | p.Arg787X | Bil | No | RT | Sarcoma | Lipo (12) |
| 31 | F26 | NA | Exon 23 | g.162266delC | p.Leu797TyrfsX13 | Bil | No | RT + CH | Osteosarcoma | (26) |
| Splice mutation | ||||||||||
| 32 | F7 | 1 (29) | Exon 13 | g.73869G>A |
p.Gln444Gln exon 13 skipped |
Bil | Yes | RT | Epithelial | L (48) |
| 33 | F20 | NA | Intron 6 |
g.45867G>A/ IVS6 + 1G>A |
n.i. | Bil | No | RT | Epithelial | B (57) |
| 34 | F21 | NA | Intron 14 |
g.76491G>T/ IVS14 + 5G>T |
n.i. | Bil | No | RT | Melanoma | (51) |
| 35 | F19 | NA | Intron 17 |
g.149997G>A/ IVS17 − 1G>A |
n.i. | Bil | No | RT | Sarcoma | Rhab (8) |
| 36 | F1 | 0 | Intron 20 |
g.160729G>C/ IVS20 − 1G>C |
n.i. | Bil | Yes | RT | Epithelial | Bl (62) |
| 37 | F1 | Intron 20 |
g.160729G>C/ IVS20 − 1G>C |
n.i. | Bil | No | RT | Sarcoma | His (31) | |
| 38 | F1 | Intron 20 |
g.160729G>C/ IVS20 − 1G>C |
n.i. | Bil | No | RT | Epithelial | B (43) | |
| Large rearrangements | ||||||||||
| 39 | F15 | 1 (5) | Duplica- tion exon 3 | g.39446-?_39561 + ?dup | n.i. | Ul | Yes | Enucl | Melanoma | (31) |
| 40 | F3 | 2 (45, 9) |
Deletion RB1 |
c.-138-?_27841 + ?del | – | Bil | No | RT | Sarcoma | Rhab (11) |
| 41 | F2 | 2 (17, 15) | Deletion exon 10/11 | Del exon 10 or 10 and 11 | n.i. | Bil | Yes | RT | Melanoma | (31) |
| 42 | F29 | NA | Deletion exon 3-17 | g.39446-?_78279 + ?del | n.i. | Bil | No | RT | Sarcoma | LMS (32) |
| 43 | F30 | NA | Deletion exon 6-17 | g.45799-?_78279 + ?del | n.i. | Bil | No | RT | Epithelial | Seb (38) |
| 44 | F18 | 2 (68, 42) |
Deletion exon 9–27 |
g.61730-?_g177078 + ?del | n.i. | Ul | Yes | Enucl | Epithelial | C (57) |
RB retinoblastoma, NA not applicable (i.e. sporadic retinoblastoma patient, n.i. not investigated), Bil bilateral disease, Ul unilateral disease, RT radiation therapy, CH chemotherapy, Enucl enucleation
B breast cancer, Bl bladder cancer, Pa pancreatic cancer, L lung cancer, C colon cancer, LMS leiomyosarcoma, O ovarian cancer, Lipo liposarcoma, Rhab rhabdomyosarcoma, His histiocytoma, Seb sebaceous adenocarcinoma
a Carriers of a recurrent stop mutation
b Italicized cases represent family members also affected with a second primary malignancy
c Family numbers 1–18: have more affected family members with RB; family numbers 19–31: sporadic retinoblastoma patients
d Reference sequence GenBank #L11910
e Underlined types of second primary malignancies represent tumours diagnosed inside the field of radiation; defined as tumor in the eye lids, orbits, periocular sinuses, temporal bones, or skin overlying the temporal bone region
f Died of other cause than second primary cancer
We assessed the risk of second malignancy in relation to type of mutation by multivariable Cox model analysis, adjusted for age and therapy. This showed that subjects carrying one of the recurrent nonsense mutations had a significantly elevated risk of developing second malignancies, compared to subjects carrying other mutations (hazard ratio [HR], 3.53; [95% confidence interval (CI), 1.82–6.84]; P = .000). Since 5 members of family 8 developed a second malignancy, we did the same analysis while excluding this family. This did not significantly change the outcome (HR, 3.17; [95% CI, 1.50–6.69]; P = .002). Leaving both family 8 and all low penetrance mutations out of the analysis, showed a lower but still statistically significantly increased risk for recurrent nonsense mutations as compared to other mutations (HR, 2.46; [95% CI, 1.14–5.28]; P = .02).
Table 3 shows the recurrent nonsense mutations known in the RB1 gene, and displays which of these mutations are found in our cohort in relation to the number of patients and the number of second primary cancers in these patients.
Table 3.
List of the 11 recurrent RB1 nonsense mutations, the number of patients in our cohort carrying the mutation and the number of patients with this mutation who developed a second primary tumor (SPT)
| Exon | Mutation | Protein | Number of patients in our cohort | Number of patients who developed a SPT |
|---|---|---|---|---|
| 8 | g.59683C>T | Arg.251X | 2 | 0 |
| 8 | g.59695C>T | Arg.255X | 1 | 1 |
| 10 | g.64348C>T | Arg.320X | 5 | 2 |
| 11 | g.65386C>T | Arg.358X | 3 | 1 |
| 14 | g.76430C>T | Arg.445X | 11 | 5 |
| 14 | g.76460C>T | Arg.455X | 8 | 0 |
| 15 | g.76898C>T | Arg.467X | 1 | 1 |
| 17 | g.78238C>T | Arg.552X | 3 | 2 |
| 17 | g.78250C>T | Arg.556X | 5 | 0 |
| 18 | g.150037C>T | Arg.579X | 7 | 3 |
| 23 | g.162237C>T | Arg.787X | 3 | 2 |
| Total | 49 | 17 |
A statistically significantly decreased risk for a second primary malignancy was found in the 52 patients with a low penetrance mutation when compared to other mutations (HR, .19; [95% CI, .05–.81]; P = .025). Of all 34 patients carrying a splice mutation, eleven were carrier of a low penetrance mutation according to our definition, i.e. a DER ≤1.5. None of the carriers of a low penetrance splice mutation developed a second primary cancer. Out of 21 carriers of a deletion involving the whole RB1 gene, just one developed a sarcoma and one out of seven carriers of a mutation in exon 1 developed breast cancer at the age of 59. No second malignancies were observed in carriers of a RB1 promoter or missense mutation (n = 13).
Discussion
Our study is the first to examine the association between specific RB1 germline mutations and the risk of second primary malignancies in a nationwide well-documented cohort. Adjusting for age and therapy, we found a higher second primary malignancy risk for retinoblastoma subjects carrying a recurrent nonsense mutation, and a lower risk for carriers of a low penetrance mutation.
We first compared the risk of second primary malignancies for carriers of recurrent nonsense mutations to all other mutations in the RB1 gene. Because one family (F8) with many family members affected by a second malignancy may have influenced the outcome too much, we excluded this family from the analysis. This still showed a statistically significantly increased risk. It is remarkable that 4 members of this family developed a melanoma. This could be due to common genetic background, though this phenomenon does not hold true for other families. As far as we are aware, the family does not display any signs of dysplastic nevus syndrome. We further hypothesized that the increased risk of second malignancies for recurrent mutations compared to all other mutations may have been caused by a substantial contribution of the lower risk of second malignancies for low penetrance mutations, included in the comparison. Therefore we also left the low penetrance mutations out of the analysis. This still showed a significantly increased risk of second primary cancers for recurrent nonsense mutations. Three recurrent nonsense mutations did not demonstrate any second malignancies in our study cohort (Table 3). Whether these mutations do not lead to a higher second malignancy risk, needs to be clarified in future studies.
What could be the cause of the higher risk for second malignancies in carriers of recurrent nonsense mutations? Nonsense and frameshift mutations are associated with bilateral retinoblastoma and high (>90%) penetrance [19], irrespective of the location of the premature stop mutation. This is attributed to nonsense mediated mRNA decay (NMD): a mechanism of mRNA surveillance that prevents the expression of truncated proteins, by breaking down mutant mRNA containing a premature termination codon [24]. NMD can be beneficial, eliminating truncated transcripts that could lead to proteins with possible dominant negative or gain-of function effects, but may also be harmful when preventing translation of truncated protein that would otherwise still be partly functional [25, 26]. Studies have also shown that NMD efficacy may vary between tissues [27, 28]. An explanation for our findings could be that specific nonsense mutations escape NMD in certain tissues, which will result in the expression of a truncated protein. This truncated protein may either have residual activity resulting in a milder effect or may have a dominant negative effect, as has been described for other tumor-suppressor genes [25, 29]. The higher risk in recurrent nonsense mutation carriers may then be explained by a differential effect of NMD between these mutations and other truncating mutations. How such a differential effect would specifically exist between the two different types of truncating mutations remains to be determined. Alternatively, while the recurrent nonsense mutations result in the loss of a 5-methylcytosine within the gene, this may affect the chromatin structure and/or expression of the gene and thereby increasing the chance of transformation. However, in spite of a clearly elevated risk for recurrent nonsense mutations in our cohort, we cannot rule out that the elevated risk is a chance finding.
Genotype-phenotype correlations of RB1 mutations have been described for specific types of mutations: certain splice mutations, promoter, exon 1 and missense mutations lead to reduced expressivity (unilateral retinoblastoma) and incomplete penetrance (unaffected carriers) of retinoblastoma [18, 19, 21, 23, 30]. This is attributed to a reduction in the amount of normal protein that is produced or to residual activity of mutant protein [18, 19]. Reduced expressivity and incomplete penetrance have also been described for deletions of the complete RB1 gene. This is thought to be caused by co-deletion of adjacent unknown genes leading to a greater chance of apoptosis, when the wildtype allele is lost as the second hit in the tumor [20]. In line with reduced expressivity in the retina, we demonstrated a lower risk of second primary cancers for carriers of these low penetrance mutations. In our study group none of the carriers (n = 24) of a missense, promoter or low penetrance splice RB1 mutation developed a second malignancy, and just one out of 7 carriers with a mutation in exon 1 developed another cancer, i.e. breast cancer at age 59. The latter may also be attributed to the high population risk of breast cancer. A lower risk for second primary malignancies was noted for carriers of a complete deletion of the RB1 gene as well: only one out of 21 carriers developed a second malignancy (i.e. rhabdomyosarcoma at age 11).
Strength of our study is the unique large data set of genotyped survivors of retinoblastoma from a population based cohort. Very few studies on genotype-phenotype relations of RB1 mutations mention second primary cancers. Two studies on mutations in de RB1 gene stated that they could not detect an association between the mutation and manifestation of a second primary cancer or tumor type [21, 31]. These studies included only a few patients with second primary cancers, however. Some limitations of our study should be considered. First, our mutation detection rate was 90% for all familial and bilateral cases at the time of the study. Leaving 10% of hereditary patients out of the analysis may have influenced the outcome. Some mutations may have been missed, because they are present in a mosaic state; others because they might be located deep in an intron. Second, some types of mutations (e.g. promoter mutations) are relatively rare and therefore some mutation type subgroups were small, making it difficult to draw firm conclusions. A third limitation is that RB1 mutation detection has become available just 20 years ago. Although many patients from the Dutch retinoblastoma cohort have been genotyped in the past years, this study still comprises a relatively young group of retinoblastoma subjects; almost 50% of all known patients with hereditary retinoblastoma could be included. Quite a few patients diagnosed with (osteo)sarcoma had already died before it was possible to perform DNA analysis. The exclusion of these (osteo)sarcomas may have limited our ability to detect possible associations between specific mutations and sarcoma risk.
In conclusion, our results suggest a genotype-phenotype correlation for second primary malignancies of retinoblastoma survivors and may impact on long-term surveillance protocols of patients with hereditary retinoblastoma, if confirmed by future studies.
Acknowledgments
The authors would like to thank all patients who participated and Q. Waisfisz, PhD, Department of Clinical Genetics, VU University Medical Center, professor dr. H.P.J. te Riele, Netherlands Cancer Institute and professor dr. D.R. Lohmann, Institut für Humangenetik, Universitätsklinikum Essen, Germany for their valuable contributions to the manuscript. The study was supported by grand VU 2004–3046 from the Dutch Cancer Society.
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Charlotte J. Dommering and Tamara Marees contributed equally to the manuscript.
References
- 1.Mahoney MC, Burnett WS, Majerovics A, Tanenbaum H. The epidemiology of ophthalmic malignancies in New York State. Ophthalmology. 1990;97:1143–1147. doi: 10.1016/s0161-6420(90)32445-4. [DOI] [PubMed] [Google Scholar]
- 2.Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 3.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leiderman YI, Kiss S, Mukai S. Molecular genetics of RB1–the retinoblastoma gene. Semin Ophthalmol. 2007;22:247–254. doi: 10.1080/08820530701745165. [DOI] [PubMed] [Google Scholar]
- 6.Moll AC, Imhof SM, Bouter LM, Kuik DJ, den OW, Bezemer PD, Koten JW, Tan KE. Second primary tumors in patients with hereditary retinoblastoma: a register-based follow-up study, 1945–1994. Int J Cancer. 1996;67:515–519. doi: 10.1002/(SICI)1097-0215(19960807)67:4<515::AID-IJC9>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004;96:357–363. doi: 10.1093/jnci/djh058. [DOI] [PubMed] [Google Scholar]
- 8.Kleinerman RA, Tucker MA, Tarone RE, Abramson DH, Seddon JM, Stovall M, Li FP, Fraumeni JF., Jr Risk of new cancers after radiotherapy in long-term Survivors of retinoblastoma: an extended follow-up. J Clin Oncol. 2005;23:2272–2279. doi: 10.1200/JCO.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 9.Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008;100:1771–1779. doi: 10.1093/jnci/djn394. [DOI] [PubMed] [Google Scholar]
- 10.Woo KI, Harbour JW. Review of 676 second primary tumors in patients with retinoblastoma: association between age at onset and tumor type. Arch Ophthalmol. 2010;128:865–870. doi: 10.1001/archophthalmol.2010.126. [DOI] [PubMed] [Google Scholar]
- 11.Eng C, Li FP, Abramson DH, Ellsworth RM, Wong FL, Goldman MB, Seddon J, Tarbell N, Boice JD., Jr Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst. 1993;85:1121–1128. doi: 10.1093/jnci/85.14.1121. [DOI] [PubMed] [Google Scholar]
- 12.Yu CL, Tucker MA, Abramson DH, Furukawa K, Seddon JM, Stovall M, Fraumeni JF, Jr, Kleinerman RA. Cause-specific mortality in long-term survivors of retinoblastoma. J Natl Cancer Inst. 2009;101:581–591. doi: 10.1093/jnci/djp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marees T, van Leeuwen FE, de Boer MR, Imhof SM, Ringens PJ, Moll AC. Cancer mortality in long-term survivors of retinoblastoma. Eur J Cancer. 2009;45:3245–3253. doi: 10.1016/j.ejca.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Mancini D, Singh S, Ainsworth P, Rodenhiser D. Constitutively methylated CpG dinucleotides as mutation hot spots in the retinoblastoma gene (RB1) Am J Hum Genet. 1997;61:80–87. doi: 10.1086/513898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lohmann DR. RB1 gene mutations in retinoblastoma. Hum Mutat. 1999;14:283–288. doi: 10.1002/(SICI)1098-1004(199910)14:4<283::AID-HUMU2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 16.Valverde JR, Alonso J, Palacios I, Pestana A. RB1 gene mutation up-date, a meta-analysis based on 932 reported mutations available in a searchable database. BMC Genet. 2005;6:53. doi: 10.1186/1471-2156-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560–561. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- 18.Harbour JW. Molecular basis of low-penetrance retinoblastoma. Arch Ophthalmol. 2001;119:1699–1704. doi: 10.1001/archopht.119.11.1699. [DOI] [PubMed] [Google Scholar]
- 19.Lohmann DR, Gallie BL. Retinoblastoma: revisiting the model prototype of inherited cancer. Am J Med Genet C Semin Med Genet. 2004;129C:23–28. doi: 10.1002/ajmg.c.30024. [DOI] [PubMed] [Google Scholar]
- 20.Albrecht P, Ansperger-Rescher B, Schuler A, Zeschnigk M, Gallie B, Lohmann DR. Spectrum of gross deletions and insertions in the RB1 gene in patients with retinoblastoma and association with phenotypic expression. Hum Mutat. 2005;26:437–445. doi: 10.1002/humu.20234. [DOI] [PubMed] [Google Scholar]
- 21.Taylor M, Dehainault C, Desjardins L, Doz F, Levy C, Sastre X, Couturier J, Stoppa-Lyonnet D, Houdayer C, Gauthier-Villars M. Genotype-phenotype correlations in hereditary familial retinoblastoma. Hum Mutat. 2007;28:284–293. doi: 10.1002/humu.20443. [DOI] [PubMed] [Google Scholar]
- 22.Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94:349–354. doi: 10.1007/BF00201591. [DOI] [PubMed] [Google Scholar]
- 23.Scheffer H, van der Vlies P, Burton M, Verlind E, Moll AC, Imhof SM, Buys CH. Two novel germline mutations of the retinoblastoma gene (RB1) that show incomplete penetrance, one splice site and one missense. J Med Genet. 2000;37:E6. doi: 10.1136/jmg.37.7.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 25.Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 26.Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE. NMD: RNA biology meets human genetic medicine. Biochem J. 2010;430:365–377. doi: 10.1042/BJ20100699. [DOI] [PubMed] [Google Scholar]
- 27.Resta N, Susca FC, Di Giacomo MC, Stella A, Bukvic N, Bagnulo R, Simone C, Guanti G. A homozygous frameshift mutation in the ESCO2 gene: evidence of intertissue and interindividual variation in Nmd efficiency. J Cell Physiol. 2006;209:67–73. doi: 10.1002/jcp.20708. [DOI] [PubMed] [Google Scholar]
- 28.Linde L, Boelz S, Neu-Yilik G, Kulozik AE, Kerem B. The efficiency of nonsense-mediated mRNA decay is an inherent character and varies among different cells. Eur J Hum Genet. 2007;15:1156–1162. doi: 10.1038/sj.ejhg.5201889. [DOI] [PubMed] [Google Scholar]
- 29.Karam R, Carvalho J, Bruno I, Graziadio C, Senz J, Huntsman D, Carneiro F, Seruca R, Wilkinson MF, Oliveira C. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene. 2008;27:4255–4260. doi: 10.1038/onc.2008.62. [DOI] [PubMed] [Google Scholar]
- 30.Zhang K, Nowak I, Rushlow D, Gallie BL, Lohmann DR. Patterns of missplicing caused by RB1 gene mutations in patients with retinoblastoma and association with phenotypic expression. Hum Mutat. 2008;29:475–484. doi: 10.1002/humu.20664. [DOI] [PubMed] [Google Scholar]
- 31.Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. The spectrum of RB1 germ-line mutations in hereditary retinoblastoma. Am J Hum Genet. 1996;58:940–949. [PMC free article] [PubMed] [Google Scholar]