

Abstract
Herein we report the discovery and SAR of a novel metabotropic glutamate receptor 3 (mGlu3) NAM probe (ML289) with 15-fold selectivity versus mGlu2. The mGlu3 NAM was discovered via a ‘molecular switch’ from a closely related, potent mGlu5 positive allosteric modulator (PAM), VU0092273. This NAM (VU0463597, ML289) displays an IC50 value of 0.66 μM and is inactive against mGlu5. 2012
Keywords: metabotropic glutamate receptor 3, mGlu3, molecular switch, NAM
The metabotropic glutamate receptors (mGlus) are members of the GPCR family C, characterized by a large extracellular amino-terminal agonist (venus fly-trap) binding domain.1,2 Eight mGlus have been cloned, sequenced and assigned to three groups (Group I: mGlu1 and mGlu5; Group II: mGlu2 and mGlu3; Group III: mGlu4,6,7,8) based on their sequence homology, pharmacology, and coupling to effector mechanisms.1,2 Highly subtype-selective allosteric ligands (both PAMs, positive allosteric modulators, and/or NAMs, negative allosteric modulators) have been developed for mGlu1, mGlu2, mGlu4, mGlu5 and mGlu7.3-11 However, aside from mGlu2 PAMs, most Group II ligands do not discriminate between mGlu2 and mGlu3; a necessary requirement as these two receptors have highly divergent expression and function.12-15 Thus, due to a lack of selective small molecule probes, it has been difficult to discern distinct pharmacological roles for mGlu3, though numerous studies suggest mGlu3 is involved in glial-neuronal communication and may have therapeutic potential for the treatment of schizophrenia, Alzheimer’s disease, and depression.3-5,12,16-18
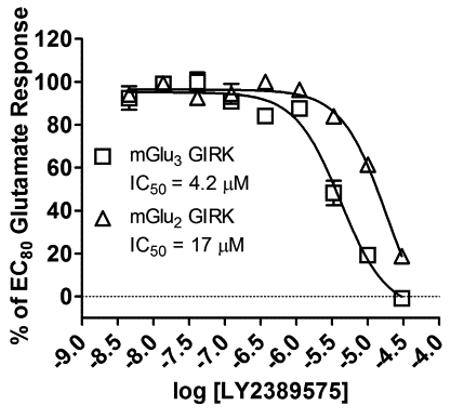
To date, only two mGlu3 NAMs have been reported (Fig. 1).19,20 The first, reported by Addex, is RO4491533 (1), a dual mGlu2/mGlu3 NAM (mGlu2 IC50 = 296 nM, mGlu3 IC50 = 270 nM) based on a benzodiazepinone nucleus that was efficacious in preclinical cognition and depression models.19 At about the same time, Lilly disclosed LY2389575 (2) as a selective mGlu3 NAM;20 however, when measuring native coupling of these receptors to G protein-coupled inwardly-rectifying potassium (GIRK) channels via thallium flux,21 we have observed that 2 is only ~4-fold selective for mGlu3 over mGlu2 (mGlu2 IC50 = 17 μM, mGlu3 IC50 = 4.2 μM) 22. Thus, there is a critical need for potent and selective mGlu3 ligands.
Figure 1.
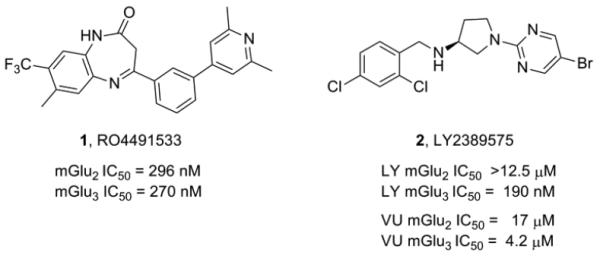
Structures of mGlu3 NAMs RO4491533 (1) and LY2389575 (2), both dual mGlu2/mGlu3 NAMs.
In the absence of an HTS campaign to identify novel mGlu3 NAMs, we elected to take advantage of the propensity of certain mGlu5 PAM chemotypes to easily modulate the mode of pharmacology or mGlu subtype selectivity with subtle structural alterations, ie. ‘molecular switches’.23-27 One such chemotype that we26,27 and others28 have reported on with a high propensity for displaying ‘molecular switches’ is represented by VU0092273 (3), a potent MPEP-site mGlu5 PAM (Fig. 2).27 Compound 3 also possessed weak mGlu3 NAM activity (IC50 ~ 10 μM, inhibits EC80 by 72%, Fig. 2B), but otherwise showed no activity at the six other mGlu subtypes.
Figure 2.
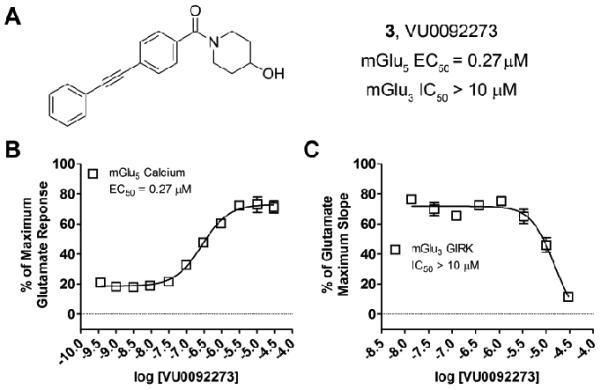
A) Structure of VU0092273 (3), a potent mGlu5 PAM (pEC50 = 6.57±0.09, EC50 = 0.27 μM). B) mGlu5 PAM concentration-response curve (CRC) in presence of an EC20 of glutamate. C) mGlu3 antagonist CRC. 3 displayed weak NAM activity at mGlu3 (IC50 >10 μM, inhibits EC80 ~ 72%).
Thus, 3 became our lead compound from which to develop a potent and selective mGlu3 NAM. As we have previously reported, due to the steep nature of allosteric modulator SAR (especially in series prone to ‘molecular switches’), we pursued an iterative parallel synthesis approach for the chemical optimization of 3.3,4 Previous work in this scaffold indicated that mGlu5 PAM activity could be greatly diminished with substitution other than fluorine on the distal aryl ring, as well as with modifications to the amide moiety.26 Therefore, our first generation library design (Fig. 3) initially held the 4-hydroxypiperidine amide constant, while surveying a diverse array of functionalized aryl/heteroaryl rings as well as other aliphatic groups. Once mGlu3-preferring modifications were identified, these would be maintained while an amide scan would be performed to improve mGlu3 NAM activity while eliminating mGlu5 PAM activity.
Figure 3.
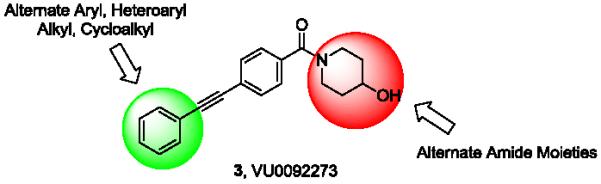
Library optimization strategy for VU0092273 (3) to improve mGlu3 NAM activity while simultaneously eliminating mGlu5 PAM activity.
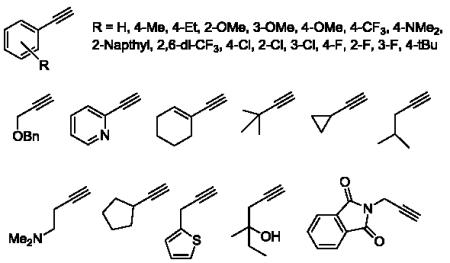
Our first 48-member library was prepared as shown in Scheme 1, and purified, to >98% purity, by reverse phase chromatography.29 Commercial 4-iodobenzoic acid 4 was coupled to 4-hydroxypiperdine, under standard EDC/HOBt conditions, to provide amide 5 in 95% yield. Once synthesized, 5 then underwent Sonogashira coupling reactions with a diverse array of 48 functionalized terminal acetylenes to provide analogs 6.30 True to allosteric modulator SAR, 47/48 of the analogs were either inactive on mGlu3 (IC50 >10 μM) or only afforded modest inhibition (5-50% Glu Min) of the glutamate EC80. Only one compound, 7 (VU0457299), possessing a 4-methoxyphenyl moiety, displayed mGlu3 NAM potency below 10 μM (mGlu3 IC50 = 3.8 μM, % Glu Min = 10.4±2.1). Interestingly, the regioisomeric 2-OMe and 3-OMe congeners were inactive.
Scheme 1.
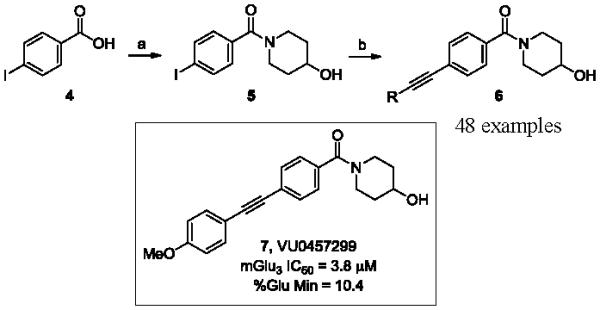
Reagents and conditions: (a) EDC, DMAP, DCM, DIPEA, 95%; (b) 20% CuI, 5% Pd(PPh3)4, 48 acetylenes (1.1 equiv.), DMF, DIEA, 60 °C, 1 h, 15-90%.
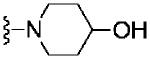
Based on these data, the next round of library synthesis held the 4-methoxyphenyl moiety in 7 constant, and 48 amines32 were employed to survey alternative amides. This library, prepared according to Scheme 2, was far more productive, providing several analogs 10 with mGlu3 NAM potencies below 10 μM; however, SAR was still quite steep (Table 1). In general, polar (10a-e) and basic substituents (10f and 10g) were the most efficacious. Of great interest was the enantioselective mGlu3 inhibition displayed by the (S)-piperidine carboxylic acid 10c (IC50 = 5.7 μM) and the (R)-enantiomer 10d (IC50 >>10 μM, essentially inactive). This result led us to resolve racemic 3-hydroxymethyl analog 10e (IC50 = 2.1 μM), which afforded a full block of the EC80. Following Scheme 2, both the (R)- and (S)-enantiomers of 10e, 11 (VU0463597) and 12 (VU0463593) were prepared and assayed in the mGlu3 GIRK assay (Fig. 4). Here, 11 (pIC50 = 5.83±0.05, IC50 = 1.5 μM) was 2-fold more potent than 12 (pIC50 = 5.49±0.02, IC50 = 3.3 μM), but both afforded full blockade (% Glu Mins of −4.4±1.9 and −1.7±1.3 respectively). Efforts now shifted towards more fully characterizing 11 (VU0463597).
Scheme 2.
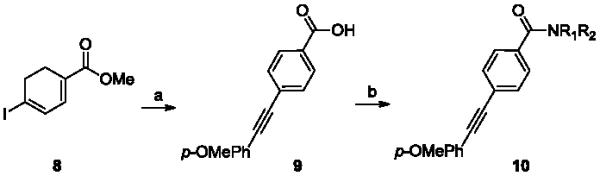
Reagents and conditions: (a) i.) 20% CuI, 5% Pd(PPh3)4, 4-OMePh acetylene (1.1 equiv.), DMF, DIEA, 60 °C, 1 h, 82%, ii) KOH, aq. MeOH, 95%; (b) HNR1R2, EDC, DMAP, DCM, DIPEA, 40-96%.
Table 1.
Structures and Activities of Analogs 10.
| ||||
|---|---|---|---|---|
|
| ||||
| Cmpd | NR1R2 | plC50a ±SEM |
IC50 (μM)a | %Glu Minb ±SEM |
| 7 |
|
5.42±0.04 | 3.8 | 10.4±2.1 |
| 10a |
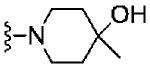
|
5.30±0.06 | 5.0 | 4.4±0.7 |
| 10b |
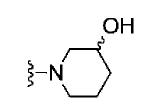
|
5.61±0.07 | 2.5 | −1.1±0.5 |
| 10c |
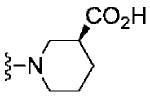
|
5.24±0.01 | 5.7 | 2.2±1.1 |
| 10d |
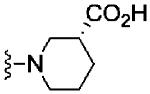
|
>10 | 16.4±2.5 | |
| 10e |
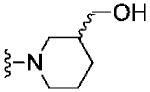
|
5.69±0.04 | 2.1 | 0.5±0.4 |
| 10f |
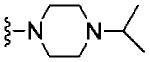
|
>10 | 7.6±3.2 | |
| 10g |
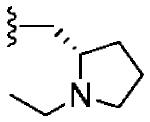
|
>10 | 9.6±2.1 | |
Measured in an mGlu3 GIRK assay.
% Glu Min is the % inhibition of the compound on an EC80 concentration of glutamate. Values represent the mean ± standard error mean for three independent experiments performed in triplicate.
Figure 4.
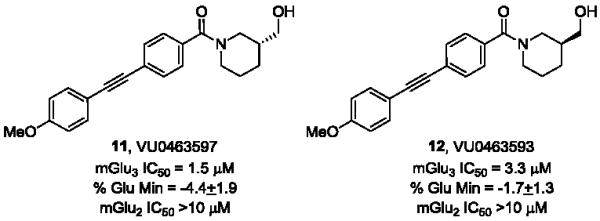
Structures and activities of (R)-11 and (S)-12, mGlu3 NAMs.
We next evaluated the selectivity of 11(VU0463597) between mGlu2 and mGlu5. Utilizing our mGlu2 GIRK line, the IC50 was much greater than 10 μM, with the CRC not reaching baseline at the highest concentration tested (30 μM) (Fig. 5A, triangles). Similarly, 11 was inactive for potentiating an EC20 concentration of glutamate (Fig. 5B, triangles) or inhibiting an EC80 concentration of glutamate (Fig. 5C, triangles) in our standard mGlu5 calcium assay. As our calcium assays typically drive our mGlu drug discovery programs, we also evaluated 11 (VU0463597) in an mGlu3 calcium assay in which mGlu3 is co-expressed with the promiscuous G protein Gα15 (Fig. 5B-C, squares). Here, we see slightly improved mGlu3 NAM potency (pIC50 = 6.18±0.03, IC50 = 0.66 μM, % Glu Min = 2.1±0.3) compared to the mGlu3/GIRK line. To verify that 11 antagonizes mGlu3 via a non-competitive (allosteric) mechanism of action, we next performed a Schild analysis. In these studies, 11 dose-dependently induced a rightward shift and decreased the maximal efficacy of the orthosteric agonist glutamate (Fig. 6), consistent with a non-competitive (allosteric) mechanism of action. Thus, starting from a very potent mGlu5 PAM (EC50 = 0.27 μM), we were able to optimize and develop a potent and selective mGlu3 NAM with high selectivity (~15-fold) versus mGlu2 and complete specificity versus mGlu5.
Figure 5.
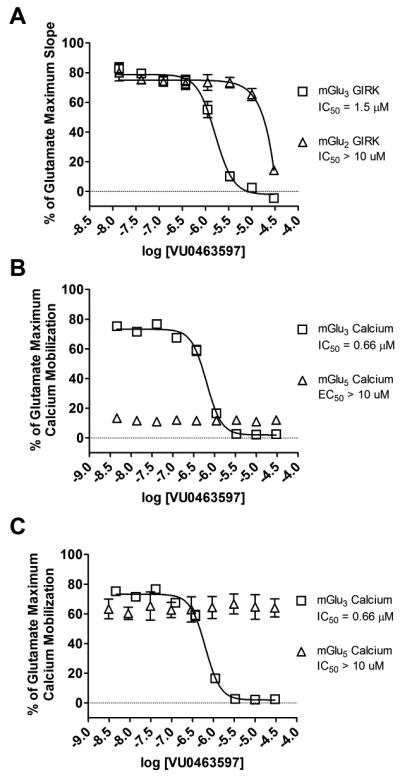
In vitro molecular pharmacology characterization of 11 (VU0463597). A) Concentration-response curves of mGlu2 and mGlu3 GIRK (antagonist mode). B) mGlu3 calcium (antagonist mode) and mGlu5 calcium (PAM mode). C) mGlu3 calcium (antagonist mode) and mGlu5 calcium (antagonist mode).
Figure 6.
Schild Analysis of 11 (VU0463597). The concentration-response of glutamate for mGlu3 GIRK is non-competitively inhibited by 11.
With this potent and selective mGlu3 NAM in hand, we began profiling 11 in a battery of ancillary pharmacology and DMPK assays to assess the quality of this probe for potential in vivo studies. A Lead Profiling Screen at Ricerca32 (68 GPCRs, ion channels and transporters screened at 10 μM in radioligand binding assays) failed to identify any off target activities for 11 (no inhibition >25% @ 10 μM). In our tier 1 in vitro DMPK screen, compound 11 displayed no P450 inhibition in human liver microsomes (IC50 >30 μM vs. 3A4, 2C9, 2D6 and 1A2), high plasma protein binding with fraction unbound (fu) levels between 1 and 2% in both rat and and human plasma, respectively; fu determined in rat brain homogenate was 1%. Intrinsic clearance (CLint) determined in rat and human liver microsomes indicated that compound 11 was rapidly cleared in vitro (rat, CLint = 240 mL/min/kg; human, CLint = 571.8 mL/min/kg). An in vitro to in vivo clearance correlation was established, as compound 11 was found to be a moderately cleared compound in rat (CL = 33 mL/min/kg) following intravenous administration (1 mg/kg); the low volume of distribution at steady state (Vss 0.6 L/kg) and moderate clearance produced a relatively short t1/2 (16.8 min) in vivo. Metabolite ID studies in rat and human liver microsomes (Fig. 7) indicated that the principle biotransformation pathway was P450-mediated O-demethylation of 11 to generate the phenol 13, a metabolite that was subsequently shown to be inactive at mGlu3 and mGlu5.
Figure 7.
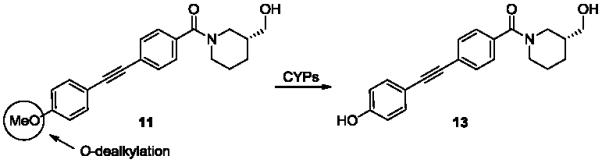
Oxidative O-dealkylation of 11 in rat and human liver microsomes.
As our earlier SAR work indicated that the methyl ether was critical for mGlu3 NAM activity, we performed an IP plasma:brain level (PBL) study to determine if we could achieve meaningful CNS exposure if first-pass metabolism was bypassed. Significantly, in a 10 mg/kg (10% Tween80 in 0.5% methylcellulose) IP plasma:brain level (PBL) study, we observed a brain (16.3 μM):plasma (9.7 μM) ratio of 1.67, indicating that 11 (VU0463597) was indeed centrally penetrant. Based on brain homogenate binding studies, this correlates to ~163 nM free in rat brain at the 10 mg/kg dose, a value below the mGlu3 IC50 (0.66 μM); thus, in order to provide adequate target engagement, a 50 mg/kg dose may be required for in vivo efficacy with this first generation mGlu3 NAM probe.
This project was an MLPCN Medicinal Chemistry FastTrack program, and based on the profile of 11, it was declared an MLPCN probe and assigned the identifier ML289.33 As such, ML289 is freely available upon request.34
In summary, we have developed a potent, selective (>15-fold vs. mGlu2) and centrally penetrant mGlu3 NAM 11 (VU0463597 or ML289) with a good overall CYP profile. ML289 is also highly selective versus mGlu5, which is notable as our lead was a 0.27 μM mGlu5 PAM, and suggests ligand cross-talk between allosteric binding sites on mGlu3 and mGlu5. Once again, a subtle ‘molecular switch’, in the form of a p-OMe moiety, conferred selective mGlu3 inhibition over mGlu5 potentiation. Further chemical optimization efforts, as well as detailed molecular pharmacological characterization of ML289, are in progress and will be reported in due course.
Acknowledgments
This work was supported by grants from the NIH. Vanderbilt is a Specialized Chemistry Center within the Molecular Libraries Probe Centers Network (U54 MH84659).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 2.Conn PJ, Pin J-P. Annu. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 3.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J. Med. Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conn PJ, Christopolous A, Lindsley CW. Nat. Rev. Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conn PJ, Lindsley CW, Jones C. Trends in Pharm. Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robichaud AJ, Engers DW, Lindsley CW, Hopkins CR. ACS Chem. Neurosci. 2011;2:433–449. doi: 10.1021/cn200043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheffler DJ, Pinkerton AB, Dahl R, Markou A, Cosford NDP. ACS Chem. Neurosci. 2011;2:382–393. doi: 10.1021/cn200008d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owen DR. ACS Chem. Neurosci. 2011;2:394–401. doi: 10.1021/cn2000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emmitte KA. ACS Chem. Neurosci. 2011;2:443–449. doi: 10.1021/cn2000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stauffer SR. ACS Chem. Neurosci. 2011;2:450–470. doi: 10.1021/cn2000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki G, Tsukamoto N, Fushiki H, Kawagishi A, Nakamura M, Kurihara H, Mitsuya M, Ohkubo M, Ohta H. J. Pharmacol & Exp, Ther. 2007;323:147–156. doi: 10.1124/jpet.107.124701. [DOI] [PubMed] [Google Scholar]
- 12.Harrision PJ, Lyon L, Sartorius LJ, Burnet PWJ, Lane TA. J. Psychopharm. 2008;22:308–322. doi: 10.1177/0269881108089818. [DOI] [PubMed] [Google Scholar]
- 13.Kew JNC, Kemp JA. Psychopharmacology. 2005;179:4–29. doi: 10.1007/s00213-005-2200-z. [DOI] [PubMed] [Google Scholar]
- 14.Woltering TJ, Wichmann J, Goetschi E, Knoflach F, Ballard TM, Huwyler J, Gatti S. Bioorg. Med. Chem. Lett. 2010;20:6969–6974. doi: 10.1016/j.bmcl.2010.09.125. [DOI] [PubMed] [Google Scholar]
- 15.Corti C, Battaglia G, Molinaro G, Riozzi B, Pittaluga A, Corsi M, Mugnaini M, Nicoletti F, Bruno V. J. Neurosci. 2007;27:8297–8308. doi: 10.1523/JNEUROSCI.1889-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moghaddam B, Adams BW. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 17.Matrrisciano F, Panaccione I, Zusso M, Giusti P, Tatarelli R, Iacovelli L, Mathe AA, Gruber SH, Nicoletti F, Girardi P. Mol. Psychiarty. 2007;12:704–706. doi: 10.1038/sj.mp.4002005. [DOI] [PubMed] [Google Scholar]
- 18.Markou A. Biol. Psychiatry. 2007;61:17–22. doi: 10.1016/j.biopsych.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 19.Campo B, Kalinichev M, Lambeng N, El Yacoubi M, Royer-Urios I, Schneider M, Legarnd C, Parron D, Girard F, Bessif A, Poli S, Vaugeois J-M, Le Poul E, Celanire S. J. Neurogenetics. 2011;24:152–166. doi: 10.3109/01677063.2011.627485. [DOI] [PubMed] [Google Scholar]
- 20.Caraci F, Molinaro G, Battaglia G, Giuffrida ML, Riozzi B, Traficante A, Bruno V, Cannella M, Mero S, Wang X, Heinz BA, Nisenbaum ES, Britton TC, Drago F, Sortino MA, Copani A, Nicoletti F. Mol. Pharm. 2011;79:618–626. doi: 10.1124/mol.110.067488. [DOI] [PubMed] [Google Scholar]
- 21.Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, Weaver CD. Mol. Pharm. 2008;73:1213–1224. doi: 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]
-
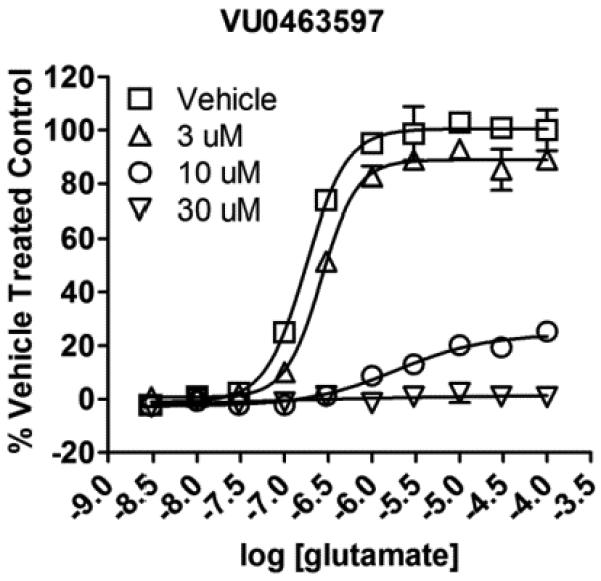
22.CRCs for LY2389575 (2) in our GIRK assays:
- 23.Sharma S, Rodriguez A, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2008;18:4098–4101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma S, Kedrowski J, Rook JM, Smith JM, Jones CK, Rodriguez AL, Conn PJ, Lindsley CW. J. Med. Chem. 2010;52:4103–4106. doi: 10.1021/jm900654c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. Biochemistry. 2011;50:2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams R, Manka JT, Rodriguez AL, Vinson PN, Niswender CM, Weaver CD, Jones CK, Conn PJ, Lindsley CW, Stauffer SR. Bioorg. Med. Chem. Lett. 2011;21:1350–1353. doi: 10.1016/j.bmcl.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon U, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Mol. Pharm. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ritzen A, Sindet R, Hentzer M, Svendsen N, Brodbeck RM, Bungaard C. Bioorg. Med. Chem. Lett. 2009;19:3275–3278. doi: 10.1016/j.bmcl.2009.04.095. [DOI] [PubMed] [Google Scholar]
- 29.Leister WH, Strauss KA, Wisnoski DD, Zhao Z, Lindsley CW. J. Comb. Chem. 2003;5:322–329. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
-
30.Representative acetylenes employed in the 48-member library:
-
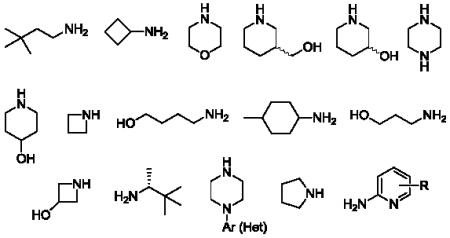
31.Representative amines employed in the 48-member library:
- 32.For full information on the targets in the Lead Profiling Screen at Ricerca, please see: www.ricerca.com
- 33.For information on the MLPCN please see: http://mli.nih.gov/mli/mlpcn/
- 34.To request your free sample of ML289, please craig.lindsley@vanderbilt.edu