

Abstract
With over 60,000 protein structures available in the Protein Data Bank, it is frequently possible use one of them to obtain starting phase information and to solve new crystal structures. Molecular replacement1–4 procedures, which search for placements of a starting model within the crystallographic unit cell that best account for the measured diffraction amplitudes, followed by automatic chain tracing methods5–8, have allowed the rapid solution of large numbers of protein structures. Despite extensive work9–14, molecular replacement or the subsequent rebuilding usually fail with more divergent starting models based on remote homologues with less than 30% sequence identity. Here we show that this limitation can be substantially reduced by combining algorithms for protein structure modeling with those developed for crystallographic structure determination. An approach integrating Rosetta structure modeling with Autobuild chain tracing yielded high-resolution structures for 8 of 13 X-ray diffraction datasets that could not be solved in the laboratories of expert crystallographers and that remained unsolved after application of an extensive array of alternative approaches. We estimate the new method should allow rapid structure determination without experimental phase information for over half the cases where current methods fail, given diffraction datasets of better than 3.2Å resolution, four or fewer copies in the asymmetric unit, and the availability of structures of homologous proteins with >20% sequence identity.
The limiting steps in molecular replacement are finding the location of the starting model and the interpretation of electron density maps, which are carried out using pattern recognition techniques in real and reciprocal space. The left column of Figure 1 illustrates the problem of initial model-building starting with distant comparative models (20–30% sequence identity) that have been correctly placed in the crystallographic unit cell. Automatic chain tracing methods fail on such maps because they often follow the incorrect comparative model (red) more closely than the actual structure (yellow); breaks in the density make it difficult to recover the correct backbone trace. Nevertheless, the maps contain considerable information about the native structure; for example, portions of the starting model that are not within density are generally incorrect.
Figure 1.

Examples of improvement in electron density and model quality. First row: Table 1 #6 (2.0 Å resolution); second row: Table 1 #7 (2.1 Å resolution); third row: Table 1 #12 (1.7 Å resolution). Left column: correct initial molecular replacement solution (not necessarily identifiable at this stage) using starting model and corresponding density. Middle column: energy optimized model and corresponding density. Right column: model and density following automatic building using the energy optimized model as the source of phase information. The final deposited structure is shown in yellow in each panel; the initial model, energy optimized model, and model after chain rebuilding are in red, green, and blue respectively. The sigma-A weighted 2mFo-DFc density contoured at 1.5σ is shown in gray.
Structure prediction methods such as Rosetta search for the lowest energy conformation of the polypeptide chain using physically realistic forcefields. Based on previous work showing that accurate structures could be obtained from even very sparse NMR datasets15 by using the data to guide structure prediction searches, we reasoned that structure prediction methods guided by even very noisy density maps might be able to improve a poor molecular replacement model before applying crystallographic model-building techniques. We developed an approach in which electron density maps generated from molecular replacement solutions for each of a series of starting models are used to guide energy optimization by structure rebuilding, combinatorial sidechain packing, and torsion space minimization16. New maps are generated using phase information from the energy-optimized models most consistent with the diffraction data, subjected to automatic chain tracing, and success is monitored through the free R factor.
Results on blind targets
To investigate the performance of the new method, we obtained 18 crystallographic datasets that had resisted previous attempts at structure determination. We first tested whether a comprehensive set of state-of-the-art molecular replacement approaches employing a range of full-length and trimmed templates and homology models could solve any of these structures (Supplementary Material). We were able to solve 5 of the structures with both the new method and the existing methods (Group A in Table 1), leaving 13 challenging datasets highly resistant (Supplementary Material, Section 1) to structure determination (Group B). For each of these, we identified homologous proteins of known structure17 and constructed sequence alignments and starting models9 from the five closest homologues. Starting models were used to search for up to five candidate molecular replacement solutions based on the likelihood of the experimental diffraction data2. Electron density maps were computed for each of these solutions, and used to guide energy minimization by first remodeling the unaligned regions and regions which poorly fit the density and then optimizing all backbone and sidechain torsion angles. The likelihood of the experimental diffraction data was computed for each optimized model2; if top ranked models were similar (see Methods), a map generated from the highest likelihood model was subjected to automatic chain rebuilding, density modification and refinement5. If this succeeded in building the majority of the protein and produced a model with free R factor18 significantly better than random (Rfree < 0.4), the structure was considered solved; rebuilt models were further analyzed by the crystallographers who supplied the original data. Using this approach, we were able to solve eight of the thirteen challenging cases (Table I, group B). In some cases, recognition of the correct placement of the model in the unit cell was only possible after Rosetta refinement (Supplementary Figure 2); in others the correct placement was clear but the density was too poor for chain rebuilding. In two of the cases (#12 and #13), even finding the correct molecular replacement solution first required energy based refinement12.
Table 1.
Determination of previously unsolved structures using the new approach. The seqid column gives the sequence identity to the closest homologue identified by HHpred17, and is shown in parentheses if this is an NMR structure; the sequence identity to the top crystallographic hit is also provided if there is one. The next seven columns give the Rfree of the model produced by different combinations of refinement and autobuilding approaches. The final column gives the Rfree after further refinement by the crystallographer who provided the data. Group A: Rosetta as well as one or more alternate approaches was sufficient to solve each of the structures (Rfree<0.4). Group B: only Rosetta was able to solve each structure: in subset 1, molecular replacement succeeds (in some cases ambiguously) using the template alone but model-building fails; in subset 2, refinement in Rosetta is required for molecular replacement to succeed. Targets that could not be solved by our approach are listed in Supplementary Table 1.
| id | sourcea | reso | %seqid | Rfree after Phaser MR and model-building protocol
|
Rfree (current best) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Auto- Build | Arp/Warp | SA + Autobuild | Torsion- space SA + Auto- Build | Extreme SA + Auto- Build | DEN + Auto- Build | Rosetta + Auto- Build | |||||
|
A. Solved by multiple methods
| |||||||||||
| #1 | JCSG | 2.1 | 22 | 0.31 | 0.50 | 0.30 | 0.30 | 0.30b | 0.35 | 0.31 | 0.22 |
|
| |||||||||||
| #2 | NSGC | 2.2 | 19 | 0.29 | 0.57 | 0.29 | 0.29 | 0.29b | 0.30 | 0.29 | 0.22 |
|
| |||||||||||
| #3 | UG | 2.5 | 27 | 0.34 | 0.59 | 0.29 | 0.29 | 0.29b | 0.35 | 0.27 | 0.19 |
|
| |||||||||||
| #4 | JCSG | 2.7 | 21 | 0.31 | 0.59 | 0.30 | 0.30 | 0.30b | 0.31 | 0.30 | 0.24 |
|
| |||||||||||
| #5 | ANL | 1.9 | 31 | 0.51 | 0.59 | 0.54 | 0.54 | 0.24 | 0.39 | 0.31 | 0.24 |
|
| |||||||||||
| B. Only solved by Rosetta | |||||||||||
|
1. Rosetta modeling with density required for successful model-building
| |||||||||||
| #6 | NCI | 2.0 | 30 | 0.56 | 0.59 | 0.60 | 0.55 | 0.55 | 0.50 | 0.34 | 0.20 |
|
| |||||||||||
| #7 | WI | 2.1 | 22/15 | 0.56 | 0.60 | 0.54 | 0.54 | 0.54 | 0.56 | 0.28 | 0.26 |
|
| |||||||||||
| #8 | JCSG | 2.8 | 29 | 0.52 | 0.55 | 0.50 | 0.50 | 0.51 | 0.45 | 0.36 | 0.36c |
|
| |||||||||||
| #9 | UC | 3.0 | 22 | 0.54 | 0.56 | 0.50 | 0.50 | 0.47 | 0.46 | 0.32 | 0.25d |
|
| |||||||||||
| #10 | JCSG | 3.2 | 20 | 0.54 | 0.57 | 0.51 | 0.51 | 0.53 | 0.46 | 0.39 | 0.33c |
|
| |||||||||||
| #11 | UG | 2.5 | 18 | 0.52 | 0.57 | 0.54 | 0.52 | 0.54 | 0.55 | 0.27 | 0.22 |
|
| |||||||||||
| MEAN | 0.54 | 0.57 | 0.53 | 0.52 | 0.52 | 0.50 | 0.33 | ||||
|
| |||||||||||
|
2. Rosetta homology modeling required for successful molecular replacement
| |||||||||||
| #12 | BI,HY | 1.7 | − (100) | − (0.54e) | − (0.60e) | − (0.30e) | − (0.30e) | − (0.28e) | − (0.30e) | 0.29 | 0.22 |
|
| |||||||||||
| #13f | JCSG | 2.9 | 29 | − (0.46e) | − (0.60e) | − (0.52e) | − (0.50e) | − (0.53e) | − (0.41e) | 0.39 | 0.23 |
Notes:
JCSG: Joint Center for Structural Genomics; NSCG: Northeast Center for Structural Genomics; UG: University of Graz; ANL: Argonne National Lab; NCI: National Cancer Institute; WI: Weizmann Institute of Science; UC: University of Cambridge; BI,HY: Institute Of Biotechnology, University of Helsinki
Since a single SA trajectory was sufficient to solve these cases, Extreme SA was not run. Values from the single SA run are shown for completeness
Solutions for both are essentially correct based on the selenium positions in the anomalous difference Fourier maps calculated from the experimental datae. However, structures are difficult to complete to deposition due to some MR solution model bias, poor or disordered density in numerous regions and low resolution.
Refinement ongoing.
Model-building results shown for comparison methods are based on a starting model constructed with Rosetta modeling
This structure was solved and all tests on this template were carried out using the intact template as a starting point. With this template both the molecular replacement step and subsequent rebuilding required Rosetta modeling for success. After determining the structure and completing the tests we found that it was also possible to solve the structure by molecular replacement if the template were split into two rigid subunits and the two domains were correctly chosen.
The improvement in electron density produced by density guided energy optimization and autobuilding are illustrated in Figure 1. The starting molecular replacement models are often quite inaccurate, and the density generated from these models has breaks within the backbone of the actual structure (left panels). After model rebuilding and energy guided structure optimization, backbone breaks are largely closed and both sidechains and backbone are more correctly modeled (middle panels). Automatic chain rebuilding into the improved map followed by density modification and reciprocal-space refinement further improve the model and the density (right panels). For all eight cases, the correlation between the final refined density and density from the original molecular replacement solutions is low, increases significantly after energy- and density-based structure optimization, and still further after automatic chain rebuilding (Supplementary Table 2).
Comparison to current MR methods
For each of the eight challenging cases solved with the new method we also applied a battery of existing methods (Table 1, group B and Supplementary Material, Section 1) including simulated annealing in Cartesian and torsion space in PHENIX and CNS14, DEN refinement13 in CNS, and using PHENIX Autobuild6 and ARP-WARP5 for model-building. In two cases (#12 and #13) Rosetta structure modeling was required to even place the template model, so existing methods clearly were not sufficient to obtain a correct structure. In the remaining 6 cases, final Rfree values were far lower using our new approach than with any other method tested (Table 1, Figure 2A). While conventional simulated annealing in both Cartesian and torsion space had little effect, the recently developed deformable elastic network19 (DEN) refinement protocol did improve three of the structures slightly, yielding free R values of 0.45–0.46 for these targets. Combination of DEN refinement with the method described here could lead to still more powerful approaches.
Figure 2.

Method comparison. (A) Histogram of Rfree values after autobuilding for the seven difficult blind cases solved using the new approach (Table 1, set B). For most existing approaches, none of the cases yielded Rfree values under 50%; DEN was able to reduce Rfree to 45–49% for three of the structures. For all seven cases, Rosetta energy and density guided structure optimization led to Rfree values under 40%. (B) Dependence of success on sequence identity. The fraction of cases solved (Rfree after Autobuilding < 40%) is shown as a function of template sequence identity over the 18 blind cases and 59 benchmark cases. The new method is a clear improvement below 28% sequence identity. (C) Dependence of structure determination success on initial map quality. Sigma-A weighted 2mFo-DFc density maps (contoured at 1.5σ) computed from benchmark set templates with divergence from the native structure increasing from left to right are shown in grey; the solved crystal structure is shown in yellow. The correlation with the native density is shown above each panel. The solid green bar indicates structures the new approach was able to solve (Rfree<0.4); the red bar those that torsion-space refinement or DEN refinement is able to solve, and the purple bar those that can be solved directly using the template.
Dependence of energy- and density-guided model improvement on starting model quality
To benchmark the sequence and structural divergence where the different methods break down, we studied two different protein families for which a total of 59 different template structures covering a broad range of sequence and structural similarity were available (Supplementary Tables 3–5). Each template was correctly placed in the unit cell, and then improved with either Rosetta energy- and density-based optimization, Cartesian- and torsion-space simulated annealing, or DEN refinement. For each resulting model, the correlation with the density of the deposited structure was evaluated. Automatic chain rebuilding beginning with the superimposed starting models was successful for 18 of the 59 cases, consistent with the observation that molecular replacement often fails with templates sharing less than 30% sequence identity with the target sequence. Torsion-space simulated annealing in CNS prior to autobuilding allowed solution of two additional structures, DEN refinement, three additional structures, and Rosetta energy based structure optimization, fourteen additional structures (Supplementary Figure 2; Supplementary Tables 3–5). We found the radius of convergence of the new method can be further extended by guiding energy based structure optimization by the Patterson correlation20 rather than electron density (see Supplementary Material). This allowed structure improvement and identification of the correct molecular replacement solution in two additional cases (Supplementary Figure 2, compare green to orange bar); for one of these the improvements were sufficient for autobuilding to effectively solve the structure.
Over the combined set of eighteen blind cases and the 59 benchmark cases, Rosetta refinement yielded a model with density correlation as good or better than any of the control methods for all but six structures. The dependence of success on sequence recovery over the combined set is illustrated in Figure 2B. The improvement in performance is particularly striking below 22% sequence identity, where the quality of the starting homology models becomes too low for the control methods in almost all cases. With the new method the success rate in the 15–28% sequence identity range, generally considered very challenging for molecular replacement, is over 50%.
Origins of failures
One possible origin of failure is that a homology model is simply too distant to provide useful structural information. Figure 2C illustrates the dependence of model-building on the quality of initial electron density. Conventional chain rebuilding requires a map in which the connectivity is largely correct (leftmost panel), whereas the new method can tolerate breaks in the chain more than other methods (panels 2–4), as long as there is sufficient information in the electron density map, combined with the Rosetta energy function, to guide structure optimization. The map on the far right contains too little information to guide energy based structure optimization and hence the new approach fails. In the five blind cases that have not yet been solved the comparative models may have been too low in quality, or there may have been complications in the X-ray diffraction datasets themselves.
Origins of success
Key to the success of the approach described here is the integration of structure prediction and crystallographic chain tracing and refinement methods. Simulated annealing guided by molecular force fields and diffraction data has played an important role in crystallographic refinement14,21. Structure prediction methods such as Rosetta can be even more powerful when combined with crystallographic data because the forcefields incorporate additional contributions such as solvation energy and hydrogen bonding, and the sampling algorithms can build non-modeled portions of the molecule and cover a larger region of conformational space than simulated annealing. The increased accuracy of the Rosetta forcefields and efficacy of the sampling algorithms make substantial improvement of homology-based models possible even in the absence of crystallographic data22. The difference between Rosetta sampling and simulated annealing sampling, both using crystallographic data, is illustrated in Figure 3. Beginning with the homology model placed by molecular replacement in the unit cell for blind case #6, we generated 100 models by simulated annealing at two starting temperatures, and 100 models with Rosetta energy- and density-guided optimization followed by refinement. The 2mFo-DFc23 electron density maps generated using phases from over 50% of the Rosetta models had correlations 0.36 or better to the final refined map, while fewer than 5% of models from simulated annealing had correlations this high. Our approach likely outperforms even extreme simulated annealing because the physical chemistry and protein structural information which guide sampling eliminate the vast majority of non-physical conformations.
Figure 3.
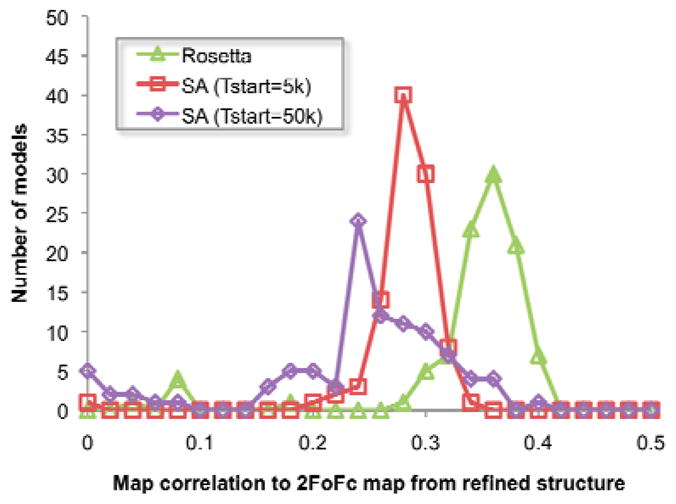
Comparison of the effectiveness of model diversification using Rosetta and simulated annealing. For blind case #6, 100 models were generated using either simulated annealing with a start temperature of 5000 K, simulated annealing with a start temperature of 50000 K, or Rosetta energy-and density-guided optimization. The correlation between 2mFo-DFc density maps computed from each structure and the final refined density was then computed; the starting model has a correlation of 0.29 and the distributions of the refined models are shown in the figure. Rosetta models have correlations better than the initial model much more often than simulated annealing.
Approaches to molecular replacement combining the power of crystallographic map interpretation and structure prediction methodology are likely to become increasingly useful in the next few years. First, the number of already-determined structures will continue increasing, making it increasingly likely that there will be a structure with the required >20% sequence identity: the chance there is a structure with a sequence identity of 20% or greater is more than twice that of finding a structure with at least 30% sequence identity24. Second, as more work focuses on proteins that cannot be expressed in E. coli, the currently preferred methods for experimental phase determination based on selenomethionine replacement may be more difficult to apply. Finally as protein structure modeling algorithms improve, better initial models should further increase the radius of convergence of the approach.
Supplementary Material
Acknowledgments
R.J.R., T.C.T., F.D, and D.B. thank the NIH, the Wellcome Trust (R.J.R.), and HHMI (F.D., D.B.) for funding this research. D.F. and A.A. acknowledge support from the Israel Science Foundation. G.O. thanks DK Molecular Enzymology (FWF-project W901) and the Austrian Science Fund (FWF-project P19858). The work of A.W. was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We thank all members of the JCSG for their general contributions to the protein production and structural work. The JCSG is supported by the NIH, National Institutes of General Medical Sciences, Protein Structure Initiative (U54 GM074898).
Footnotes
Author Contributions F.D., T.C.T., R.J.R., and D.B. developed the methods described in the manuscript; F.D., T.C.T., R.J.R., A.W., and D.B wrote the paper. A.W., G.O., U.W., E.V., A.A., D.F., H.L.A., D.D., S.M.V., H.I., and P.R.P collected data.
References
- 1.Rossmann MG. The Molecular Replacement Method. Gordon & Breach; New York: 1972. [Google Scholar]
- 2.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system (CNS): A new software system for macromolecular structure determination. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 4.Vagin A, Teplyakov A. Molrep: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 5.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature Protocols. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst D. 2007;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DePristo MA, de Bakker PIW, Johnson RJK, Blundell TL. Crystallographic refinement by knowledge-based exploration of complex energy landscapes. Structure. 2005;13:1311–1319. doi: 10.1016/j.str.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Cowtan K. The Buccaneer software for automated model building. Acta Cryst D. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 9.Schwarzenbacher R, Godzik A, Grzechnik SK, Jaroszewski L. The importance of alignment accuracy for molecular replacement. Acta Cryst D. 2004;60:1229–1236. doi: 10.1107/S0907444904010145. [DOI] [PubMed] [Google Scholar]
- 10.Rodríguez DD, Grosse C, Himmel S, González C, de Ilarduya IM, Becker S, Sheldrick GM, Usón I. Crystallographic ab initio protein structure solution below atomic resolution. Nature Methods. 2009;6:651–653. doi: 10.1038/nmeth.1365. [DOI] [PubMed] [Google Scholar]
- 11.Suhre K, Sanejouand YH. On the potential of normal-mode analysis for solving difficult molecular-replacement problems. Acta Cryst D. 2004;60:796–799. doi: 10.1107/S0907444904001982. [DOI] [PubMed] [Google Scholar]
- 12.Qian B, Raman S, Das R, Bradley P, McCoy AJ, Read RJ, Baker D. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259–64. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schröder G, Levitt M, Brünger AT. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464:1218–1222. doi: 10.1038/nature08892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brünger AT, Kuriyan J, Karplus M. Crystallographic R-factor refinement by molecular dynamics. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 15.Raman S, Lange OF, Rossi P, Tyka M, Wang X, Aramini J, Liu G, Ramelot TA, Eletsky A, Szyperski T, Kennedy MA, Prestegard J, Montelione GT, Baker D. NMR structure determination for larger proteins using backbone-only data. Science. 2010;327:1014–8. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 77:363–82. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 17.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 18.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 19.Schröder GF, Brunger AT, Levitt M. Combining efficient conformational sampling with a deformable elastic network model facilitates structure refinement at low resolution. Structure. 2007;15:1630–41. doi: 10.1016/j.str.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brünger AT. Extension of molecular replacement: a new search strategy based on Patterson correlation refinement. Acta Cryst A. 1990;46:46–57. [Google Scholar]
- 21.Brünger AT, Karplus M, Petsko GA. Crystallographic refinement by simulated annealing: application to crambin. Acta Cryst A. 1989;45:50–61. [Google Scholar]
- 22.Kryshtafovych A, Fidelis K, Moult J. CASP8 results in context of previous experiments. Proteins. 2009;77:217–228. doi: 10.1002/prot.22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Read RJ. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Cryst A. 1986;42:140–149. [Google Scholar]
- 24.Vitkup D, Melamud E, Moult J, Sander C. Completeness in structural genomics. Nature Structural Biology. 2001;8:559–566. doi: 10.1038/88640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.