

The paper by Tomita and colleagues (1) completes a series of experiments carried out in many laboratories (2–4), which all lead to the same conclusion. Mutations in all three of the known pathogenic genes, the amyloid precursor protein (APP) gene (5), the presenilin 1 (PS1) gene (6), and the presenilin 2 (PS2) gene (7), have in common the fact that they alter APP processing such that an increased amount of the 42(43) amino acid peptide Aβ42(43) is produced (1–4, 8). The effects of the APP mutations (summarized in Table 1) have been elucidated over the past 5 years. However, the data implicating a link between the presenilins (PS) and APP began with the seminal observation that serum from individuals with PS-encoded Alzheimer disease (AD) contained, and that fibroblasts from such individuals produced, more Aβ42(43) (8). Since these observations were made in tissues from affected individuals, the effects of mutant PS on APP processing have been reproduced in a remarkably diverse series of tissues, in a large number of independent laboratories. Thus the effects have been seen in a range of transfected cells and in a variety of transgenic mice (some containing only the endogenous APP, and others doubly transgenic for both APP and PS wild-type and mutations) (1–4).
Table 1.
The known routes to Alzheimer disease
| Primary cause | Route | Final result |
|---|---|---|
| Down syndrome | More APP production | More Aβ42(43) and more Aβ40 |
| APP670/1 (Swedish) | Potentiation of β-secretase | More Aβ42(43) and more Aβ40 |
| APP692 (Flemish) | Inhibition of α-secretase | More Aβ42(43) |
| APP717 (London) | Alteration of site of γ-secretase cut | More Aβ42(43) |
| PS1 mutations | Subtle alteration of APP processing | More Aβ42(43) |
| PS2 mutations | Subtle alteration of APP processing | More Aβ42(43) |
What are the implications of these data, and what questions now need to be addressed?
Perhaps the most obvious and important implication is that these data provide the strongest possible evidence that the “amyloid cascade hypothesis” for the etiology and pathogenesis of AD is correct, at least in outline (9, 10). While there have been a large number of hypotheses concerning the pathogenesis of this tragic and prevalent disorder, the genetic evidence, together with work such as that described by Tomita and colleagues (1), and pathological analyses showing that Aβ42(43) is deposited early and selectively in the plaques that are characteristic of the disease (11) and in vitro data showing that this form of Aβ is particularly fibrillogenic (12), all combine to give a single, coherent picture of the pathogenesis of the disease. In this scheme, Aβ deposition, precipitated by Aβ42(43) initiates the disease pathogenesis. All the other features of the disease: the neurofibrillary tangles, the synapse and cell loss, and the dementia follow from this critical initiating event. While the mutations in these genes account for only a small proportion of the total number of cases of AD, it is impressive that all known causes of AD, including Down syndrome, appear to share this common mechanism. The available data on AD in general would suggest that similar increases in Aβ42(43) may occur in a proportion of other, more “typical” AD cases (8): however, other mechanisms (such as decreased clearance of Aβ deposits) also may play a pathogenic role in some cases. This is important, not only from a purely scientific perspective, but also because it leads to the suggestion that therapies aimed at or downstream of APP processing have every chance of having general applicability. Of course, the existence of transgenic animals that replicate some of the features of the disease and constructed based on these genetic findings offers the potential to test these therapies in a comparatively rapid and efficient manner (13, 14).
However, the nature of the connection between the PS and APP processing remains obscure, and it is of importance to try and define the biochemistry of this relationship, not only for purely scientific reasons, but also because it may offer some novel therapeutic targets.
The first question relates to the nature of the effects of the PS mutations: do they represent a gain in function, a loss of function, or a gain of a novel function? Because all but one of the mutations have been missense mutations (15) [the single exception being an in-frame deletion of part of the hydrophilic loop of PS1 (see Fig. 1) (16)], the consensus was that these mutations most probably caused gains of misfunction (see, for example, ref. 15). This supposition was strengthened by the fact that mice expressing mutant PS genes (but still with their endogenous PS genes) showed the Aβ phenotype, suggesting that normal levels of the wild-type endogenous protein could not compensate for the mutant (2–4). However, more recent work, particularly on the Caenorhabditis elegans isologues of the PS, has cast doubt on this explanation.
Figure 1.
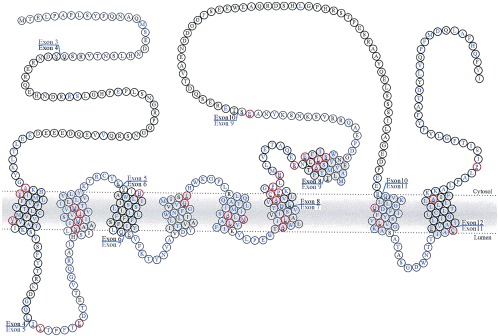
The structure of the PS1 protein derived from the author’s interpretation of ref. 20, showing the positions of the exon boundaries (21) and mutations (reviewed in ref. 22).
Two C. elegans isologues of the PS have been identified: spe-4 and sel-12 (17, 18). Both are recessive phenotypes. spe-4 mutants have a defect in spermatogenesis believed to be caused by a problem with protein trafficking in the Golgi (17), and sel-12 mutants have an egg-laying defect in which the notch pathway is implicated (18). Molecular analysis of the latter with respect to the PS has been particularly informative, because, in this pathway, wild-type PS rescues the phenotype, but several missense mutations do not (ref. 19, †). Also, work in which PS expression is reduced in cells transfected with antisense constructs has shown that this reduction of PS expression leads to an increase in Aβ42(43) production.‡ These data strongly suggest that there is a loss of function component to the effects of the mutations (19), although it is clear that complete loss of function is extremely unlikely because mice in which the PS1 gene has been knocked out, die in utero.§ If loss of PS function is critical to the AD phenotype, then understanding the normal roles of this family of proteins becomes key. To date, we have only the sketchiest of ideas as to what these roles might be. Clearly, further genetic analysis of C. elegans is warranted, specifically to determine other components of the spe-4 and sel-12 pathways so that their human homologues can then be tested for any role in the expression of the Aβ phenotype. Thus, it may well be that assessment of Aβ42(43) will be used as a surrogate marker for AD, and that egg-laying defects in C. elegans will be used as a surrogate for the Aβ phenotype. The development of powerful molecular screens, such as these, for identifying components of the early stages in the pathogenesis of the disease, can only speed the process of understanding the disease.
The cliché that many reviews (and grant applications) on AD used to start with was “Alzheimer disease is a dementia of unknown cause and inexorable progression.” Over this last couple of years, this cliché has become no longer true. We now know several causes of the disease (all genetic so far), and all of these “causes” have pointed toward the path of APP metabolism and Aβ production and deposition as being the key early event in disease pathogenesis. Unfortunately, the “inexorable progression” part of the cliché remains true; however, genetics and molecular biology now are revealing credible drug targets, and it is to be hoped that relatively soon we will be able to turn this leap in understanding into effective therapy. The unofficial “goal” of the National Institute on Aging was to have some form of effective therapy by the year 2000. This ambitious goal may yet be realized.
Footnotes
Haass, C. & Baumeister, R., Data presented at Society for Neuroscience Meeting, Washington, DC, November 1996.
Younkin, S. & Refolo, L., Data presented at Society for Neuroscience Meeting, Washington, DC, November 1996.
Sisodia, S. & Wong, C., Data presented at Society for Neuroscience Meeting, Washington, DC, November 1996.
References
- 1.Tomita T, Maruyama K, Saido T C, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J, Grunberg J, Haass C, Iwatsubo T, Obata K. Proc Natl Acad Sci USA. 1997;94:2025–2030. doi: 10.1073/pnas.94.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duff K, Eckman C, Zehr C, Yu X, Prada C M, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon M N, Holcombe L, Refolo L, Zenk B, Hardy J, Younkin S. Nature (London) 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 3.Borchelt D, Thinakaran G, Eckman C B, Lee M K, Davenport F, et al. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 4.Citron M, Westaway D, Xia W, Carlson G, Diehl G, et al. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 5.Goate A, Chartier-Harlin M C, Mullan M C, Brown J, Crawford F, et al. Nature (London) 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 6.Sherrington R, Rogaev E I, Liang Y, Rogaeva E A, Levesque G, et al. Nature (London) 1995;375:754–760. [Google Scholar]
- 7.Levy-Lahad E, Wysman E M, Nemens E, Anderson L, Goddard K A B, Weber J L, Bird T D, Schellenberg G D. Science. 1995;269:973–977. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- 8.Scheuner D, Eckman C, Jensen M, Song X, Citron M, et al. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 9.Hardy J, Allsop D. Trends Pharm Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 10.Selkoe D. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 11.Mann D M A, Iwatsubo T, Cairns N J, Lantos P L, Nochlin D, Sumi S M, Bird T D, Poorkaj P, Hardy J, Hutton M, Prihar G, Crook R, Rossor M N, Haltia M. Ann Neurol. 1996;40:149–156. doi: 10.1002/ana.410400205. [DOI] [PubMed] [Google Scholar]
- 12.Jarrett J T, Berger E T, Lansbury P T. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 13.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, et al. Nature (London) 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 14.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 15.Cruts M, Hendriks L, Van Broeckhoven C. Hum Mol Genet. 1996;5:1449–1455. doi: 10.1093/hmg/5.supplement_1.1449. [DOI] [PubMed] [Google Scholar]
- 16.Perez-Tur J, Froelich S, Prihar G, Crook R, Baker M, et al. NeuroReport. 1995;7:297–301. [PubMed] [Google Scholar]
- 17.L’Hernault S W, Arduengo P M. J Cell Biol. 1992;119:55–68. doi: 10.1083/jcb.119.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levitan D, Greenwald I. Nature (London) 1996;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 19.Levitan D, Doyle T G, Brousseau D, Lee M K, Thinkaran G, Slunt H H, Sisodia S S, Greenwald I. Proc Natl Acad Sci USA. 1996;93:14940–14944. doi: 10.1073/pnas.93.25.14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Greenwald I. Neuron. 1996;17:1015–1021. doi: 10.1016/s0896-6273(00)80231-7. [DOI] [PubMed] [Google Scholar]
- 21.Alzheimer Collaborative Group. Nat Genet. 1996;11:219–222. [Google Scholar]
- 22.Hardy, J. (1997) Trends Neurosci., in press.