

Abstract
In bacterial chemotaxis two-component signaling system, the histidine-containing phosphotransfer domain (the “P1” domain) of CheA receives a phosphoryl group from the catalytic domain (P4) of CheA and transfers it to the cognate response regulator (RR) CheY, which is docked by the P2 domain of CheA. Phosphorylated CheY then diffuses in cytoplasm and interacts with the FliM moiety of the flagellar motors, thereby modulating the direction of the flagella rotation. Structures of various histidine phosphotransfer domains (HPt) complexed with their cognate RR domain have been reported. Unlike the E. coli chemotaxis system, however, these systems lack the additional domains dedicated to binding to the response regulators, and the interaction of an HPt domain with an RR in the presence of such a domain has not been examined on the structural basis.
In this study, we used modern NMR techniques to construct a model for the interaction of the E. coli CheA P1 domain (HPt) and CheY (RR) in the presence of the CheY-binding domain, P2. Our results indicate that the presence of P2 may lead to a slightly different relative orientation of the HPt and RR domains from those seen in such complex structures previously reported.
Keywords: Two Component Signaling system (TCS), Chemotaxis, Histidine phosphotransfer domain (HPt), Response Regulator (RR), Chemical Shift Perturbation (CSP), Nuclear Overhauser Effect spectroscopy (NOESY), Paramagnetic Relaxation Enhancement
B. Introduction
Motile bacteria propel themselves by the rotary motion of flagella, and they have the tendency to swim toward attractants and away from repellents, the behavior known as chemotaxis. The direction of the bacteria movement is determined by the sense of rotation of the flagellar motor, which is controlled by a complex two-component signal transduction system that senses the temporal concentration gradient of the attractants and repellents in their surroundings. The binding of such molecules by transmembrane chemoreceptors modulates the autophosphorylation activity of CheA (the histidine kinase, HK), the central kinase in the bacterial chemotactic signaling system. The increased autophosphorylation of CheA in the presence of negative stimuli results in elevated phosphorylation of CheY, one of the two CheA substrates. Phosphorylation of CheY enhances its affinity to the FliM moiety of the flagellar motor. This interaction enhances the probability that the motor will rotate clockwise, which leads to the tumbling motion of the cell and the resulting change of direction. In the presence of attractants, the CheA phosphorylation activity is inhibited, and the majority of CheY remain unphosphorylated, ensuring the counter-clockwise rotation of the motor, that results in the smooth swimming. (1, 2, 3)
CheA is a five-domain protein (Figure 1) (2, 4). The functional unit of CheA is thought to be the dimeric form, and the dimerization is mediated by the P3 domain. The environmental signal input sensed by the chemoreceptor is transduced to CheA through its regulatory domain P5, which mediates the interaction of CheA a phosphoryl group from the ATP bound to the P4 domain. The P1 domain subsequently transfers the phosphoryl group to the cognate response regulator (RR) CheY. In E. coli and related systems CheY is bound to P2. Conformational change in CheY upon phosphorylation facilitates its dissociation from the P2 domain into the cytoplasm for the eventual interaction with the flagellar motors. (5, 6)
Figure 1.

Histidine kinase CheA and CheY in E. coli. Function and the residue numbers of each domain are shown. The solid arrows show the flow of phosphate transfer. The broken arrow shows the interaction between the P2 domain and CheY.
While the structures and functions of the receptor, CheW, CheY, and the five CheA domains have been well established, the nature of the interactions through which the signal is relayed between these participants has been poorly understood. One of the unresolved issues is how the activated (phosphorylated) CheA P1 domain enhances the rate of CheY phosphorylation. It is known that CheY can be phosphorylated by small-molecule phosphodonors such as acetyl phosphate and phosphoimidazole, but the rates are slower by three orders of magnitude than the phosphorylation rate by CheA (7). The observed rate enhancement by CheA seems to be at least partially due to the enhanced local concentration of CheY around the phosphotransfer domain, P1. In CheA, P1 is connected to the CheY-binding domain (P2) by a 23-residue linker (8). The P2 domain binds tightly to CheY, with the dissociation constant in the low-micromolar range, and it can be estimated that the effective local concentration of CheY for the P1 domain is roughly 4 mM (see discussion). However, the rate of phosphotransfer by CheA is still much faster than that by the small-molecule phosphodonors at a comparable concentration, and it seems obvious that the P1 domain plays an active role in the enhanced phosphotransfer rate by CheA. The goal of this study is to provide a structural basis for the mechanism by which P1 accomplishes this.
Structures of various histidine phosphotransfer domains (HPt) complexed with their cognate RR have been reported (9, 10, 11, 12). Among them, R. sphaeroides CheA3P1 and CheY6 are orthologs of E. coli P1 and CheY, and yeast Ypd1 and SLN1 (R1) are their structural homologs, and possible clues for the P1-CheY interaction mechanism have come from these crystal structures. In both cases, two major factors stabilizing the complex appear to be (1) the interaction between the α-helix 1 of the RR domain and the helices equivalent to the α-helix 1 of E. coli P1 and (2) the interaction of the helix containing the active site histidine of the HPt domain with the β5-α5 loop of the RR. Do E. coli P1 and CheY employ a similar mode of interaction? One thing that sets the E. coli CheA-CheY system apart from these systems is the presence of the CheY-binding domain, P2, in E. coli CheA. This may affect the way P1 can orient itself against CheY in at least two ways. First, unlike the HPt domains in the above systems used in the crystallographic studies, where the HPt and RR domains provide all the affinity and specificity of the interaction, E. coli system has an additional CheY binding domain contributing specificity and affinity to the inherently low affinity of the P1-CheY interaction. Second, the linker connecting the two domains, while enhancing the effective concentration of CheY for P1, may also have a restricting effect on how P1 can sample the space around CheY.
To address these issues, we used the modern NMR techniques to construct a structural model for the interaction between CheY and CheA P1P2 (where P1 is connected to P2 by the native linker). In this analysis, the P1 domain and CheY were docked as rigid bodies based on the short-range distance constraints provided by the NOE measurements and long-range distance constraints provided by the paramagnetic relaxation enhancement measurements using the nitroxide spin-label MTSL (13, 14). We observed that the interaction between CheY and P1 is mediated through the interaction surfaces largely consistent with those observed in the crystal structures of the yeast Ypd1-SLN1(R1) complex and the R. sphaeroides CheA3P1-CheY6 complex. Our model indicates, however, in the presence of the linker and the P2 domain, the relative orientation of the HPt and RR may be slightly different from those seen in the previously reported HPt-RR crystal structures. We also show that the mutations affecting the P1-CheY interaction surface as observed by NMR had detrimental effects in the rate of phosphotransfer, as assessed by a single turn-over kinetics method, which suggests that the mode of interaction reported here is biochemically relevant.
C. Material and Methods
Protein expression and purification
Wild-type E. coli CheY (residues 1–129) was expressed in an E. coli K38 strain carrying the pCW expression plasmid (15). CheA P1 domain (residues 1–134) was cloned into pET28a (Novagen) at the NcoI- and XhoI sites in frame with the C-terminal his6-tag and expressed in BL21(DE3) (Novagen). CheA P2 domain (residues 156–229) was also cloned into pET28a at the NcoI- and XhoI sites in frame with the C-terminal his6-tag and expressed in BL21(DE3). CheA P1P2 fragment was expressed in BL21(DE3) carrying pREP4 and pRS1-4 (pQE12: CheA residues 1–223, C-terminal his6-tag) (8, 16). CheA P3P4P5 fragment (residues 261–654) was cloned into pET28a with the N-terminal his6-tag and expressed in BL21(DE3). All proteins were purified following the published protocols (8, 15, 17) or by Ni2+- NTA chromatography (Qiagen).
P1 mutants F8A, E15A, P1P2 single mutant C213A, and double mutant C213AF8A were made by following Quick Change mutagenesis protocol using whole-plasmid PCR. CheY single-cysteine mutants (E37C, N62C, E89C, K91C) were made by following overlap extension PCR protocol. P1 Δ 9 (residues 10–134) mutant was made by following regular PCR protocol. Pfu Turbo DNA polymerase (STRATAGENE) was used for all PCR reactions. Restriction enzymes and T4 DNA ligase were purchased from New England BioLabs. All mutations were confirmed by DNA sequencing and expressed and purified with the same protocols as for the wild-type proteins.
Site-directed spin-labeling of CheY
CheY cysteine mutants in 50 mM sodium phosphate, 150 mM sodium chloride pH7.9, were treated with 5 mM dithiothreitol (DTT) for 1 hour to prevent the dimerization one hour prior to spin-labeling. DTT was removed by Sephadex G-25 (Sigma) spin-column immediately before the modification with MTSL [(1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)-methanethiosulfonate], in which the nitroxide spin label was attached to the single cysteine through disulfide bond (13). 1–2% (v/v%) of the concentrated MTSL stock in ethanol was added to the sample at a molar ratio of 10 MTSL to 1 protein and incubated at room temperature for 2 hours, and G-25 spin-column was used to remove the excess free MTSL and exchange the sample into the NMR buffer (50 mM sodium phosphate, pH 6.5). The color test developed by Murray et al. was used to ensure the completion of the MTSL modification (18). The modified protein was concentrated and used at 100 μM for NMR experiments. Modified CheY mutant was mixed with 100 μM of deuterated P1P2 C213A, and an 1H-15N HSQC spectrum was acquired (19). As a reference, another 1H-15N HSQC spectrum was acquired after the MTSL in the sample was reduced by the incubation with 200-fold excess of ascorbic acid at room temperature in the dark for at least 10 hours (13, 14). Control experiments were performed in the same method, but used P1P2 F8AC213A instead of P1P2 C213A.
NMR spectroscopy
All the data reported in this study were collected with a Varian Inova 600 MHz spectrometer equipped with a cryoprobe at 25°C. NMR data were analyzed with the nmrPipe package and assignments were made with ANSIG3.3 (20, 21). All NMR samples (except for spin label experiments) were in 50 mM sodium phosphate, pH 6.5. 5 mM DTT, 0.2% sodium azide and 8% D2O were added prior to NMR experiments. Samples used in experiments requiring D2O-based buffers were lyophilized after dialysis and dissolved in 99.8% D2O.
The backbone and side-chain sequential assignments were obtained from the following 2D and 3D experiments: 1H-15N-HSQC (19), 1H-13C-HSQC (22), HNCACB (23), HNCO (18,23), (H)C(CO)NH-TOCSY (24,25,26), H(C)(CO)NH-TOCSY (24,25,26).
The P1-CheY/P2 complex chemical shift assignments were made by tracing the complex resonances through titration experiments back to the resonances of their unbound states. Titration experiments were done with the following samples: 200 μM of 15N/2H P1 was titrated with the mixture of 3 mM unlabeled CheY and 3.2 mM2H P2. 200 μM of 15N/2H CheY and 250 μM 2H P2 were titrated with 3 mM 2H P1.
Additionally, multidimensional NOESY spectra (27) were collected for inter-proton distance restrains: 3D 13C-separated/12C-filtered NOESY using a 1H/15N/13C -labeled sample (3 mM CheY or P1) and their respective unlabeled partner in the D2O buffer; 4D 15N-separated/13 C-filtered NOESY using a 2H/15N-labeled sample (3 mM CheY or P1) and their respective 2H/13C-labeled partner in the H2O buffer (28). 2 or 3 mM of 2H/14N/12C/-labeled P2 sample was used in all of these experiments.
Kdcalculation
To estimate the affinity of the P1-CheY complex, the extent of the interaction was monitored by the chemical shift perturbation of the representative residues of 15N-labeled proteins as a function of the concentration of their interaction partner (29). The resulting data points (CSP, chemical shift perturbation) were fit with a model that consists of the product of the chemical shift perturbation for a given residue at the saturation point (CSPmax) and the fraction of the bound species (fbound), with the dissociation constant (Kd) and the CSPmax as adjustable parameters:
The fraction of the bound species was expressed as follows (in cases where P1 is the titrant):
Structure Calculation
Distance constraints derived from the intermolecular NOE experiments were conservatively classified into three levels, 1.8–4.2 Å, 1.8–5.2 Å, and 1.8–7.2 Å, corresponding to strong, medium, and weak NOEs. Long-range distance constraints derived from the site-directed spin-labeling experiment. Based on the ratio of the intensity of the oxidized protein spectrum to that of the reduced protein spectrum (Iox/Ired,), the distance constraint of 1.8–15 Å was assigned for the peaks that were broadened more than 35% (30–38).
The starting structures were generated by linking the P2 domain of the E. coli CheY/P2 complex (1EAY) to the structure of the Salmonella typhimurium P1 domain (1I5N) by the E. coli linker sequence, since structure of E. coli P1 is not available (15, 39). Residue 1–3 and 132–134, which are not visible in 1I5N were also added. The sequence identity between the Salmonella typhimurium and E. coli P1 (residue 1–134) is 94%, and all nuclei for which we observed NOEs belonged to residues that were identical in both proteins. Residues 60 and 61, for which we observed the paramagnetic broadening in the spin-label experiment, are not identical, but they were backbone amides and therefore deemed equivalent in both proteins. 200 of such structures were generated starting from random initial orientations and separations, and 4 rounds of refinement were made using the 26 NOE constrains and 14 spin-label constraints listed in Table 2. The simulated annealing protocol of the program XPLOR-NIH was used to calculate the structures (40). The P1 and CheY/P2 complex were docked as rigid bodies, with following exceptions; the side chains of residues responsible for the interaction based on NOEs and the chemical shift perturbation were allowed to move freely in the torsion angle dynamics simulation (P1 residues 1–20, 50–60, CheY residues 92–6, 57–59, 86–91, 111–114). Both the side chains and backbone of the first three and the last three residues of P1, whose coordinates were not shown in the crystal structure due probably to the flexibility of the termini, the 23 residue linker between P1 and P2, and residues surrounding these regions (CheA residues 1–7, 132–159) were also allowed to move.
Table 2.
Summary of the observed intermolecular NOEa and long-range distance constraintsb obtained from MTSL experiments
| aCheY nuclei | P1 nuclei |
| T16 Hγ2# | T11 HN, F12 HN, F13 HN, D14 HN, E15 HN, A16 HN |
| R18 HN | T11 Hγ2# |
| R19 Hβ# | T11 Hγ2#, E15 HN |
| R19 HN | T11 Hγ2# |
| 120 Hδ1# | M3 Hε#, F8 HN, T11 Hγ2#, F12 HN, T56 Hγ2# |
| 120 HN | T11 Hγ2# |
| N23 Hδ# | T11 Hγ2# |
| N23 HN | M3 Hε#, T11 Hγ2# |
| L24 HN | M3 Hε# |
| K109 HN | M3 Hε# |
| F111 HN | M3 Hβ1, M3 Hγ1, M3 Hε#, 15 Hγ2#, 15 Hδ# |
| bMTSL position on CheY | P1 residues broadened by MTSL |
| E89C | 52HN, 53HN, 55HN, 58HN, 60–66HN, 68HN |
| K91C | Q63 Hε#, N71 Hδ# |
only the results used in the structure calcuation are shown here
indicates the three equivalent protons on methyl groups
Sopped-Flow Observation of the CheY W58 Fluorescence
Activation of the P1 domain by transphosphorylation was done by incubating 100 μM 15N-labeled P1 with 10 μM CheA P3P4P5 (residues 261–654) and 10 mM ATP in 50 mM TRISHCl, 5 mM MgCl2, 100 mM KCl, 0.5 mM DTT, pH 8.2 for more than 4 hours. 1H-15N HSQC experiments were used to assess the extent of phosphorylation.(41)
The time course for CheY phosphorylation was monitored by the decrease of the intrinsic tryptophan fluorescence of CheY (7). CheY was mixed with various phosphorylated P1 constructs in a stopped-flow spectrofluorometer (Applied Photophysics SX. 18MV, deadtime ~ 2 ms). The reactions were maintained at 24-25°C in the observation chamber of the instrument with a thermostat, and the samples were excited at 295 nm (~7 nm slit width on the excitation monochromator), and fluorescence emission was monitored at a combination of wavelengths above 320 nm with a cutoff filter. All phosphotransfer reactions were carried out in 50 mM TRIS-HCl, 5 mM MgCl2, 0.5 mM DTT, pH 7.5 (42).
Data were fit to the following kinetic scheme:
Where A-P is the phosphorylated form of the P1 domain of CheA, Y is CheY, Y-P is the phosphorylated form of CheY. Phosphorylation of CheY results in a decrease in tryptophan fluorescence. In the presence of excess CheY, the phosphorylation time course as observed from the CheY perspective can be described as the disappearance of free CheY fluorescence due to phosphorylation followed by its recovery due to auto-dephosphorylation:
Where the phosphotransfer step becomes pseudo first order and equal to the product k1[CheY] where k1 is the apparent second order rate constant for phosphotransfer from P1 to CheY. The change in CheY fluorescence F(t) as a function of time is then
Where c is the fluorescence at t = 0, d = k2, b = k1[CheY], a = FCheY-PA0k1[CheY]/(k2-k1[CheY]), in which FCheY-P represents the fluorescence of fully phosphorylated CheY, A0 is the initial phosphorylated P1 concentration.(43)
D. Results
CheA P1 interacts with CheY weakly yet specifically
To identify the residues of CheA P1 responsible for its interaction with CheY, we titrated 15N-labeled P1 with unlabeled CheY. The extent of interaction was monitored by the 1H-15-NHSQC spectrum of P1, and the residues with significant chemical shift perturbations were mapped onto the structure of P1 (Figure 2A, C). The P1 residues that showed the largest chemical shift perturbation were residues 4–20 (the beginning of α1) and residues 50 and 53–60 (the end of α2 and the turn that connects it to α3), which together form a continuous surface on the P1 structure. These results suggest that there is one specific CheY-binding surface on the P1 domain.
Figure 2.

(a). Chemical shift perturbation of the P1 domain by CheY. 200 μM 15N-labeled P1 was titrated with 2 mM unlabeled CheY, and the chemical shift difference, ((Δδ1H × 600)2 + (Δδ15N × 60.8)2)1/2, at the titration endpoint for each P1 residue was plotted.
Figure 2.

(c). The chemical shift perturbation in the presence of CheY shown in (b) was color-coded and plotted onto the P1 structure (PDB code 1I5N). The red color indicates larger chemical shift difference. Residues in blue color showed smaller differences. The active site histidine and its side chain are shown in green.
Chemical shift perturbations of 15N-labeled CheY by unlabeled P1, on the other hand, showed that there were two separate surfaces on CheY that interact with P1 (Figure 3A, B). The first interaction surface is centered around the active site Asp 57. The second surface overlapped the interface between CheY and the CheA P2 domain as evidenced by the crystal structure of the CheY-P2 complex (15, 44, 45). CheY is known to bind to P2 with the dissociation constant (Kd) in the low micromolar range, which is much weaker than the Kd for the P1-CheY interaction (see below). We asked if the second mode of interaction between CheY and P1 could be eliminated if the experiment was repeated in the presence of saturating amount of P2 (45, 46). Not surprisingly, when high levels of P2 were present, the CheY residues involved in the interaction with P2, which comprise the majority of the second P1 interaction surface, did not exhibit significant chemical shift changes upon the addition of P1, and the P1 interaction was now limited to the surface surrounding the site of phosphotransfer (Figure 3C, D). This indicates the second mode of interaction observed in the absence of P2 was likely a non-productive one. The CheY residues showing the largest chemical shift perturbations by P1 included residues 9–20 (the turn following β1 and the beginning of α1), residues 57–59 (the end of β3, the active site), residues 86–90 (the end of β4 and the turn leading to α4), residues 111-114 (the turn following β5 and the beginning of α5). This is largely consistent with the mode of interaction seen in the crystal structures of the yeast Ypd1-SLN1 (R1) complex and the CheA3P1-CheY6 complex in the R. sphaeroides discussed above (11, 12).
Figure 3.

(a). Chemical shift perturbation of CheY by the P1 domain. 200 μM 15N-labeled CheY was titrated with 2.4 mM unlabeled P1 and the chemical shift difference at the titration endpoint for each CheY residue was plotted.
Figure 3.

(b). The chemical shift perturbation in the presence of P1 shown in (a) was color-coded and plotted onto the CheY structure (PDB code 3CHY). The red color indicates larger chemical shift difference. Residues in blue color showed smaller differences. The active site aspartate and its side chain are shown in green.
Figure 3.
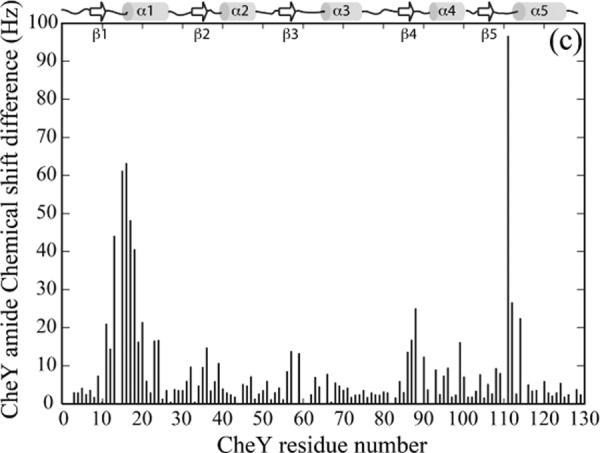
(c). Chemical shift perturbation of CheY complexed with the P2 domain by P1. 200 μM 15N-labeled CheY complexed with deuterated, unlabeled P2 was titrated with 2.4 mM deuterated, unlabeled P1 and the chemical shift difference at the titration endpoint for each CheY residue was plotted.
Figure 3.
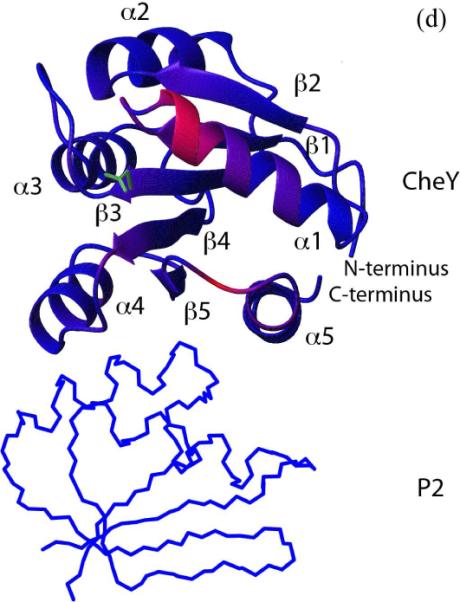
(d). The chemical shift perturbation of CheY complexed with P2 (PDB code 1EAY) in the presence of P1 shown in (c) was color-coded and plotted onto the CheY structure. The red color indicates larger chemical shift difference. Residues in blue color showed smaller differences. The active site aspartate and its side chain are shown in green.
Based on the concentration dependence of the chemical shift perturbation, we estimated the dissociation constant, Kd, between the P1 domain and CheY in the presence of the unlinked P2 domain to be about 5 mM (Figure 4). Also, the presence of P2 did not seem to affect the CheY-interaction surface of P1, as the residues that showed largest chemical shift perturbation were mostly the same with or without P2 (Figure 2B, C). Taken together, these results suggest there is a weak but specific mode of interaction between CheY and P1 in the presence of P2, even when the linker between P1 and P2 is absent.
Figure 4.

The affinity of the P1-CheY interaction. For both P1 (a) (b) and CheY (c) (b), titration curves were constructed for representative residues by monitoring their amide chemical shift perturbations throughout the course of the titrations shown in Figure 2 and 3 and fitting them with a model described in Materials and Methods. The dissociation constant was estimated by taking their average value (~4.5 mM).
Figure 2.

(b). Chemical shift perturbation of the P1 domain by CheY complexed with the P2 domain. 200 μM 15N-labeled P1 was titrated with 3 mM deuterated, unlabeled CheY and P2, and the resulting chemical shift difference for each P1 residue was plotted.
The observed interaction between P1 and CheY-P2 is biochemically relevant
The above results indicate that only a small number of residues are involved in the P1-CheY interaction and that the affinity is very weak. We wondered whether the interaction we detected was the one that leads to phosphorylation. To address this question, we mutated selected P1 residues within the proposed binding surface and examined their effects on the rate of phosphotransfer from P1 to CheY by monitoring the fluorescence quenching of CheY W58 upon phosphorylation of D57 (42). All mutants are folded correctly as confirmed by 15N HSQC spectrums (data not shown). Mutation of P1 residues F8 and E15 to alanine dramatically weakened its interaction with the CheY-P2 complex, as evidenced by the lack of chemical shift perturbation, and deletion of the first 9 residues of P1 N-terminus also nearly abolished the interaction (data not shown). The phosphotransfer rate was 5–30 times slower for these mutants compared to wild type P1 (Figure 5 and table 1). In total, we made 8 separate mutations in the proposed binding surface of P1, but we were unable to isolate a mutant whose binding was compromised but phosphotransfer rate was not. These results suggest that the mode of interaction between P1 and CheY we detected is the one that leads to phosphotransfer.
Figure 5.
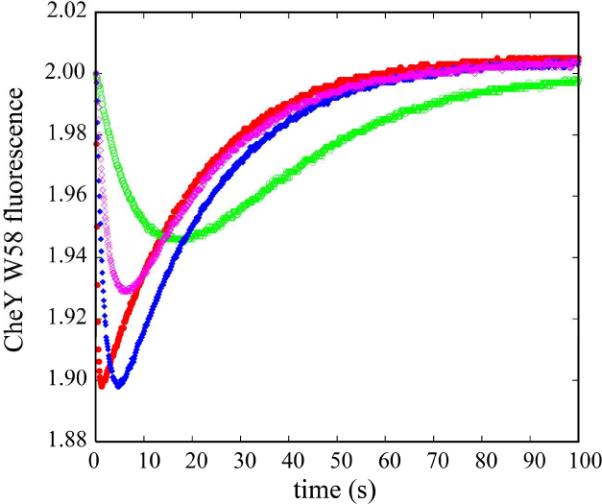
Time course of CheY phosphorylation monitored by CheY W58 fluorescence quenching. The fluorescence emission intensity of 1 μM CheY was monitored as it was mixed with 0.25 μM of various P1 constructs (red: WT P1, blue: F8A, magenta: E15A, green: P1 Δ9), which had been activated prior to mixing. For each P1 construct, the measurement was repeated at least 3 times, and the data were averaged. The initial value for CheY fluorescence for each time course was normalized for comparison. Results for the first 100 s are shown.
Table 1.
Kinetic parameters for phosphotransfer by P1 mutants
| Constructs | kphos (M−1s−1) | kdephos (S−1) |
|---|---|---|
| P1 WT | 2.5 × 106 | 0.050 |
| P1 Δ9 | 0.075 × 106 | 0.046 |
| P1 F8A | 0.55 × 106 | 0.050 |
| P1 E15A | 0.36 × 106 | 0.049 |
NOE-based short-range distance constraints
Having seen that the observed P1-CheY interaction was biochemically relevant, we proceeded to construct a structural model for this interaction. Multidimensional Nuclear Overhauser Effect spectroscopy (NOESY) was used to provide the short-range distance constraints between the P1 domain and CheY complexed with the P2 domain. The unlinked P2 was used to eliminate the non-specific interaction between P1 and CheY discussed above. Examples of the resulting NOESY spectra are shown in Figure 6A and 6B, and the residues responsible for the intermolecular NOEs are mapped onto the P1- and CheY with the receptor and the coupling protein, CheW. Signals from the receptor upon the repellent ligand binding or attractant desorption activates the kinase activity of CheA, and the histidine-containing phosphotransfer domain (HPt) P1which receives structures (Figure 7B, C). In total, twenty-six intermolecular NOEs between eleven P1 residues and nine residues of CheY were observed (Table 2). The P1 residues that gave the intermolecular NOEs were 3, 5, 8, 11–16, and 56, and most of them are located at the N-terminus of α1. CheY residues that contributed to the intermolecular NOEs were 16, 18–20, 23, 24, 109, 111. These residues are located at the N-terminus of α1, and the loop connecting β5 and α5. The results from the NOE experiment and the chemical shift perturbation results of the 15N HSQC titration experiments are in excellent agreement.
Figure 6.
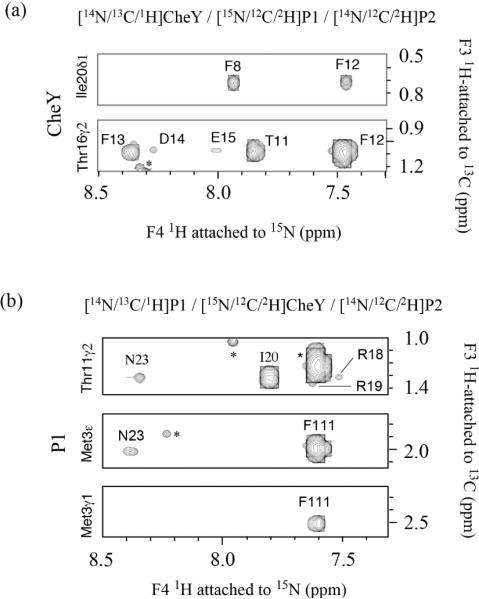
(a), (b). Observed NOEs between P1 and CheY in the P1-CheY/P2 complex. NOEs were obtained from four dimensional 15N-separated/13C-filtered NOE experiments that provide the intermolecular NOE between protons attached to 15N on one protein and protons attached to 13C on the other protein. The P2 domain is deuterated and unlabeled in both cases. In the experiments shown in (a), CheY is 13C-labeled, and P1 is 15N-labeled and deuterated. The top panel shows the plane at 13.7 ppm in 13C and 121 ppm in 15N, which shows the NOEs between the CheY I20 δ1 methyl protons and P1 F12- and F8 backbone amide protons. The bottom panel shows the NOEs from CheY T16 γ2 methyl protons to several P1 amides on the plane at 22.1 ppm in 13C and 118 ppm in 15N. In (b), the P1 domain is 13C-labeled, and CheY is deuterated and 15N-labeled. The top panel shows the plane at 22.3 ppm in 13C and 118 ppm in 15N, which shows the NOEs between the P1 T11 δ2 methyl protons and CheY R18-, R19-, I20- and N23 backbone amide protons. The middle panel shows the NOEs from P1 M3 ε methyl protons to CheY N23- and F111 amides on the plane at 17.6 ppm in 13C and 119 ppm in 15N. The bottom panel shows the NOEs from P1 M3 γ1 methyl protons to P1 F111 amides on the plane at 32.2 ppm in 13C and 116 ppm in 15N.
Figure 7.

(b), (c). The observed intermolecular NOEs shown in Table 2 are shown in dark red and plotted onto the structure of P1 (b) and CheY (c). The paramagnetic broadening of P1 amide resonances by CheY E89C are in yellow (b). The location of the spin label at the position 89 was indicated in yellow (c). The active site histidine and aspartate and their side chain are shown in red.
Long-range distance constraints based on site-directed spin labeling
Results of the titration and NOESY experiments showed that P1- and CheY residues responsible for the direct interaction were clustered into a small region in both proteins. To supplement the NOE-based short-range distance information during the structure calculation, we used site-directed spin labeling to obtain long-range distance constraints from the parts of the proteins that did not have NOE.
Two single-cysteine mutants of CheY were constructed for site-direct nitroxide spin labeling with MTSL (Table 2). The positions of the mutation (residues 89 and 91) were chosen to be close to the active site Asp 57 and away from the binding interface determined by the chemical shift perturbation mapping and NOEs (Figure 7C). These mutations are in loops and exposed to the solvent and not expected to disrupt the three-dimensional structure of CheY. The 1H-15N HSQC spectra of these mutants showed only minor chemical shift perturbations around the sites of mutations, suggesting their effects on the global structure are negligible. Also, the mutations did not seem to affect the interaction with the P1 domain, as the chemical shift perturbation in the 1H-15N HSQC spectra of P1 bound to these mutants were indistinguishable from those caused by the wild-type protein (data not shown).
To identify the P1 residues in proximity to the spin-labeling sites, each CheY mutant labeled with MTSL was mixed with the P1P2 construct of CheA (residue 1–233, the P1 domain and the P2 domain connected with the native linker), and an 1H-15N HSQC spectrum was collected, and the resulting resonance intensity for each P1P2 backbone amide (Ioxidized) was recorded. To quantify the paramagnetic broadening of the resonances, the same experiment was repeated after the MTSL in the sample was inactivated by the addition of excess ascorbic acid to yield the baseline value (Ireduced). To eliminate the effects of possible nonspecific interactions, a control experiment for each CheY mutant was performed by using P1P2F8A, a P1P2 mutant with reduced affinity to CheY. We reasoned that non-specific interactions were unlikely to be changed by this mutation but the specific interactions should be significantly weakened.
Figure 7A shows the resulting peak intensity ratio (Ioxidized/Ireduced) plotted against the P1 residue numbers when P1P2 was mixed with the CheY mutant labeled with MTSL at residue 89. It indicates P1 amide resonances broadened by MTSL at this position were those of residues 8, 9, 12, 52, 53, 55, 57–68, 82, 85, 108, 126. Among them, residue 12 and residues 85, 108, and their adjacent residues were also shown to be broadened in the control experiment with P1P2F8A. The results for these residues were not used as distance constraints in the structure calculation (Figure 7B). P1 residues affected by CheY K91C labeled with MTSL at different positions were also determined likewise (Table 2). These results provided additional information about the distance between the second and third helices of P1 and the active site surface of CheY.
Figure 7.

(a). Paramagnetic broadening of P1 amide resonances by CheY mutants spin-labeled with MTSL. 15N-labeled P1P2 was mixed with CheY constructs labeled with MTSL at various positions, including E89C used in the experiment shown here. The ratio of P1 amide intensity with the oxidized- (active) and the reduced (inactive) spin-label was plotted across the P1 sequence. An arbitrary intensity ratio of 1.40 was assigned to residues for which the intensity ratio could not be calculated due to resonance overlap or lack of assignment. Assignments of the distance restraints, as well as the results obtained with CheY constructs containing the spin-labels at other positions are summarized in Table 2.
The structural model for the P1P2-CheY complex
The crystal structures of the isolated P1 domain (PDB code 1I5N), P2 domain (1FWP), and CheY (3CHY), as well as the CheY-P2 complex (1EAY) have been solved previously (39, 47, 48, 15). To initiate the model building for the P1P2-CheY complex, we generated a starting structure, in which the structure of P1 was connected by the native linker to P2 in the CheY-P2 complex structure. 200 of such structures were refined by four successive rounds of simulated annealing and torsion angle dynamics, using the distance constraints obtained from the NOE experiments and the spin-label experiments with MTSL. The NOE-based distance constraints were classified into three levels, 1.8–4.2 Å, 1.8–5.2 Å, and 1.8–7.2 Å, corresponding to strong, medium, and weak NOEs. The MTSL-based longer distance constraints were assigned to 1.8–15 Å for the peaks that were broadened more than 35%, based on the ratio of the intensity of the oxidized spectrum to that of the reduced spectrum (Ioxidized/Ireduced). During the refinement, the flexible N-terminal residues of P1, the residues in the P1–P2 linker region, and the side chains of the residues responsible for the interactions as determined by NOE and chemical shift perturbation (see Materials and Methods), were allowed to move freely in the torsion angle space, but otherwise each domain was treated as a rigid body, for the chemical shift perturbation in our titration experiments suggested that there were little global structural changes.
Superposition of the 50 lowest-energy structures with no distance violation is shown in Figure 8A. The P1 backbone rmsd was ~1.3 Å when the backbone of the CheY/P2 complex was aligned. Residues responsible for interactions that collectively hold these two domains in the observed orientation are confined in seemingly small regions. The CheY-interaction surface of P1 appears to be located mainly in the first half of α1 and the end of α2. On the CheY side, the interacting residues are mostly in its α1 and the adjacent loop between β5 and α5. Figure 10A shows a detailed view of the interface between these two regions. A thread of interaction takes place as two faces of CheY α1 (T16-I20-L24 and R19-N23) interlock with the side chains of the D7-F8-T11-E15 face of P1 α1. Additionally, P1 I5 (on a different face of α1) and M3 (in the N-terminal loop) interact with two CheY residues (F111 and K109) in the β5-α5 loop, which completes the trapezoidal plane of an interacting surface. The interaction between P1 T56 (at the end of α2) and CheY I20 should provide another source of stability to the complex.
Figure 8.

(a) (b). Structural model for the P1P2-CheY complex in solution. (a) Superposition of the 50 lowest-energy results from the calculation with no distance constraint violation is shown. The backbone rmsd for the P1 domain in this bundle, when the backbone of the CheY-P2 complex was aligned, was 1.3 Å. The average Cα-Cα distances between the active site H48 and D57 is 11.7 Å. The average distances between the H48Nε to D57O1/2 are 5.91/5.52 Å. (b) The representative of 50 bundle shown in (a). P1 and the linker are shown in blue, CheY in red, and P2 in grey. The active site side chains are shown in green. The spin-labeled positions E89 and K91 are indicated.
Figure 10.
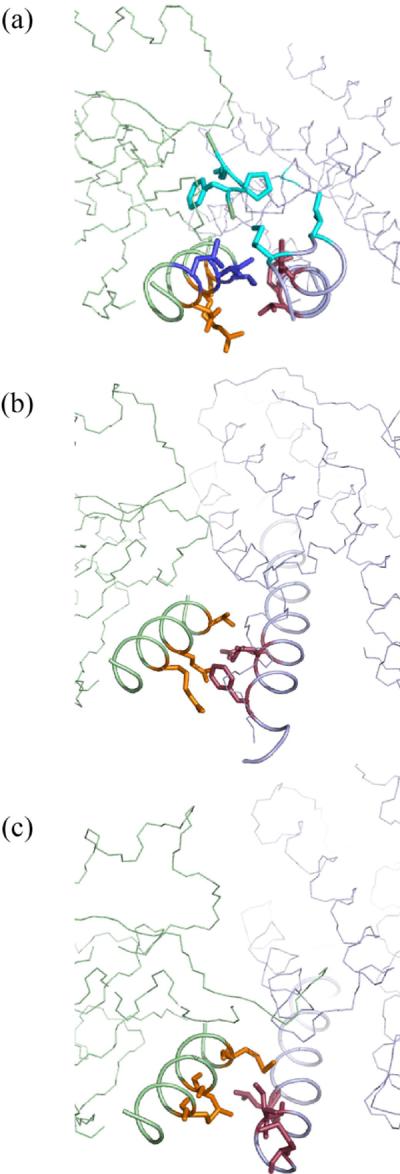
(a) (b) (c). Comparison of HPt-RR interaction surface in the E. coli P1-CheY complex (a), the yeast Ypd1-Sln1(R1) complex (b), and the R. sphaeroides CheA3P1-CheY6 complex (c). HPt domains are shown on the right in light blue, and RRs are shown on the left in light green. The residues responsible for the HPt interaction in yeast Sln1(R1) and R. sphaeroides CheY are colored orange. The interacting residues in yeast Ypd1 and R. sphaeroides CheA3P1 are shown in dark red. The equivalent positions in the E. coli proteins are shown in the same colors. E. coli CheY observed to be interacting with P1 are shown in blue. An additional interaction between CheY loop (109–111) and P1 M3 and I5 are shown in cyan.
This small web of interactions leads to such a degree of convergence among the calculated results that the Cα-Cα distance between the active site of P1 and CheY ranges from 11.3–12.3Å for the 50 structures (Figure 8A). This proximity alone should make phosphorylation of CheY by P1 much more favorable than phosphorylation by small molecule phosphodonors with no specific affinity to CheY. The distance between the C-terminus of the P1 domain (residue 131) to the N-terminus of the P2 domain (residue 160) was approximately 45 Å, which is expected from a flexible linker of this length (see discussion).
E. Discussion
In this study, we constructed a structural model for the interaction of the E. coli chemotactic proteins CheY and CheA P1, in the presence of the CheY-binding domain of CheA, P2. Although complex structures of their homologs (R. sphaeroides CheA3P1-CheY6) and the structural homologs (yeast Ypd1-Sln1) have been reported, these systems differ from the E. coli system in that they lack the additional domain dedicated to binding to the response regulator, and the interaction of a histidine phosphate transfer domain (HPt) with a response regulator domain (RR) in the presence of such a domain has not been examined on the structural basis. Here we discuss the overall evaluation of the resulting model, its relation to the previously reported structures of the HPt-RR complexes, and its biological significances.
Evaluation of the P1-CheY/P2 complex structure
In our structural calculation, CheY and P1 were treated as rigid bodies and docked based on the distance information gained from the NOESY experiments and site-directed spin-label experiments with MTSL. Our approach to the model-building, based on the distance information alone, is an abbreviated version of the procedure pioneered by the Clore group in their work on the sugar transport systems (30–38). The backbone rmsd for the P1 domain in the final 50 lowest-energy structures, when the backbone of the CheY-P2 complex was held constant, was ~1.3 Å (Figure 8). This degree of convergence is significantly lower compared to the standard set by the work of the Clore group, which typically includes the torsion angle constraints and constraints derived from the residual dipolar coupling, in addition to the NOE-based distant constraints, with the reported convergence rates ranging from 0.1 Å for the mannitol transporter system to 0.5 Å for the mannose transporter system (30–38). Our approach is based only on the conservative distance constraints and lacks residual dipolar coupling data and as a result does not match the apparent resolution reported by the Clore group. The lower resolution in our work represents valid alternative in addressing issues when lower resolution structural information answers the questions at hand with a significantly smaller investment in machine and data analysis time.
The low degree of convergence in the P1-CheY model is also due to that the interaction appears to be mediated by a single, small surface on both proteins. Our NOE results show that the major interaction is mediated by a plane defined by a part of a helix (residues 14–24) and a few residues in the adjacent loop (residues 109–112) in CheY, and only a handful of P1 residues, mostly in its N-terminal loop and α1, make contacts to these residues. The importance of the P1 residues lining the proposed interface was attested by our observation that alanine mutation at many of these positions severely compromises the interaction, and this seems to indicate that this surface is largely, perhaps solely, responsible for the interaction. The interactions between P1 and CheY, as outlined above, contained enough information to align the side chains of the two active site residues, P1 H48 and CheY D57, at a close distance. In the majority of the 50 calculated structures, the distance for the catalytic H48Hε2 of P1 to CheY D57Oδ1/2 was between 4–7 Å (Figure 8). P1 is known to phosphorylate CheY a few thousand fold faster than the small-molecule phosphate donors at a comparable concentrations, presumably because of participation of its active site residues (7).
Comparison of the P1-CheY model with the previously reported HPt-RR complex structures
A few high resolution HK-RR complex crystal structures have been solved previously: the Spo0B-Sp0F complex in the Bacillus subtilis sporulation regulation system, the hypothetical HPt-RR combination, HK853-RR468, in Thermotoga maritima, the Ypd1-SLN1 (R1) complex in the yeast environmental stress response system, and the CheA3P1-CheY6 complex in the Rhodobacter sphaeroides chemotaxis two-component system (9, 10, 11, 12, 49, 50). While the structure of the RR domains are highly conserved, the structural feature of the HPt domains is loosely defined as a helix bundle with the active site histidine that interacts with the active site aspartate of the CheY-like response regulators, and its members are not always structural homologs, and their sequences are often not conserved beyond the small regions around the catalytic histidine (41). The domain architecture of Spo0B and HK835cp is a dimer of two-helix bundles with the active site histidine in each monomer, and these are not homologous to E. coli P1 (9, 10). On the other hand, yeast Ypd1 is a monomeric HPt protein that has a 4-helix bundle with an additional short helix at the N-terminus and is a structural homolog of P1 (11). Also, CheA3P1 is a 5-helix bundle monomeric HPt protein, and it is one of the three Rhodobacter sphaeroides orthologs of E. coli CheA P1 (12). We compared these two structures with our model, and the results showed that, while E. coli P1 and CheY employ a mode of interaction similar to those seen in these structures, the presence of P2 and the linker might lead to a slightly different orientation of the HPt domain with respect to the RR domain (Figure 9).
Figure 9.
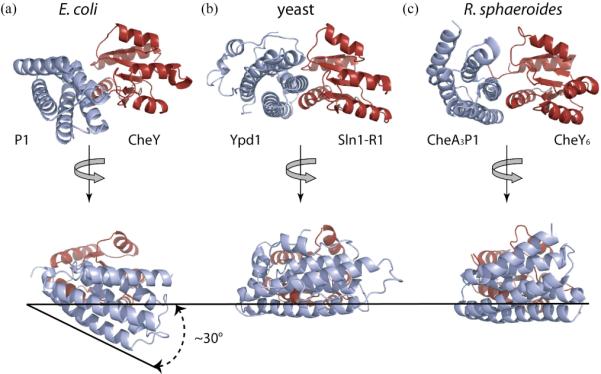
(a) (b) (c). Comparison of the E. coli P1-CheY complex (a) with the yeast Ypd1-Sln1(R1) complex (b) and the R. sphaeroides CheA3P1-CheY6 complex (c). The HPt domains are in light blue, and the RR domains, in red, are held constant to show the difference in the relative orientation of the two domains among these structures. E. coli P1 appears to be rotated downward (~30°) compared to yeast Ypd1 and R. sphaeroides CheA3P1 such that the top of the active site surface of CheY is more exposed in the E.coli model (bottom figures). The extent of rotation was estimated by comparing the orientation of E.coli P1 α1 to its equivalent helices in the other two structures.
In all three cases, two major factors stabilizing the complex appear to be (1) the interaction between the α-helix 1 of the RR domain and the helices equivalent to the α-helix 1 of E. coli P1 and (2) the interaction of the helix containing the active site histidine of the HPt domain with β5-α5 loop of the RR (11, 12). However, the resulting relative orientation of the HPt domain with respect to the RR domain is slightly different. When the CheY- and CheY6 parts are held constant (the alignment rmsd ~ 1.18Å), E. coli P1 is rotated (by about 30°) with respect to R. sphaeroides CheA3P1 such that the part of the P1-interaction surface of CheY close to its P2-interface is more exposed in the E. coli model (Figure 9). The orientation of the HPt domain with respect to the RR domain in the yeast Ypd1-SLN1(R1) complex is similar to the R. sphaeroides complex. The average Cα- Cα distance between the active sites in E. coli structures is within the range of the other two systems (11.7 Å in E. coli, 14 Å in R. sphaeroides, and 10.2Å in yeast), as well as their average catalytic side chain atoms (H48 Nε2 to D57 Oδ1/2, 5.9/5.5 Å in E. coli, 7.3/7.04 Å in R. sphaeroides, and 4.58/3.91 Å in yeast) (12).
What is the meaning of such differences? There are reasons to think that our results might represent a biologically relevant species. A recent statistical analysis indicates that a pairs of interacting proteins can show different assembly orientations when they have less than 30–40% sequence identity (51). For instance, there are two distinct families, one designated as the E. coli family and the other, Bacillus subtilis family, among the known structures of the CheY-P2 complexes. Both families share similar binding regions of P2 and CheY, but the P2 domain in one family is rotated by almost 90°with respect to that of the other family when the CheY parts of the complex structures are aligned (51). There is little sequence identity between E. coli P1 and Ypd1 (28%) or CheA3P1 (25%), and between E. coli CheY and SLN1 (R1) (14%) or CheY6 (26%). Given the low sequence identities (even within their binding surfaces) E. coli P1-CheY shares with the yeast Ypd1-SLN1 complex or the R. sphaeroides CheA3P1-CheY6 complex, it is not inconceivable that these complexes should show different interaction orientations.
Moreover, careful examination of our NOE results suggests that the E. coli complex should show a different relative orientation of the HPt domain with respect to the RR domain. While the positions of the HPt residues responsible for interaction are equivalent in all three cases, the positions of their interaction partners on the RR side appear to be different in the E. coli complex. In our E. coli model, P1 residues F8, T11, and E15 (Figure 10A: residues color in dark red) align with the face of CheY α1 that consists of residues L24, I20, and T16 (Figure10A: residues colored in blue). In the crystal structures of the yeast complex and the R. sphaeroides complex, the HPt residues at the same positions (although not conserved) appear to interact mostly with the RR residues equivalent to positions 16, 19, and 23 in E. coli CheY (Figure 10: residues in orange), which comprise the face of the RR α1 adjacent to that which interacts with E. coli P1. This mode of interaction also enables the interactions between P1 residues M3 and I5 and the CheY loop between β5 and α5 (Figure 10A: residues color in magenta) evidenced by multiple NOEs. This requires that the C-terminal end of E. coli P1 α1 be rotated downward with respect to CheY compared to the R. sphaeroides CheA3P1 or yeast Ypd1, which accounts for the differences in the orientation shown in Figure 9. This argument hinges on the notion that the arrangement of the RR α1 with respect to the rest of the domain in these complexes is similar, which appears to be the case.
If the complex reported in this study indeed is a complex that takes place during the phosphate relay from CheA to CheY, what might be its meaning? One obvious hypothesis would be that this is the complex that takes place as the reaction happens. As discussed above, the distance between the active site residues clearly makes the transfer possible. Another possibility would be that phosphotransfer reaction is actually carried out by a complex much closer to the yeast- or the R. sphaeroides complex (or other unknown complex) and that our structure represents a kind of a stand-by complex leading to such a complex. If that is the case, the fact that it eluded our detection suggests that the lifetime of such a hypothetical complex is shorter still than that of the observed complex, whose affinity is already very weak. One reason for our inability to observe such a complex would be that the HPt domain used in the NMR experiments was not activated due to the long-term instability of the phosphorylated E. coli P1. In others HPt-RR systems where this is less of an issued, it has been reported that the complex structure of the activated proteins resembles the `inactive' complex except for the rigid-body shift of 2–3Å that brings the domains closer (9, 22, 12, 49, 50).
The interaction surface between P1 and CheY as seen in our model (~700Å2, calculated by PISA Webserver) is smaller than the equivalent surfaces seen in the structure of yeast Ypd1-Sln1 (R1) (970Å2) complex, but larger than in the structure of R. sphaeroides CheA3P1-CheY6 (530 Å2) complex (Figure 10). However the P1-CheY interaction is much weaker than these complexes. This may be a reflection of the difference in the mechanisms by which phosphotransfer is accomplished. In cases where phosphate is transferred between the HPt domain and the RR domain without the P2-like domains, it would take either high intrinsic affinity between the participants or high concentrations of them to carry out such a bimolecular reaction in a timely manner. The binding between P1 and CheY is weak, with the Kd in the 5 mM range, but the overall signaling response in E. coli chemotaxis may not suffer from it because of the presence of a covalently linked CheY-binding domain (P2). P1 is connected to P2 by a 23 residue linker and 5 flexible terminal residues. The length of a linker of this size (given by r2=Cnl2, where n is the number of residues, C, the characteristic ratio, is close to 7 for n=28, and l is 3.8Å for the average Cα-Cα bond length) is expected to be ~53Å, which agrees with our result of ~45Å (30). CheY and the P2 domain form a stable complex (2 μM Kd), and the linker should constrain CheY to a space surrounding P1, with the estimated effective concentration of each component of ~4 mM (30). This means roughly twenty percent of the P1 population interacts with CheY at any time, despite the low intrinsic affinity. The interaction between P1 and CheY, as we detected, align the sidechains of the two active site residues, P1 H48 and CheY D57, in close proximity to each other. This should enhance the rate of phosphotransfer greatly compared to the small-molecule phosphate donors with no specific affinity to CheY.
It should also be noted that, before interacting with CheY, P1 binds to the catalytic domain of CheA (the P4 domain) to receive the phosphoryl group at the same active site histidine, and it is almost certain that the relative affinity of P1 to P4 and CheY has been fine-tuned to meet the biological demand of chemotaxis signaling. The nature and affinity of the interaction between P1, CheY, and P2 reported here show how and how likely this interaction is designed to happen. Understanding how they fit with the previous round of phosphotransfer, however, would require a study conducted on a CheA construct including the kinase domain and is out of the scope of this report.
Acknowledgments
This work was supported by NIH grant 9 R01 GM59544.
REFERENCES
- 1.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu. Rev. Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 2.Hamel DJ, Zhou H, Starich MR, Byrd A, Dahlquist FW. Chemical-shift-perturbation mapping of the phosphotransfer and catalytic domain interaction in the histidine autokinase CheA from Thermotoga maritima. Biochemistry. 2006;45(31):9509–9517. doi: 10.1021/bi060798k. [DOI] [PubMed] [Google Scholar]
- 3.Baker MD, Wolanin PM, Stock JB. Systems biology of bacterial chemotaxis. Current Opinion in Microbiology. 2006;9:187–192. doi: 10.1016/j.mib.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Baker MD, Wolanin PM, Stock JB. Signal transduction in bacterial chemotaxis. BioEssays. 2006;28:9–22. doi: 10.1002/bies.20343. [DOI] [PubMed] [Google Scholar]
- 5.Schuster M, Silversmith RE, Bourret RB. Conformational coupling in the chemotaxis response regulator CheY. Proc. Natl. Acad. Sci. U. S. A. 2001;98(11):6003–6008. doi: 10.1073/pnas.101571298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shukla D, Matsumura P. Mutation leading to altered CheA binding cluster on a Face of CheY. J. Biol. Chem. 1995;270(41):24414–24419. doi: 10.1074/jbc.270.41.24414. [DOI] [PubMed] [Google Scholar]
- 7.Mayover TL, Halkides CJ, Stewart RC. Kinetic characterization of CheY phosphorylation reactions: comparison of P-CheA and small-molecule phosphodonors. Biochemistry. 1999;38(8):2259–2271. doi: 10.1021/bi981707p. [DOI] [PubMed] [Google Scholar]
- 8.Zhou HZ, McEvoy MM, Lowry DF, Swanson RV, Simon MI, Dahlquist FW. Phosphotransfer and CheY-binding domains of the histidine autokinase CheA are joined by a flexible linker. Biochemistry. 1996;35(2):433–443. doi: 10.1021/bi951960e. [DOI] [PubMed] [Google Scholar]
- 9.Zapf J, Sen U, Madhusuda, Hoch JA, Varughese KI. A transient interaction between two phosphorelay proteins trapped in a crystal lattice reveals the mechanism of molecular recognition and phosphotransfer in signal transduction. Structure. 2000;8(8):851–862. doi: 10.1016/s0969-2126(00)00174-x. [DOI] [PubMed] [Google Scholar]
- 10.Casino P, Rubio V, Marina A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell. 2009;139:325–336. doi: 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 11.Xu Q, Porter SW, West AH. The yeast YPD1/SLN1 complex: insights into molecular recognition in two-component signaling systems. Structure. 2003;11:1569–1581. doi: 10.1016/j.str.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 12.Bell CH, Porter SL, Strawson A, Stuart DI, Armitage JP. Using structural information to change the phosphotransfer specificity of a two-component chemotaxis signaling complex. PLoS Biol. 2010;8(2):e1000306. doi: 10.1371/journal.pbio.1000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 14.Peterson DW, Zhou H, Dahlquist FW, Lew J. A soluble oligomer of Tau associated with fiber formation analyzed by NMR. Biochemistry. 2008;47(28):7393–7404. doi: 10.1021/bi702466a. [DOI] [PubMed] [Google Scholar]
- 15.Lowry DF, Roth AF, Rupert PB, Dahlquist FW. Signal transduction in chemotaxis. J. Biol. Chem. 1994;269(42):26358–26362. [PubMed] [Google Scholar]
- 16.Swanson RW, Schuster SC, Simon MI. Expression of CheA fragments which define domain encoding kinase, phosphotransfer, and cheY binding activities. Biochemistry. 1993;32(30):7623–7629. doi: 10.1021/bi00081a004. [DOI] [PubMed] [Google Scholar]
- 17.McEvoy MM, Hausrath AC, Randolph GB, Remington SJ, Dahlquist FW. Two binding modes reveal flexibility in kinase / response regulator interactions in the bacterial chemotaxis pathway. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7333–7338. doi: 10.1073/pnas.95.13.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grassetti DR, Murray JF., JR. The use of 2,2-dithiodipyridine in the determination of glutathione and of triphosphopyridine nucleotide by enzymatic cycling. Analytical Biochemistry. 1967;12:427–434. doi: 10.1016/0003-2697(67)90317-x. [DOI] [PubMed] [Google Scholar]
- 19.Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 1992;114(26):10663–10665. [Google Scholar]
- 20.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMR Pipe: A multidimensional spectra; processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 21.Kraulis PJ. ANSIG: A program for the assignment of protein 1H 2D NMR spectra by interactive graphics. J. Magn. Reson. 1989;24:627–633. [Google Scholar]
- 22.Vuister GW, Bax A. Resolution enhancement and spectral editing of uniformly 13C-enriched protein by homonuclear broadband 13C decoupling. J. Magn. Reson. 1992;98:428–435. [Google Scholar]
- 23.Muhandiram DR, Kay LE. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. Ser. B. 1994;103:203–216. [Google Scholar]
- 24.Montelione GT, Lyons BA, Emerson SD, Tashiro M. An efficient triple experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotropically-enriched proteins. J. Am. Chem. Soc. 1992;114:10974–10975. [Google Scholar]
- 25.Grzesiek S, Anglister J, Bax A. Correlation of backbone amide and aliphatic side-chain resonance in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J. Magn. Reson. Ser. B. 1993;101:114–119. [Google Scholar]
- 26.Logan TM, Olejiniczak ET, Xu RX, Fesik SW. A general method for assigning NMR spectra of denatured proteins using 3D HC(CO)NH-TOCSY triple resonance experiments. J. Biomol. NMR. 1993;3:225–231. doi: 10.1007/BF00178264. [DOI] [PubMed] [Google Scholar]
- 27.Clore GM, Gronenborn AM. Determingint the structures of large proteins and protein complexes by NMR. TBTECH. 1998;16:22–34. doi: 10.1016/S0167-7799(97)01135-9. [DOI] [PubMed] [Google Scholar]
- 28.Cai M, Huang Y, Zheng R, Wei S, Ghirlando R, Lee MS, Craigie R, Gronenborn AM, Clore GM. Solution structure of the cellular factor BAF responsible for protecting retroviral DNA from autointegration. Nat. Struct. Boil. 1998;5(10):903–909. doi: 10.1038/2345. [DOI] [PubMed] [Google Scholar]
- 29.Kawamura T, Le LUK, Zhou H, Dahlquist FW. Solution structure of Escherichia coli PapI, a key regulator of the Pap Pili phase variation. J Mol. Biol. 2007;365:1130–1142. doi: 10.1016/j.jmb.2006.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suh J, Cai M, Williams DC, Jr., Clore GM. Solution structure of a post-transition state analog of the phosphotransfer reaction between the A and B cytoplasmic domains of the Mannitol transporter IIMannitol of the Escherichia coli phosphotransferase system. J. Biol. Chem. 2006;281(13):8939–8949. doi: 10.1074/jbc.M513466200. [DOI] [PubMed] [Google Scholar]
- 31.Garrett DS, Seok Y, Peterkofsky A, Gronenborn AM, Clore GM. Solution structure of the 40,000Mr phosphoryl transfer complex between the N-terminal domain of enzyme I and HPr. Nat. Struct. Biol. 1999;6(2):166–173. doi: 10.1038/5854. [DOI] [PubMed] [Google Scholar]
- 32.Suh J, Iwahara J, Clore GM. Intramolecular domain- domain association/dissociation and phopsphoryl transfer in the mannitol transporter of Escherichia coli are not coupled. Proc. Natl. Acad. Sci. U. S. A. 2007;104(9):3153–3158. doi: 10.1073/pnas.0609103104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu J, Hu K, Williams DC, Jr., Komlosh ME, Cai M, Clore GM. Solution NMR structure of productive and non-productive complexes between the A and B domains of the cytoplasmic subunit of the Mannose transporter of the Eschericia coliI phosphotransferase system. J. Biol. Chem. 2008;283(16):11024–11037. doi: 10.1074/jbc.M800312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung Y, Cai M, Clore GM. Solution structure of the IIAChitobiose-IIBChitobiose complex of the N,N'-diacetylchitobiose branch of the Escherichia coli phosphertransferase system. J. Biol. Chem. 2010;285(6):4173–4184. doi: 10.1074/jbc.M109.080937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang G, Louis JM, Sondej M, Seok Y, Peterkofsky A, Clore GM. Solution structure of the phosphoryl transfer complex between the signal transducing proteins HPr and IIAGlucose of the Escherichia coli phosphoenolpyruvate: sugar phosphotransferase system. EMBO. 2000;19(21):5635–5649. doi: 10.1093/emboj/19.21.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornilescu G, Lee BR, Cornilescu CC, Wang G, Peterkofsky A, Clore GM. Solution structure of the phosphoryl transfer complex between the cytoplasmic A domain of the mannnitol transporter IIMannitol and HPr of the Escherichia coli phosphotransferase system. J. Biol. Chem. 2002;277(44):42289–42298. doi: 10.1074/jbc.M207314200. [DOI] [PubMed] [Google Scholar]
- 37.Cai M, Williams DC, Jr., Wang G, Lee BR, Peterkofsky A, Clore GM. Solution structure of the Phosphoryl transfer complex between the signal-transducing protein IIAGlucose and the cytoplasmic domain of the glucose transporter IICBGlucose of the Escherichia coli glucose phosphotransferase system. J. Biol. Chem. 2003;278(27):25191–25206. doi: 10.1074/jbc.M302677200. [DOI] [PubMed] [Google Scholar]
- 38.Williams DC, Jr., Cai M, Suh J, Peterkofsky A, Clore GM. Solution NMR structure of the 48-kDa IIAMannose-HPr complex of the Escherichia coli mannose phosphotransferase system. J. Biol. Chem. 2005;280(21):20775–20784. doi: 10.1074/jbc.M501986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mourey L, Re SD, Pedelacq J, Tolstykh T, Faurie C, Guillet V, Stock JB, Samama J. Crystal structure of the CheA histidine phosphotransfer domain that mediates response regulator phosphorylation in bacterial chemotaxis. J. Biol. Chem. 2001;276(33):31074–31082. doi: 10.1074/jbc.M101943200. [DOI] [PubMed] [Google Scholar]
- 40.Brunger AT. A System for X-ray Crystallography and NMR. X-PLOR. version 3.1 Yale University Press; New Heaven, CT: 1992. [Google Scholar]
- 41.Zhou H, Dahlquist FW. Phosphotransfer site of the chemotaxis-specific protein kinase CheA as revealed by NMR. Biochemistry. 1997;36(4):699–710. doi: 10.1021/bi961663p. [DOI] [PubMed] [Google Scholar]
- 42.Stewart RC, Jahreis K, Parkinson JS. Rapid phosphotransfer to CheY from a CheA protein Lacking the CheY – binding domain. Biochemistry. 2000;39(43):13157–13165. doi: 10.1021/bi001100k. [DOI] [PubMed] [Google Scholar]
- 43.Frost AA, Pearson RG. Kinetics and Mechanism. 2nd edition John Wiley & Sons, Inc.; NY, USA: 1965. p. 166. [Google Scholar]
- 44.Welch M, Chinardet N, Mourey L, Birck C, Samama J. Structure of the CheY – biding domain of histidine kinase CheA in complex with CheY. Nat. Strust. Biol. 1998;5(1):25–29. doi: 10.1038/nsb0198-25. [DOI] [PubMed] [Google Scholar]
- 45.Gouet P, Chinardet N, Welch M, Guillet V, Cabantous S, Birck C, Mourey L, Samama J. Further insights into the mechanism of function of the response regulator CheY from cystallorgraphic studies of the CheY-CheA124–257 complex. Acta. Cryst. 2001;D57:44–51. doi: 10.1107/s090744490001492x. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Swanson RV, Simon MI, Weis RM. The response regulators CheB and CheY exhibit competitive binding to the kinase CheA. Biochemistry. 1995;34(45):14626–14636. doi: 10.1021/bi00045a003. [DOI] [PubMed] [Google Scholar]
- 47.McEvoy MM, Muhandiran DR, Kay LE, Dahlquist FW. Structure and dynamics of a CheY- binding domain of the chemotaxis kinase CheA determined by nuclear magnetic resonance spectroscopy. Biochemistry. 1996;35(18):5633–5640. doi: 10.1021/bi952707h. [DOI] [PubMed] [Google Scholar]
- 48.Volz K, Matsumura P. Crystal structure of Escherichia coli CheY refined at 1.7-Å resolution. J. Biol. Chem. 1991;266(23):15511–15519. doi: 10.2210/pdb3chy/pdb. [DOI] [PubMed] [Google Scholar]
- 49.Varughese KI, Tsigelny I, Zhao H. The crystal structure of beryllofluoride Spo0F in complex with the phosphotransferase Spo0B represents a phosphotransfer pretransition state. J. Bacteriol. 2006;188(13):4970–4977. doi: 10.1128/JB.00160-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao X, Copeland DM, Soares AS, West AH. Crystal structure of a complex between the phosphorelay protein YPD1 and the response regulator domain of SLN1 bound to a phosphoryl analog. J. Mol. Biol. 2008;375(4):1141–1151. doi: 10.1016/j.jmb.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park S, Beel BD, Simon MI, Bilwes AM, Crane BR. In different organisms, the mode of interaction between two signaling proteins is not necessarily conserved. Proc. Natl. Acad. Sci. U. S. A. 2004;101(32):11646–11651. doi: 10.1073/pnas.0401038101. [DOI] [PMC free article] [PubMed] [Google Scholar]