

Abstract
Helix-loop-helix (HLH) family transcription factors regulate numerous developmental and homeostatic processes. Dominant-negative HLH (dnHLH) proteins lack DNA-binding ability and capture basic HLH (bHLH) transcription factors to inhibit cellular differentiation and enhance cell proliferation and motility, thus participating in patho-physiological processes. We report the first structure of a free-standing human dnHLH protein, HHM (Human homologue of murine maternal Id-like molecule). HHM adopts a V-shaped conformation, with N-terminal and C-terminal five-helix bundles connected by the HLH region. In striking contrast to the common HLH, the HLH region in HHM is extended, with its hydrophobic dimerization interfaces embedded in the N- and C-terminal helix bundles. Biochemical and physicochemical analyses revealed that HHM exists in slow equilibrium between this V-shaped form and the partially unfolded, relaxed form. The latter form is readily available for interactions with its target bHLH transcription factors. Mutations disrupting the interactions in the V-shaped form compromised the target transcription factor specificity and accelerated myogenic cell differentiation. Therefore, the V-shaped form of HHM may represent an autoinhibited state, and the dynamic conformational equilibrium may control the target specificity.
Keywords: carcinogenesis, TGF-β signal, transcription factor, X-ray crystallography
Introduction
The helix-loop-helix (HLH) proteins are central regulators in a wide variety of developmental and homeostatic processes (Olson and Klein, 1994). Especially, basic helix-loop-helix (bHLH) transcription factors play key roles in regulating gene expression, cell cycle control, and developmental processes, by binding to the ‘E box’ in the promoters of tissue-specific genes through homo- and heterodimer formation (Murre et al, 1989; Blackwell et al, 1990; Kreider et al, 1992; Zebedee and Hara, 2001). The HLH proteins are divided into seven classes, based on the presence of a DNA-binding region as well as other appended motifs/domains (Massari and Murre, 2000). The class-I bHLH proteins, such as E12, E47, HEB, and E2-2, are ubiquitously expressed in many tissues. In contrast, the class-II bHLH proteins, such as MyoD, NeuroD, and Hes, exhibit tissue-specific expression and form heterodimers with the class-I bHLH proteins to regulate distinct developmental pathways, such as myogenesis, neurogenesis and lymphopoiesis (Weintraub et al, 1990; Lassar et al, 1991; Weintraub et al, 1991; Weintraub, 1993; Parkhurst and Meneely, 1994; Lee et al, 1995; Shen and Kadesch, 1995; Ma et al, 1996; Rawls and Olson, 1997). The class-III HLH proteins include the Myc family of transcription factors, which contain a leucine zipper (LZ) adjacent to the HLH motif. The class-IV HLH proteins include Mad, Max, and Mxi, which are capable of dimerizing with the Myc proteins and with each other (Blackwood and Eisenman, 1991; Ayer et al, 1993; Zervos et al, 1993). The class-V HLH proteins are represented by the Id family proteins (Id1, Id2, Id3, and Id4), which do not contain the basic region prior to the HLH motif and lack DNA-binding ability. The Id family proteins form heterodimers with the bHLH proteins, and inhibit their functions in a dominant-negative manner (Benezra et al, 1990; Sun et al, 1991; Yokota and Mori, 2002; Perk et al, 2005). The class-VI HLH proteins specifically contain a Pro residue in their basic region (Klambt et al, 1989; Rushlow et al, 1989). The class-VII HLH proteins are characterized by the bHLH–PAS domain, and include the aromatic hydrocarbon receptor and its nuclear translocator, as well as HIF1α, SIM, AhR, ARNT, and circadian clock-related factors (Crews, 1998).
The Id family proteins of the dominant-negative class-V HLH (dnHLH) have attracted strong medical interest, based on the findings that deregulated Id activity is tumourigenic and contributes to malignancy, such as loss of differentiation, unrestricted proliferation, enhanced cell motility, and neoangiogenesis (Perk et al, 2005). Elevated expression of Id genes has been observed in carcinomas of various origins as well as melanomas and leukaemias (Ishiguro et al, 1995; Fong et al, 2004; Han et al, 2004; Wang et al, 2004). While the Id genes themselves are not canonical oncogenes, their overexpression affects key oncogenic pathways involving Ras, Myc, and ETS (Perk et al, 2005). However, the structures of the Id proteins and the mechanisms controlling their activities have remained unclear.
Human homologue of murine maternal Id-like molecule (HHM), also known as cyclin D1-binding protein (DIP1), is a member of the dnHLH family, but it is larger than the Ids and includes a putative LZ motif and an acidic C-terminal region (Terai et al, 2000). By definition, HHM associates with cyclin D1 to regulate the G1/S-phase progression of hepatocytes, probably through the Rb pathway (Xia et al, 2000). Intriguingly, HHM exerts opposite effects on cells, depending on the cellular contexts. HHM was shown to be involved in the growth regulation of hepatocytes and the progression of hepatocellular carcinomas. In HHM-knockout mice, liver regeneration after partial hepatectomy was attenuated, as compared with that in wild-type mice (Ma et al, 2006). In addition, HHM expression is increased in the early phases of hepatocarcinogenesis, and HHM accelerates S-phase entry in HepG2 hepatocellular carcinoma cells (Terai et al, 2000). These observations indicate positive regulatory roles of HHM in proliferation of liver cells. In contrast, HHM-knockout mice often develop liver tumours (Sonnenberg-Riethmacher et al, 2007), and transgenic mice overexpressing HHM in the liver are less susceptible to chemical hepatocarcinogenesis (Ma et al, 2006). These observations indicate that HHM has tumour-suppressor functions in the liver. However, the mechanism underlying these opposite roles of HHM in the regulation of cell proliferation and tumour progression, which are dependent on the cellular context, remains to be elucidated. Recently, HHM was found to disrupt the physical interaction of specific transcription factors with R-Smads, to inhibit TGF-β signalling in a cellular response-specific manner (Ikushima et al, 2008). Oligodendrocyte transcription factor 1 (Olig1), a class-II bHLH protein, was identified as one of the Smad-binding transcription factors inhibited by HHM (Ikushima et al, 2008). We have observed that Olig1 regulates the expression of the TGF-β controlled genes that enhance cell motility and migration (Motizuki M & Miyazawa K, unpublished results). In contrast to the Id proteins, which interact with the ubiquitously expressed class-I bHLH transcription factors, HHM associates with the tissue-specific class-II bHLH transcription factor Olig1 to regulate Smad-dependent transcription, suppressing tumour progression.
To understand the regulation mechanism of the dnHLH proteins, we solved the crystal structure of full-length HHM at 2.5 Å resolution. This is the first structure of a dnHLH, as well as the first presentation of the free-form of an HLH transcription regulator, devoid of its dimeric counterpart. The HHM structure adopts a V-shaped conformation composed of N- and C-terminal five-helix bundles. The hydrophobic dimerization interfaces of the HLH region are embedded in these helix bundles, and are thereby protected from nonspecific interactions. Combined with biochemical, physicochemical, and cell biological analyses, we propose that the present crystal structure of HHM represents an autoinhibited state, and the slow equilibrium with the partially unfolded conformation enables the fine-tuning of the specificity for the target transcription factor.
Results and Discussion
Structure determination
The crystal structure of HHM (360 residues, molecular weight 40 kDa) was determined by the multiple anomalous diffraction method, using a selenomethionine-labelled crystal. We first obtained experimental phases to 4.0 Å resolution using a SeMet-labelled crystal, as previously described (Seto et al, 2009), but the quality of the resulting electron density map was quite low and the assignment of the amino-acid residues was virtually impossible. The addition of a sulfhydryl-specific reagent, p-chloromercuribenzoic acid (PCMB), to the protein sample prior to crystallization improved the resolution of the crystal to 3.5 Å, which enabled the preliminary interpretation of the electron density map. The exposed Cys residues may disturb the crystal packing, thus reducing the crystal quality. On the basis of this preliminary structural model, we substituted the Cys residues exposed on the molecular surface (Cys198 and Cys300; Supplementary Figure S1) with Ser, which further improved the resolution to 2.5 Å. The final model was refined against the diffraction data extending to 2.5 Å resolution with crystallographic R/R free=22.2/26.1%, by refinement of the individual B-factor and TLS tensor parameters. The model contains one molecule in the asymmetric unit, in which residues 1–15, 41–44, 139–150, 201–228, and 329–333 are structurally disordered.
Overall structure
HHM adopts an all α-helical structure consisting of two-helix bundles, which are arranged in a V-shaped conformation (Figure 1). Hereafter, we divide the HHM structure into the following three regions: the N-terminal helix bundle (N-bundle, residues 16–138), the HLH region (residues 151–200), and the C-terminal helix bundle (C-bundle, residues 229–360; Figure 1). The HLH region of HHM shares sequence homology with the other HLH proteins and consists of helices α5 and α6, which correspond to the first and second α-helices, respectively, of the canonical HLH motif and are connected by the short loop L5. The N-bundle consists of helices α1 to α4 and forms extensive hydrophobic interactions with helix α5 of the HLH region (Figure 2A). The C-bundle consists of helices α7 to α10 and forms extensive hydrophobic interactions with helix α6 of the HLH region (Figure 2B). The loop regions connecting the N-bundle (residues 139–150) and the C-bundle (residues 201–228) to the HLH region are structurally disordered.
Figure 1.

Structure of HHM. (A) Schematic representation of the HHM domain architecture. (B) Overall structure of HHM. (C) Schematic diagram of the secondary structures of HHM. In all of the panels, the same colour code as in panel A is used.
Figure 2.

Intramolecular interactions in free-standing HHM. (A) Hydrophobic interface between α5 and the N-bundle. (B) Hydrophobic interface between α6 and the C-bundle. (C) Intramolecular interactions between the N terminus of α6 and the N- and C-bundles at the edge of the V-shape. (D) van der Waals interactions involving the conserved NKAAA motif, stabilizing the V-shaped conformation to support the interactions between the HLH region and the N- and C-bundles. In all of the panels, the same colour code as in panel A is used.
Previous studies suggested that the acidic domain and the putative LZ motif immediately follow the HLH motif, and these regions of HHM may be involved in intermolecular interactions (Hwang et al, 1997; Terai et al, 2000). This acidic domain is included in loop L6, which is between the HLH region and the C-bundle, and is mostly disordered in the present crystal structure. Moreover, the conserved Leu and Ser/Cys residues (Leu240, Leu247, Cys254, and Leu261) in this putative LZ motif on helix α7 participate in hydrophobic interactions with the core of the C-bundle, and do not form a canonical LZ structure (Supplementary Figure S2).
Interactions between the HLH and helix bundles in the free-standing HHM
In the present structure of HHM, helix α5 and loop L5 of the HLH region only interact with the N-bundle, while the N terminus of helix α6 forms extensive interactions with both the N- and C-bundles (Figure 2A and C, and Supplementary Figure S3). Especially, the N terminus of helix α6 harbours a sequence conserved in the HHM orthologues from various species, representing the 169NKAAA173 motif, which is likely to be important for the interactions between the HLH region and the N- and C-bundles (Figure 2C and D).
This conserved NKAAA motif reinforces the interactions between the N- and C-bundles. The Nδ atom of the Asn169 side chain, which is located at the N terminus of helix α6, hydrogen bonds with the side-chain carboxyl group of Asp275 and the main chain carbonyl oxygen of Val271 in helix α8 of the C-bundle (Figure 2C). On the other hand, the Oδ atom of Asn169 hydrogen bonds with the main chain amide group of Leu114 in helix α4 of the N-bundle (Figure 2C). Furthermore, the side-chain amino group of Lys170 hydrogen bonds with the C-terminal carbonyl oxygen atoms of helix α7 in the C-bundle (Figure 2D). Finally, the side-chain methyl groups of the three consecutive Ala residues (Ala171, Ala172, and Ala173), which form the first turn of helix α6, closely pack against the hydrophobic core of the C-bundle (Figure 2D). Therefore, these interactions seem to be important for HHM to adopt the V-shaped conformation.
In addition to the interactions described above, minor interactions that are independent of the HLH region occur between the N- and C-bundles (Figure 2C). The side-chain carboxyl group of Asp275 hydrogen bonds with the Oγ atom of Thr113 and the main chain amide groups of Ile112 and Thr113. The Cγ atom of Val271 makes a van der Waals contact with the Cα atom of Gly111. Gly111, Val271, and Asp275 are also conserved in the HHM orthologues from various species. These interactions anchor the N termini of helices α4 and α8, thereby stabilizing the V-shaped structure.
Structural comparison between the HLH motifs of HHM and canonical bHLH transcription factors
The canonical structures of the bHLH transcription factors reported to date form homo- or heterodimers through the conserved hydrophobic residues on the amphiphilic α-helices, H1 and H2 (Longo et al, 2008; Figure 3A). The C-terminal halves of helices H1 and the N-terminal halves of helices H2 form a short four-helix bundle, while the N-terminal halves of helices H1 provide a basic DNA-binding interface and the C-terminal halves of helices H2 form a two-helix bundle structure (Figure 3B). These dimeric DNA-bound structures of the bHLH transcription factors are considered as the ‘active forms’, which can activate the transcription of specific genes.
Figure 3.

Active dimer formation of HLH proteins. (A) The canonical heterodimer structure of the E47 and NeuroD1 bHLH transcription factors bound to DNA. (B) Typical dimerization interface of the E47 and NeuroD1 bHLH transcription factor complex, divided into five sections based on the hydrophobic interactions. (C) Sequence alignment of HLH proteins. Sections in the dimerization interface are coloured grey. The basic residues that interact with DNA are coloured blue. (D) Schematic diagram of each section in the dimerization interface between E47 and NeuroD1. (E) Putative schematic diagram of each section in the dimerization interface between HHM and Olig1.
In contrast to these dimeric HLH proteins, the crystal structure of the free-standing HHM revealed that HHM does not form a dimer, and the conserved hydrophobic residues of helices α5 and α6 (corresponding to H1 and H2 in the canonical bHLH, respectively) separately participate in the hydrophobic core formation with the N- and C-bundles (Figure 2A and B). The arrangement of helices α5 and α6 is quite different from that of H1 and H2 observed in the canonical HLH transcription factors (Figures 1B and 3A). There is no contact between helices α5 and α6, whereas helices H1 and H2 of the dimerized HLH transcription factors form intramolecular interactions. Instead, the HLH region of HHM bridges the N- and C-bundles, stabilizing the V-shaped conformation (Figure 1B).
Conservation in the HLH region allows active heterodimer formation in HHM
It is unlikely that the HLH region in the present V-shaped HHM can interact with another HLH protein without undergoing a structural change, as its molecular surface is mainly hydrophilic, and there is no hydrophobic cluster on the surface suitable for the interactions (Supplementary Figure S4). On the basis of the sequence similarity between the bHLH domains of transcription factors and the dnHLH domains of the Id family proteins, the dnHLH and bHLH domains are considered to form a heterodimer, similar to the active forms of the bHLH transcription factors (Wibley et al, 1996). By analogy, can the HLH region of HHM also form a similar heterodimer to those of the transcription factors?
Chavali et al (2001) proposed some of the important positions for the residues involved in the dimeric stability and the functional specificity of the DNA-bound ‘active form’ of HLH proteins. Especially, positions 8′ and 11′ in helix H1 and 4″, 5″, 8″, 11″, 12″, 15″, and 19″ in helix H2 are important for the stable packing of the core residues, at the interface of the short four-helix bundle and two-helix bundle structures (Figure 3C). Here, we divided the dimerization interface into five sections, from I to V (Figure 3B and C). These sections are depicted schematically in Figure 3D, using the complex structures of NeuroD1 and E47 (Longo et al, 2008) as an example.
The HLH region of HHM shares amino-acid sequence similarity with the bHLH transcription factors, as well as other dnHLH transcriptional regulators (Figure 3C). In HHM, sections I to V are also occupied by similar amino acids to those observed in the canonical HLH proteins (Figure 3E). In sections I and II, positions 4″ and 8″ of HHM are replaced by larger residues (Met and Asn) as compared to the canonical HLH proteins, while the corresponding interaction partners (positions 8′ and 11′) are replaced by smaller residues (Val and Ala; Figure 3D and E). Therefore, upon binding to the target transcription factors, the HLH region of HHM can form the heterodimeric ‘active’ structure, with the interface stabilized by these complementary residues. Again, it should be noted that these putative interface residues of HHM are involved in the core formation of the N- and C-bundles in the present free-standing structure.
In addition, position 1″ in helix H2 is highly conserved as Lys or Arg in the bHLH transcription factors (Figure 3C), which coincides with the observations that the basic residue at this position interacts with the backbone phosphates of DNA in the reported crystal structures. In contrast, this position is replaced by Ala (the last Ala173 in the NKAAA motif) in HHM. This substitution seems to be reasonable, as HHM no longer interacts with DNA. In the case of Id3, another member of the dnHLH family, this position is occupied by a non-basic Gln residue.
Furthermore, in many HLH proteins, including Myc, Max, and the Id family proteins, the amino-acid sequences of the loop region between helices H1 and H2 also share significant similarity, although they are not directly involved in dimer formation. The third and penultimate positions of the loop region are conserved as small hydrophobic residues (Figure 3C), forming the core of the loop region in the three-dimensional structure (Figure 3A). Thus, these conserved residues are important for the active dimer formation. In HHM, these positions are also conserved as small hydrophobic residues (Ile165 and Ala171). Moreover, the last four residues of the loop region (169NKAA174, the first four residues of the NKAAA motif), which form the N-terminal turn of helix α6 (H2) in the present structure, share considerable sequence similarity with the loop region of the bHLH transcription factors, including v-Myc (EKAA) and Max (EKAS) (Figure 3C). Although HHM is bound to the class-II bHLH transcription factors and thus may correspond to the class-I HLH proteins, we hypothesize that this loop region of HHM may form a similar structure to those of the class-III and IV transcription factors, such as Myc and Max (Nair and Burley, 2003). Therefore, the present helical structure (α6) of these four N-terminal residues of HHM may be restructured into the loop conformation upon binding with the target transcription factors.
On the basis of these observations, we constructed a docking model of Olig1–bHLH and the HLH region of HHM (Figure 4A). In HHM, helix H2 is longer than that of the canonical HLH structure, suggesting that the C terminus of helix H2 of HHM may become disordered upon complex formation. Similarly, the basic region of Olig1 in the complex may be disordered without bound DNA. Previous in vitro experiments demonstrated that the HLH region of HHM alone is sufficient to exclusively interact with the HLH region of the class-II bHLH transcription factor, Olig1 (Ikushima et al, 2008), supporting this docking model. Therefore, although the complex structure of the HLH regions of HHM and Olig1 is still not available, we propose that HHM disrupts the Smads–Olig1 complex by forming an HHM–Olig1 heterodimer via the HLH regions, as in this docking model. Furthermore, this model suggests that the V-shaped form of HHM should undergo a drastic structural change to form this heterodimeric complex.
Figure 4.

Conformational transition of HHM upon association with the target transcription factors. (A) Docking model of Olig1–bHLH and the HLH region of HHM. The regions that are presumably disordered are semitransparent. (B) Model of the conformational transition of HHM upon association with and dissociation from the target transcription factors.
Equilibrium between the V-shaped and relaxed conformations
To investigate the stability of the V-shaped form of HHM in solution, we incorporated two TEV protease recognition sites into the disordered loops L4 and L6 of the N-terminally GST-tagged HHM (Figure 5A), to enable the separation of the GST-tagged N-bundle, the HLH region, and the C-bundle. We then performed a GST pull-down assay following proteolysis with TEV protease (Figure 5B). If the tertiary interactions between the HLH region and the N- and C-bundles are tight enough to form the stable V-shaped structure, then the C-bundle (and HLH region) will be pulled down together with the GST-tagged N-bundle. Unexpectedly, the HLH region and the C-bundle were not co-precipitated with the N-bundle after the cleavage at loops L4 and L6 (Figure 5B). Although we cannot exclude the possibility that the cleavage of the loops increases the entropy of the unfolded state, this result suggests that the N- and C-bundles and the HLH region spontaneously dissociate, despite the absence of the binding target.
Figure 5.

Conformational equilibrium of HHM. (A) Schematic representation of the intact GST–HHM and that with TEV sites. (B) The GST pull-down assay. After the pull-down, the samples were resolved by SDS–PAGE and the protein bands were stained with Coomassie Brilliant Blue. (C) The sedimentation coefficient profiles of wild-type (top), P166Y mutant (middle), and N169E mutant (bottom) HHM. (D) Representative results of the sedimentation equilibrium experiment. The absorbance at 280 nm versus the radius and the best-fit curve are shown in the bottom panel. The residuals of the fit from the data are shown in the upper panel.
To further investigate the dynamics of the HHM molecule in solution, we performed analytical ultracentrifugation experiments of HHM (Figure 5C and D, and Supplementary Figure S5). The results of the sedimentation velocity experiment revealed the presence of two molecular species, with sedimentation coefficients of 2.6 S and 3.3 S in solution (Figure 5C, top panel). The sedimentation coefficient depends on both the molecular weight and shape of a protein molecule, and thus the result suggested two possibilities: HHM exists in equilibrium between monomer and dimer, or between two distinctive conformations of monomers. To clarify this point, we further performed sedimentation equilibrium experiments, which would determine the exact molecular weight of the protein. The results indicated that HHM exists as a particle with a molecular weight of 39.4±1.3 kDa (Figure 5D), which is in good agreement with the monomeric mass of HHM calculated from its amino-acid sequence (40.26 kDa). Therefore, these results clearly indicated that the HHM molecule exists in slow equilibrium between two distinctive conformations of monomers, with sedimentation coefficients of 2.6 S and 3.3 S, respectively.
Next, to identify these HHM conformers, we estimated the sedimentation coefficient of the V-shaped crystal structure of HHM, using the programme HYDROPRO (de la Torre et al, 2000). The result indicated that the theoretical sedimentation coefficient of V-shaped HHM is 3.5 S, which is consistent with the second peak (i.e., 3.3 S) in the sedimentation velocity experiment (Figure 5C, top panel). Furthermore, given the results of the pull-down assay of TEV-cleaved HHM, the first peak may correspond to a partially unfolded conformation, lacking both of the interactions between the HLH region and the N- and C-bundles (Figure 4B). Without these interactions, HHM may adopt a flexible and extended conformation with a higher frictional coefficient, which is consistent with the smaller sedimentation coefficient than that of the V-shaped form. Hereafter, we denote this partially unfolded conformation of HHM as the ‘relaxed’ form. Taken together, the HHM molecule exists in slow equilibrium between the V-shaped and relaxed forms. In the relaxed form, the HLH region is released from the N- and C-bundles and may readily interact with the target transcription factors, such as Olig1 (Figure 4B).
We also performed MD simulations of the N- and C-bundles lacking the HLH region, which suggested a potential domain rearrangement following the relaxed state of HHM (Supplementary Figure S7 and Supplementary Discussion).
Autoinhibited form of HHM as the transcription regulating factor
On the basis of the results from the above docking model, the pull-down assays, and the ultracentrifugation analyses, the V-shaped form of HHM may represent an inactive, autoinhibited state. In solution, this V-shaped autoinhibited form of HHM is spontaneously and reversibly converted into the relaxed form, which can readily interact with its target bHLH proteins (Figure 4B, upper panel). Upon binding to class-II bHLH transcription factors, the HLH region of HHM then changes its conformation to adopt the canonical HLH active dimer structure (Figure 4B, lower panel). In the autoinhibited form, the conserved hydrophobic residues in the HLH region, which may compose the molecular interface in the active heterodimeric complex, alternatively interact with the N- and C-bundles to form their hydrophobic cores. Therefore, these N- and C-bundles may act as a cradle to prevent the hydrophobic residues of the HLH region from nonspecifically binding with other proteins. The dynamic equilibrium between the autoinhibited and relaxed forms of HHM may modulate the population of the complex with the target transcription factor, thereby tuning the expression levels of the target genes (Figure 4B).
Previous analyses revealed that HHM is widely expressed in differentiated cells in adults. Thus, the transcription suppressing activity of HHM may not be controlled by its expression level, but by the dynamic equilibrium between the autoinhibited and relaxed forms. In contrast, the expression of the Id family proteins is robustly regulated, and is induced in response to extracellular signalling molecules, including bone morphogenetic proteins (Miyazono et al, 2010). As the Id family proteins are stronger antagonists of the DNA-binding activity of the target transcription factors than HHM (Terai et al, 2000), their activities may be principally controlled at the expression level. Consistently, the amino-acid sequences of the Id family proteins suggest that they lack the extra domain that may act as the cradle in HHM.
Mutations that disrupt the interactions between HLH and the N- and C-bundles compromise the transcription factor specificity of HHM
To functionally examine the above hypothesis, we created four HHM mutants designed to weaken the interactions between the HLH and the N- and C-bundles. In the first mutant, Pro166, which is located in the L5 loop connecting helices α5 and α6 (Figure 2D), was mutated to a bulky Tyr (HHM-P166Y) to destabilize the V-shaped conformation. In the second mutant, Asn169, which resides at the N terminus of helix α6 and interacts with both the N- and C-bundles (Figure 2C), was mutated to Glu (HHM-N169E) to destabilize the association between α6 and α4/8. Sedimentation velocity experiments were performed to verify that these mutants do not form aggregates (Figure 5C, middle and lower panels). The results also showed that the mutants have sedimentation coefficients of 2.5–2.6 S, corresponding to that of the relaxed form of wild-type HHM. Thus, these mutants may originally adopt the relaxed form, as expected. In the third and fourth mutants, Val271 and Val278 on α8 in the C-bundle were mutated to Phe and Arg, respectively (HHM-V271F, HHM-V278R). The expression levels of these mutants in COS7 cells were similar to that of the wild type, suggesting that the overall structure or stability is maintained. We then examined the physical interactions of these mutants with HLH proteins (Olig1, NeuroD1, and Id2) as well as cyclin D1 (Figure 6). The wild-type HHM and all of the mutants almost equally interacted with Olig1 and cyclin D1. In contrast, the wild-type HHM only weakly interacted with NeuroD1 and Id2, while the four mutants more efficiently interacted with these non-cognate HLH proteins. These mutations thus relaxed the binding specificity for HLH transcription factors.
Figure 6.

Physical interactions of HHM mutants with HLH proteins and cyclin D1. COS7 cells were transfected with the indicated plasmids (FLAG-tagged HHM or HHM mutants and 6myc-tagged Olig1, NeuroD1, Id2, or cyclin D1). HHM and its mutants were immunoprecipitated (IP), and the co-precipitated proteins were visualized by immunoblotting (top panel). Input of each target protein (second panel), HHM and its mutants (third panel) are also shown. Representative data from three independent experiments are presented. HHM-bound proteins are quantified at the bottom. Error bars represent s.d.
We next examined whether the loss of strict binding specificity in HHM mutants affects cellular process using an in vitro cell differentiation system. Myoblastic C2C12 cells undergo myogenic differentiation when they are cultured with a low concentration of serum (Bains et al, 1984). The adenoviral constructs bearing HHM or its mutants were used to infect the cells for ectopic expression, followed by induction of differentiation. Myogenic differentiation was assessed by mRNA expression of myosin heavy chain as well as myogenin (Figure 7). At 12 h after the induction of differentiation, marker expression was not significantly different among samples. At 24 h after induction, wild-type HHM did not significantly affect myogenic differentiation compared with empty vector control. In contrast, the P166Y, N169E, V271F, and V278R mutants all enhanced expression of myosin heavy chain and myogenin. Thus, these HHM mutants appear to accelerate myogenic cell differentiation, possibly by acting as dnHLH proteins with relaxed binding specificity. At present, the precise mode of action remains to be elucidated. Myogenic differentiation is driven by the active complex formation between class-I E12/47 and class-II MyoD (Lassar et al, 1991). We observed that HHM and its mutants failed to interact with MyoD (Supplementary Figure S6). In contrast, HHM mutants, but not wild-type HHM, efficiently interacted with Id2 (Figure 6). Id proteins are known to disrupt E12/E47–MyoD complex to inhibit its transcriptional activity (Benezra et al, 1990). Thus, the HHM mutants are likely to bind and sequester Ids, which originally suppress the E12/47–MyoD complex, accelerating myogenic differentiation by the reformation of the active complex between E12/47 and MyoD. At 36 h, difference was less clear in expression of myogenin and not evident in expression of myosin heavy chain (data not shown).
Figure 7.

Accelerated myogenic differentiation by HHM mutants. C2C12 cells were infected with adenoviruses carrying empty vector, HHM or its mutants. After 24 h, the cells were cultured in differentiation medium. Twelve or twenty-four hours later, the cells were collected and measured for expression of myogenic markers, myosin heavy chain (MHC), and myogenin. Expression of HHM at 24 h is also shown. Data were normalized by expression of hypoxanthine phosphoribosyltransferase 1. Error bars represent s.d. *P<0.01 compared with both Adeno-null and HHM wild type.
Altogether, these findings suggest that the V-shaped conformation of the free-form HHM not only has an autoinhibitory function, but also controls the transcription factor binding activity to enhance the factor specificity.
Concluding remarks
The present structural and functional studies on HHM suggested that the V-shaped form of HHM is the autoinhibited state for transcriptional regulation, and that the dynamic equilibrium between the V-shaped and relaxed forms controls the transcription suppressing activity of HHM (Figure 4B). Thus, HHM appears to play important roles in cellular regulation, including suppression of tumour progression by controlling activities of HLH transcription factors, such as Olig1, through conformational transition. Our present study also highlights the impact of regulation of binding specificities of HLH proteins, to ensure successful coordination of cellular responses mediated by a network of HLH proteins, total understanding of which may explain the patho-physiological processes of dnHLH proteins.
Materials and methods
Sample preparation and crystallization
The selenomethionine-derivatized HHM (SeM-HHM) proteins were prepared as previously described (Seto et al, 2009), with slight modifications as follows: (i) the reducing reagent (5 mM β-mercaptoethanol) was replaced by 10 mM tris(2-carboxyethyl)phosphine (TCEP), and (ii) the final gel filtration chromatography purification step was omitted. The purified protein sample was treated with PCMB, to prevent the formation of nonspecific intermolecular disulphide bonds. The stoichiometry of the mercury and the sulphydryl groups in the protein monomer was adjusted to 1:1. Subsequently, the PCMB-treated protein solution was concentrated to 10 mg/ml. The crystals of PCMB-treated SeM-HHM were prepared as previously described (Seto et al, 2009).
The C198S/C300S mutant of HHM (HHM-CS) was prepared by site-directed mutagenesis using a QuikChangeTM Site-Directed Mutagenesis kit (Stratagene), according to the manufacturer’s instructions. The HHM-CS protein was also prepared as described above, without the PCMB treatment step. The crystals of HHM-CS were obtained by the sitting-drop vapour diffusion method. The drops were prepared by mixing equal volumes of a 10 mg/ml HHM-CS solution and a reservoir solution, containing 100 mM imidazole (pH 8.0), 500 mM (NH4)2HPO4, and 100 mM NaCl. The crystals grew to a size of 0.3 mm×0.3 mm×0.5 mm within a week. The crystals were transferred stepwise to the harvesting solution, 200 mM potassium citrate, 10 mM TCEP and 13% (w/v) PEG3350, containing 35% (v/v) xylitol as a cryoprotectant, and were flash-cooled in a nitrogen stream at 100 K.
Data collection, processing, and structure determination
All diffraction data sets were collected at the station NW12A at KEK PF-AR (Tsukuba, Japan). Data sets were processed with the HKL2000 suite (HKL Research). The statistics of the data processing are summarized in Tables I and II. The structure determination of wild-type SeM-HHM by the MAD method was performed as previously described (Seto et al, 2009). The phase calculation statistics are summarized in Table I. The resulting initial model was manually modified to fit into the electron density maps by the programs O (Jones et al, 1991) and Coot (Emsley and Cowtan, 2004).
Table 1. Data collection and phasing statistics.
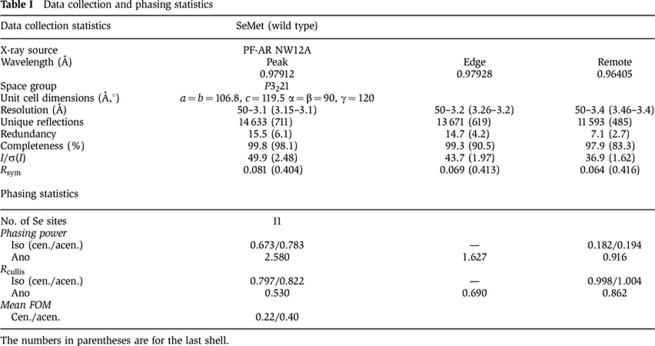
The numbers in parentheses are for the last shell.
Table 2. Data collection and structure refinement statistics.
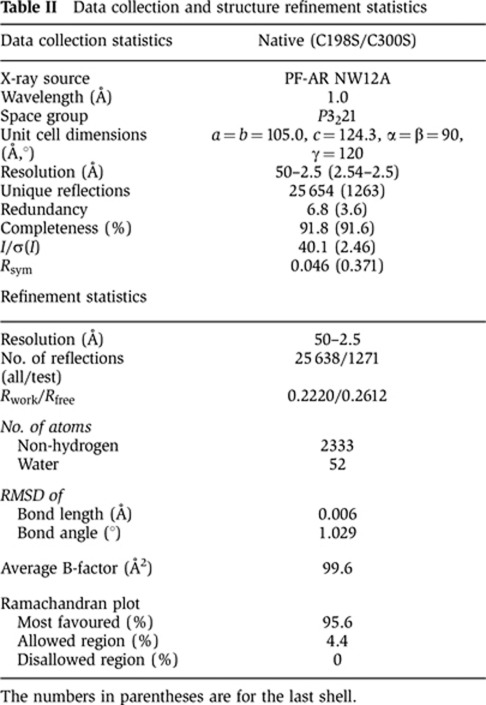
The numbers in parentheses are for the last shell.
The crystals of the HHM-CS mutant belonged to the same space group as the wild-type protein crystals. The structure of the HHM-CS mutant was determined by the molecular replacement method with the program MOLREP (Vagin and Teplyakov, 2000), using the structure of wild-type SeM-HHM as the search model. No significant differences between the wild-type and mutant HHM structures were observed. Finally, the atomic models of the HHM-CS mutant were refined against reflections up to 2.5 Å resolution, using the program PHENIX (Adams et al, 2002). The final round of refinement treated the N-terminal (1–167) and C-terminal (168–360) regions as independent TLS groups, which significantly lowered the value of R free. The structural refinement statistics are summarized in Table II. Molecular graphics were illustrated using CueMol ( http://www.cuemol.org/).
GST pull-down assay
Equal amounts of GST–HHM(wt), GST–HHM(TEV), and GST–HHM(TEV) following TEV protease cleavage were incubated with Glutathione Sepharose 4B beads. The beads were washed three times in the binding/washing buffer (50 mM Tris–HCl, pH 8.0, containing 150 mM NaCl, 1 mM EDTA, and 0.1% NP-40). The bound proteins were eluted with the SDS–PAGE sample buffer, resolved by SDS–PAGE, and stained by Coomassie Brilliant Blue.
Analytical ultracentrifugation
Prior to analytical ultracentrifugation, the samples were purified by gel filtration chromatography, in 20 mM Tris–Cl buffer (pH 7.5) containing 200 mM NaCl and 7 mM TCEP. The same buffer was used as the reference solution. Sedimentation velocity experiments were performed with an Optima XL-I analytical ultracentrifuge (Beckman-Coulter). Sample (400 μl) and reference (420 μl) solutions were loaded into double sector centrepieces mounted in a Beckman An-50Ti rotor. The concentration profiles of the samples were monitored by the absorbance at 280 nm, without intervals between successive scans. The data were analysed by the SEDFIT program (Schuck, 2000) to obtain the c(s) profiles. On the basis of the assumption that the frictional ratio f/f
0 was common to all of the molecular species, c(s) was converted to obtain the molecular mass distribution function, c(M), using the implemented function in SEDFIT. The partial specific volume 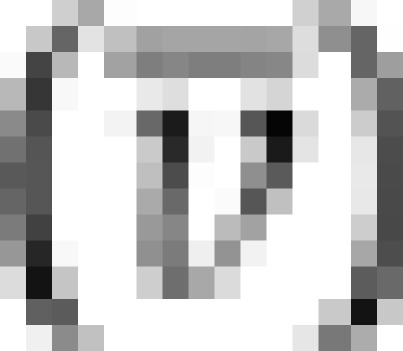 of HHM from the amino-acid sequence, the buffer density (ρ), and viscosity (η), based on the solvent composition, were calculated by the SEDNTERP program (Laue et al, 1992).
of HHM from the amino-acid sequence, the buffer density (ρ), and viscosity (η), based on the solvent composition, were calculated by the SEDNTERP program (Laue et al, 1992).
Sedimentation equilibrium experiments were carried out in an Optima XL-I analytical ultracentrifuge, using a 4-hole An60Ti rotor at 20 °C with a standard double sector centrepieces and quartz windows. A quantity of 120 μl of each sample with the absorption of 0.2, 0.3, and 0.5 at 280 nm was applied in the sample hole, and the corresponding dialysate was loaded in the reference hole of the cell. The concentration gradients of the samples were monitored by absorption at 280 nm. The rotor speeds were 8800 r.p.m. for 24 h, 12 600 r.p.m. for 20 h, and 22 000 r.p.m. for 20 h. Scans were made every 2 h, and the equilibrium of the system was assumed when the last three scans were identical. Totally, nine data sets were globally fitted to a single species model to determine the weight-average-molecular weight by the nonlinear least-square fitting, as implemented in the Beckman-Coulter software package.
Protein binding assay
The expression constructs encoding HHM, human Olig1, and human Id2 were described previously (Ikushima et al, 2008). The full-length cDNA encoding mouse NeuroD1 was synthesized and cloned into the mammalian expression vector pcDNA3. The cDNAs encoding HHM mutants were generated using a PCR-based approach. COS7 cells were obtained from the American Type Culture Collection, and were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 50 U/ml penicillin, and 50 μg/ml streptomycin. Cells were transfected with various plasmids, using the FuGENE6 transfection reagent (Roche Diagnostics) according to the manufacturer’s recommendations. Cell lysis, immunoprecipitation, and immunoblotting were performed as described previously (Mizutani et al, 2010). An anti-FLAG M2 antibody (Sigma-Aldrich) and an anti-Myc 9E10 antibody (Pharmingen) were used for immunological detection of proteins. Immunoblotting images were obtained using a Luminescent Image Analyzer (LAS-4000, Fujifilm). Bands on immunoblots were quantified using the Image-J software (National Institutes of Health, USA). All luminescent images were acquired and quantified under unsaturated conditions.
Myogenic differentiation assay
cDNAs encoding HHM and its mutants were cloned into a modified pENTR vector (Invitrogen) carrying CAG promoter and polyA signal (excised from pAxCAwt, Takara Bio), and introduced into the adenoviral genome via recombination between pENTR vector and the pAd/PL/DEST vector using LR Clonase (Invitrogen). HEK293A cells were transfected with pAd/PL/HHM after linearization of it with PacI. Viral particles were isolated by three freeze-thaw cycles and amplified by reinfection in HEK293A cells. C2C12 cells were obtained from the American Type Culture Collection, and were maintained in DMEM containing 20% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin. C2C12 cells were plated on a six-well culture dish (5×104 cells/well), infected with adenoviruses, and cultured for 24 h. The cells were then cultured in differentiation medium (DMEM containing 2% FBS) for 12–24 h. Myogenic marker expression was determined by quantitative real-time PCR analysis as described previously (Koinuma et al, 2011). Primer sequences used are as follows: mouse myosin heavy chain (sense, 5′- CCGGTGCTGTGATGCATTATG-3′; antisense, 5′-CAGCAACTTCGGTGCCATCT-3′); mouse myogenin (sense, 5′-CCAGGAGATCATTTGCTCGC-3′; antisense, 5′-TGATGCTGTCCACGATGGAC-3′); HHM (sense, 5′-GCAGTCCCCACCCTGGCTTC-3′; antisense, 5′-GAGCAACCGCAGCTCCTCCG-3′); mouse hypoxanthine phoshoribosyltransferase 1 (sense, 5′-CAGGCCAGACTTTGTTGGAT-3′; antisense, 5′-AACTTGCGCTCATCTTAGGC-3′).
Multiple comparisons of the data were performed using the Tukey–Kramer method.
Accession codes
The atomic coordinates have been deposited in the Protein Data Bank under accession code 3AY5.
Supplementary Material
Acknowledgments
We are grateful to Dr Nakamura (University of Tokyo) for helpful discussions regarding the construction of the atomic model of HHM. We thank the beam-line staffs at BL41XU of SPring-8 and NW12A of KEK PF-AR for assistance in data collection. We thank the RIKEN Integrated Cluster of Clusters (RICC) for providing computational resources. This work was supported by a grant from the Japan Society for the Promotion of Science (JSPS) through its ‘Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST program)’ to ON, by Core Research for Evolutional Science and Technology (CREST) Program ‘The Creation of Basic Medical Technologies to Clarify and Control the Mechanisms Underlying Chronic Inflammation’ of Japan Science and Technology Agency (JST) to ON, by a Grant-in-Aid for Scientific Research on Innovative Areas from MEXT to KoM, RI, and ON, by the Kurata Memorial Hitachi Science and Technology Foundation grant to ON, and by a grant from the Fugaku Memorial Foundation and the Mitsubishi Science Foundation to KeM.
Ryohei Ishii, Daizo Koinuma, Kohei Miyazono, Keiji Miyazawa, Ryuichiro Ishitani, and Osamu Nureki designed the experiments and analysed the data; Ryohei Ishii, Kazunobu Isogaya, Azusa Seto, Daizo Koinuma, Yuji Watanabe, So-ichi Yaguchi, Hiroaki Ikushima, Naoshi Dohmae, Ryuichiro Ishitani, and Osamu Nureki performed the most experiments; Fumio Arisaka designed and performed the analytical ultracentrifugation experiments; Ryohei Ishii, Daizo Koinuma, Keiji Miyazawa, Ryuichiro Ishitani, and Osamu Nureki wrote the paper; Keiji Miyazawa and Osamu Nureki supervised the project.
Footnotes
The authors declare that they have no conflict of interest.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D58: 1948–1954 [DOI] [PubMed] [Google Scholar]
- Ayer DE, Kretzner L, Eisenman RN (1993) MAD—a heterodimeric partner for max that antagonizes Myc transcriptional activity. Cell 72: 211–222 [DOI] [PubMed] [Google Scholar]
- Bains W, Ponte P, Blau H, Kedes L (1984) Cardiac actin is the major actin gene-product in skeletal-muscle cell-differentiation in vitro. Mol Cell Biol 4: 1449–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H (1990) The protein Id—A negative regulator of helix-loop-helix DNA-binding proteins. Cell 61: 49–59 [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H (1990) Sequence-specific DNA-binding by the C-Myc protein. Science 250: 1149–1151 [DOI] [PubMed] [Google Scholar]
- Blackwood EM, Eisenman RN (1991) Max—a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with myc. Science 251: 1211–1217 [DOI] [PubMed] [Google Scholar]
- Chavali GB, Vijayalakshmi C, Salunke DM (2001) Analysis of sequence signature defining functional specificity and structural stability in helix-loop-helix proteins. Proteins 42: 471–480 [DOI] [PubMed] [Google Scholar]
- Crews ST (1998) Control of cell lineage-specific development and transcription by bHLH-PAS proteins. Genes Dev 12: 607–620 [DOI] [PubMed] [Google Scholar]
- de la Torre JG, Huertas ML, Carrasco B (2000) Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J 78: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D-Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fong S, Debs RJ, Desprez PY (2004) Id genes and proteins as promising targets in cancer therapy. Trends Mol Med 10: 387–392 [DOI] [PubMed] [Google Scholar]
- Han SA, Guo CC, Hong L, Liu H, Zheyi H, Liu CJ, Wang J, Wu KC, Ding J, Fan DM (2004) Expression and significance of Id1 helix-loop-helix protein overexpression in gastric cancer. Cancer Lett 216: 63–71 [DOI] [PubMed] [Google Scholar]
- Hwang SY, Oh B, Fuchtbauer A, Fuchtbauer EM, Johnson KR, Solter D, Knowles BB (1997) Maid: a maternally transcribed novel gene encoding a potential negative regulator of bHLH proteins in the mouse egg and zygote. Dev Dyn 209: 217–226 [DOI] [PubMed] [Google Scholar]
- Ikushima H, Komuro A, Isogaya K, Shinozaki M, Hellman U, Miyazawa K, Miyazono K (2008) An Id-like molecule, HHM, is a synexpression group-restricted regulator of TGF-beta signalling. EMBO J 27: 2955–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro A, Spirin K, Shiohara M, Tobler A, Norton JD, Rigolet M, Shimbo T, Koeffler HP (1995) Expression of Id2 and Id3 mRNA in human lymphocytes. Leuk Res 19: 989–996 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr Sect A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Klambt C, Knust E, Tietze K, Camposortega JA (1989) Closely related transcripts encoded by the neurogenic gene-complex enhancer of split of Drosophila melanogaster. EMBO J 8: 203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koinuma D, Shinozaki M, Nagano Y, Ikushima H, Horiguchi K, Goto K, Chano T, Saitoh M, Imamura T, Miyazono K, Miyazawa K (2011) RB1CC1 protein positively regulates transforming growth factor-β signaling through the modulation of Arkadia E3 ubiquitin ligase activity. J Biol Chem 286: 32502–32532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreider BL, Benezra R, Rovera G, Kadesch T (1992) Inhibition of myeloid differentiation by the helix-loop-helix protein Id. Science 255: 1700–1702 [DOI] [PubMed] [Google Scholar]
- Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H (1991) Functional-activity of myogenic hlh proteins requires hetero-oligomerization with E12/E47-Like proteins in vivo. Cell 66: 305–315 [DOI] [PubMed] [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM, Pelletier SL (1992) Computer-aided interpretation of analytical sedimentation data for proteins. InAnalytical Ultracentrifugation in Biochemistry and Polymer Science Harding SE, Rowe AJ, Horton JC (eds) pp90–125Cambridge, UK: Royal Society of Chemistry, [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H (1995) Conversion of xenopus ectoderm into neurons by neurod, a basic helix-loop-helix protein. Science 268: 836–844 [DOI] [PubMed] [Google Scholar]
- Longo A, Guanga GP, Rose RB (2008) Crystal structure of E47-NeuroD1/beta2 bHLH domain-DNA complex: heterodimer selectivity and DNA recognition. Biochemistry 47: 218–229 [DOI] [PubMed] [Google Scholar]
- Ma QF, Kintner C, Anderson DJ (1996) Identification of neurogenin, a vertebrate neuronal determination gene. Cell 87: 43–52 [DOI] [PubMed] [Google Scholar]
- Ma W, Xia X, Stafford LJ, Yu C, Wang F, LeSage G, Liu M (2006) Expression of GCIP in transgenic mice decreases susceptibility to chemical hepatocarcinogenesis. Oncogene 25: 4207–4216 [DOI] [PubMed] [Google Scholar]
- Massari ME, Murre C (2000) Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 20: 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K, Kamiya Y, Morikawa M (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147: 35–51 [DOI] [PubMed] [Google Scholar]
- Mizutani A, Saitoh M, Imamura T, Miyazawa K, Miyazono K (2010) Arkadia complexes with clathrin adaptor AP2 and regulates EGF signalling. J Biochem 148: 733–741 [DOI] [PubMed] [Google Scholar]
- Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, Cabrera CV, Buskin JN, Hauschka SD, Lassar AB, Weintraub H, Baltimore D (1989) Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA-sequence. Cell 58: 537–544 [DOI] [PubMed] [Google Scholar]
- Nair SK, Burley SK (2003) X-ray structures of Myc-Max and Mad-Max recognizing DNA: Molecular bases of regulation by proto-oncogenic transcription factors. Cell 112: 193–205 [DOI] [PubMed] [Google Scholar]
- Olson EN, Klein WH (1994) bHLH factors in muscle development—dead lines and commitments, what to leave in and what to leave out. Genes Dev 8: 1–8 [DOI] [PubMed] [Google Scholar]
- Parkhurst SM, Meneely PM (1994) Sex determination and dosage compensation—lessons from flies and worms. Science 264: 924–932 [DOI] [PubMed] [Google Scholar]
- Perk J, Iavarone A, Benezra R (2005) ID family of helix-loop-helix proteins in cancer. Nat Rev Cancer 5: 603–614 [DOI] [PubMed] [Google Scholar]
- Rawls A, Olson EN (1997) MyoD meets its maker. Cell 89: 5–8 [DOI] [PubMed] [Google Scholar]
- Rushlow CA, Hogan A, Pinchin SM, Howe KM, Lardelli M, Ishhorowicz D (1989) The Drosophila hairy protein acts in both segmentation and bristle patterning and shows homology to N-MYC. EMBO J 8: 3095–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 78: 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto A, Ikushima H, Suzuki T, Sato Y, Fukai S, Yuki K, Miyazawa K, Miyazono K, Ishitani R, Nureki O (2009) Crystallization and preliminary X-ray diffraction analysis of GCIP/HHM transcriptional regulator. Acta Crystallogr F-Struct Biol Cryst Commun 65: 21–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CP, Kadesch T (1995) B-cell-specific DNA-binding by an E47 homodimer. Mol Cell Biol 15: 4518–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg-Riethmacher E, Wustefeld T, Miehe M, Trautwein C, Riethmacher D (2007) Maid (GCIP) is involved in cell cycle control of hepatocytes. Hepatology 45: 404–411 [DOI] [PubMed] [Google Scholar]
- Sun XH, Copeland NG, Jenkins NA, Baltimore D (1991) ID proteins ID1 and ID2 selectively inhibit DNA-binding by one class of helix-loop-helix proteins. Mol Cell Biol 11: 5603–5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai S, Aoki H, Ashida K, Thorgeirsson SS (2000) Human homologue of maid: a dominant inhibitory helix-loop-helix protein associated with liver-specific gene expression. Hepatology 32: 357–366 [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (2000) An approach to multi-copy search in molecular replacement. Acta Crystallogr Sect D-Biol Crystallogr 56: 1622–1624 [DOI] [PubMed] [Google Scholar]
- Wang Q, Tsao SW, Fu SB, Xue WC, Meng XN, Feng HC, Wong YC, Wang XH (2004) Overexpression of Id-1 in gastric adenocarcinoma: implication for a novel diagnostic marker. Anticancer Res 24: 881–886 [PubMed] [Google Scholar]
- Weintraub H (1993) The MyoD family and myogenesis—redundancy, networks, and thresholds. Cell 75: 1241–1244 [DOI] [PubMed] [Google Scholar]
- Weintraub H, Davis R, Lockshon D, Lassar A (1990) MyoD binds cooperatively to 2 sites in a target enhancer sequence—occupancy of 2 sites is required for activation. Proc Natl Acad Sci USA 87: 5623–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscott SJ (1991) Muscle-specific transcriptional activation by MyoD. Genes Dev 5: 1377–1386 [DOI] [PubMed] [Google Scholar]
- Wibley J, Deed R, Jasiok M, Douglas K, Norton J (1996) A homology model of the Id-3 helix-loop-helix domain as a basis for structure-function predictions. Biochim Biophys Acta-Protein Struct Molec Enzym 1294: 138–146 [DOI] [PubMed] [Google Scholar]
- Xia CZ, Bao ZM, Tabassam F, Ma WB, Qiu MS, Hua SB, Liu MY (2000) GCIP, a novel human Grap2 and cyclin D interacting protein, regulates E2F-mediated transcriptional activity. J Biol Chem 275: 20942–20948 [DOI] [PubMed] [Google Scholar]
- Yokota Y, Mori S (2002) Role of id family proteins in growth control. J Cell Physiol 190: 21–28 [DOI] [PubMed] [Google Scholar]
- Zebedee Z, Hara E (2001) Id proteins in cell cycle control and cellular senescence. Oncogene 20: 8317–8325 [DOI] [PubMed] [Google Scholar]
- Zervos AS, Gyuris J, Brent R (1993) MXI1, a protein that specifically interacts with max to bind Myc-Max recognition sites. Cell 72: 223–232 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.