

Abstract
X-linked hyper-IgM syndrome (XHM) is a combined immune deficiency disorder caused by mutations in CD40 ligand. We tested CP-870,893, a human CD40 agonist monoclonal antibody, in the treatment of two XHM patients with biliary Cryptosporidiosis. CP-870,893 activated B cells and APCs in vitro, restoring class switch recombination in XHM B cells and inducing cytokine secretion by monocytes. CP-870,893 infusions were well tolerated and showed significant activity in vivo, decreasing leukocyte concentration in peripheral blood. Although specific antibody responses were lacking, frequent dosing in one subject primed T cells to secrete IFN-g and suppressed oocyst shedding in the stool. Nevertheless, relapse occurred after discontinuation of therapy. The CD40 receptor was rapidly internalized following binding with CP-870,893, potentially explaining the limited capacity of CP-870,893 to mediate immune reconstitution. This study demonstrates that CP-870,893 suppressed oocysts shedding in XHM patients with biliary cryptosporidiosis. The continued study of CD40 agonists in XHM is warranted.
Keywords: X-linked hyper-IgM syndrome, CD40 ligand, CP-870, 893, CD40-R internalization
1. Introduction
X-linked hyper-IgM syndrome (XHM) is a rare primary immune deficiency disorder caused by mutations in the gene encoding CD40 ligand (CD40L or CD154), a protein expressed primarily on the cell surface of activated CD4+ T cells1-3. T cells deficient in CD40L cannot activate the CD40 receptor molecule on B cells, resulting in diminished immunoglobulin class switch recombination (CSR) and low levels of serum IgG, IgA, and IgE with normal to elevated IgM4-9. Although the CD40-CD40L interaction was first studied in B cells, its importance in T cell interaction with other antigen-presenting cells, such as macrophages, monocytes, and dendritic cells, has now been established8,10. Upon CD40 engagement, these APCs upregulate proinflammatory cytokines and costimulatory molecules, and in turn activate naïve T cells10,11. Due to the importance of CD40L in APC activation, T cell priming, and antibody diversification, its deficiency results in the combined immunodeficiency observed in XHM.
XHM patients are susceptible to infection by opportunistic pathogens, such as Pneumocystis jiroveci and Cryptosporidium sp.12-15. These patients usually present with recurrent respiratory infections, and neutropenia, protracted diarrhea, liver disease, or CNS infections are common13-15. The standard of care for XHM patients consists of prophylactic antibiotics and immunoglobulin replacement, as well as growth factors to treat neutropenia if indicated13,16. While these therapies offer some benefit, the current survival of XHM patients is limited with only one-fifth of untransplanted patients reaching the 4Th decade of life and the rest succumbing to infectious disease or malignancy13,17.
In XHM patients, the liver is the most common extra intestinal site of Cryptosporidium infection. Cryptosporidium associated cholangiopathy is generally not responsive to antimicrobial therapy and the infection leads to biliary cirrhosis and ultimately to liver failure. Antigen primed CD4+ T cells and INF-γ are key factors in the control of cryptosporidiosis. Recently, CD8+ T cells have been shown to reduce the parasite load in infected intestinal epithelial cell cultures18. Therefore, a targeted molecular approach that could restore deficient immune function in a more specific manner and improve outcomes for XHM patients would be preferable.
CD40 agonists hold promise as a targeted therapy for XHM. Antibodies that stimulate the CD40 receptor have been shown to substitute for CD4+ T helper cells that are dependent on CD40L expression in mouse models of cytotoxic T lymphocyte priming19-21. Other investigators have found that administration of CD40-specific antibodies facilitates CTL activation and tumor rejection in mouse models of cancer22,23. CP-870,893 is a fully human IgG2 antibody that is agonistic for CD4023. It was shown to be a potent activator of dendritic cells and B cells in vitro, and to promote tumor rejection in mice23,24. In a Phase I clinical trial for patients with advanced solid tumors, CP-870,893 was found to evoke a partial response in 4 of 27 patients25.
In this study, we characterize the functional capacity of CP-870,893 to restore deficient immune responses of XHM patients in vitro and in vivo. Our results show that, in combination with IL-4 and IL-10, CP-870-893 induces immunoglobulin class switch recombination in patient B cells. CP-870,893 also induces patient dendritic cells to secrete IL-12, and induced T cells to secrete TNF-α and INF-γ. On the basis of these preclinical data, we administered CP-870,893 to two XHM patients with biliary Cryptosporidum infection that was refractory to standard medical therapy.
2. Methods
2.1. Participants
Patients were studied at the Clinical Center, NIAID, NIH (Trials registration: NCT00266513). Two XHM patients aged 16 and 19, who were related by one shared parent, were enrolled in the institutional review board approved protocol with informed consent. At the time of enrollment, both patients presented with biliary Cryptosporidium hominis infection that was refractory to medical treatment with daily nitazoxanide alone or in combination with azithromycin. We treated both patients with CP-870,893, an investigational human CD40 ligand agonist antibody developed by Pfizer, Inc. Patients were monitored weekly for evidence of drug toxicity using the National Cancer Institute (NCI) Common Toxicity Criteria version 2.0 (available from http://ctep.cancer.gov/) during treatment and every 4 weeks during the post-biologic observation period. Patients were maintained on the standard dose of immune globulin replacement therapy and no adjustments in dose or frequency of these infusions were permitted during the study period.
2.2. Immune Assessment
The patients were assessed for their ability to mount delayed type hypersensitivity reactions (DTH) by subcutaneous injection of purified Candida antigen prior to and during the course of the study. To assess for antigen-specific antibody responses, three doses of rabies vaccine (HDCV, Sanofi Pasteur) were administered on days 1, 7 and 21, while being treated with infusions of CP-870,893. Antigen-specific IgG antibody levels in serum samples obtained prior to the study and 7 days after each immunization were determined by ELISA.
In vitro cytokine responses were evaluated prior to and during the course of the study. Peripheral blood mononuclear cells (PBMC) were obtained using Ficoll-Hypaque density gradient lymphocyte separation medium (Pharmacia Biotech, Inc., Piscataway, New Jersey, USA) and standard techniques.26 PBMCs (2.5 × 105/ml) were stimulated in vitro with anti-CD3 antibody, (OKT3: gift of Ortho Biotech, Somerset, New Jersey, USA) and after 36 hours of culture, supernatants were harvested and assayed for levels of IFN-γ and TNF-α by specific ELISA (R&D Systems Inc., Minneapolis, Minnesota, USA) according to the manufacturer's instructions. PBMCs were also stimulated before and immediately after terminating CP-870,893 therapy with the super-antigen Staphylococcal enterotoxin B (SEB; Sigma Aldrich, St. Louis, MO)27 and intracellular cytokine detection was performed as previously described.28 MoDCs were prepared from elutriated monocytes using standard techniques and stimulated in vitro with recombinant CD40 ligand trimer (rCD40L; Amgen- Seattle) or CP-870,893 (Pfizer, Groton, CT) with IFN-γ (Genzyme, Cambridge, MA). For in vitro immunoglobulin production, PBMCs (2.5 × 105/ml) were stimulated with rCD40L or CP 870, 893 with IL-4 and IL-10 (both at 10 ng/ml; PeproTech Inc.). Immunoglobulin levels were determined by ELISA 14 days after stimulation by previously described methods29. Anti-human CD40 antibody (clone 5C3) was purchased from BD Bioscience (San Jose, CA); LOB7/6 was purchased from Serotec (Raleigh, NC) and FITC labeled CP-870,893 was custom labeled by Invitrogen (Carlsbad, CA).
2.3. Microbiology
Weekly stool specimens were collected and fixed in 10% formalin. Specimens were concentrated at a centrifuge speed of 800 × g and slides smears were made from the resulting pellet. Slides were stained with modified Dimethyl Sulfoxide-Modified Acid-Fast Stain and read at 1000× (oil immersion). A stool was considered Cryptosporidium-positive if typical oocysts 4-6 μm in diameter were visible and scored as heavy, moderate, light, or undetected. Cryptosporidium was also detected by PCR analysis from stool samples, as previously described30.
2.4. Confocal microscopy
Living MoDCs were washed twice in cold PBS with 1% FCS and re-suspended in cold washing buffer at ∼2 × 106 cells/ml. Cells were then incubated with FITC-labeled CP-870,893 in a 37°C water bath for the time indicated. In parallel experiments, MoDCs were incubated with anti-CD40 FITC (clone 5C3), which lacks agonist activity, at 37°C for 15 minutes. Cells were washed twice in cold washing buffer and then incubated with recombinant CD40 ligand trimer in a 37°C water bath for the time indicated. To stop staining, cells were chilled immediately on ice. After washing twice with cold PBS, cells were kept on ice until transfer to chambered coverglass (LAB-TEK, Rochester, NY) for confocal imaging. Images were collected on a Leica TCS-NT/SP1 confocal microscope (Leica Microsystems, Exton, PA, USA) using a 40× oil immersion objective NA 1.32, zoom 4×. Images were processed using Leica TCS-NT/SP software (version 1.6.587), Imaris 3.3.2 (Bitplane AG, Zurich Switzerland), and Adobe Photoshop 7.0 (Adobe systems, San Jose, CA).
3. Results
3.1. CP-870,893 induces patient dendritic cells to secrete IL-12 and TNF-α and B cells to class switch in vitro
Prior to in vivo use of CP-870,893 in our study patients, we conducted in vitro studies of CP-870,893 and recombinant CD40 ligand (rCD40L) to compare their agonistic activity. Monocyte-derived dendritic cells (moDCs) from XHM patients were stimulated with increasing concentrations (0.1, 1.0, 3.5, or 10 μg/ml) of CP-870,893 or rCD40L. Using ELISA, we measured the concentration of secreted TNF-α and IL-12, two proinflammatory cytokines that are produced by APCs in response to CD40 signaling. TNF-α secretion was observed at 1.0 and 3.5 μg/mL CP-870,893 or rCD40L, but not at 10 μg/mL (Figure 1A). In contrast, significant production of IL-12 was observed at all concentrations of CP-870,893 or rCD40L above 0.1 μg/mL (Figure 1B). For both stimuli, a concentration of 3.5 μg/mL was found to induce the maximal response. At 1.0 and 3.5 μg/mL, the response induced by CP-870,893 was 80% of that induced by rCD40L.
Figure 1.
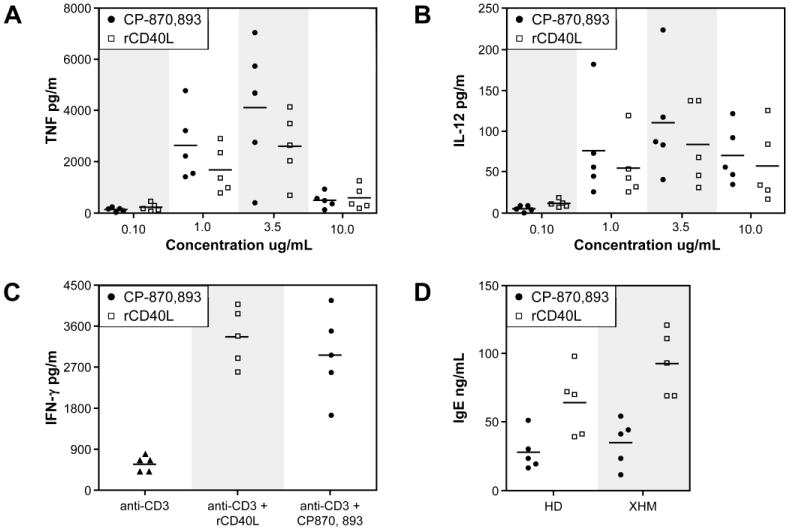
Comparison of in vitro agonist activities of CP-870,893 and recombinant CD40 ligand trimer (rCD40L). A. TNF-α production by MoDCs following in vitro stimulation with CP-870,893 or rCD40L at different concentrations. B. IL-12 production by MoDCs following stimulation with CP-870,893 or rCD40L in a manner identical to A. Cytokine measurements were determined by ELISA. C. INF-γ production by PBMC stimulated with anti- CD3 alone (represented by solid triangles), anti- CD3 with rCD40L (3.5 ug/ml), or anti- CD3 with CP870, 893 (2.5 ug/ml). For anti- CD3 stimulation, plates were pre-coated with OKT3. D. B cells from XHM patients or healthy controls were stimulated with rCD40L or CP-870,893 with IL-4 plus IL-10, and IgE production was measured by ELISA.
We previously reported that T cells from XHM patients secrete reduced levels of IFN-γ following activation through the antigen receptor31. This impairment in T cell priming was due to the lack of CD40L expression on CD4+ T cells and insufficient stimulation of APCs to synthesize IL-12 or upregulate co- stimulatory molecules. To determine whether CP-870,893 can restore IFN-γ production by patient T cells in vitro, we next stimulated PBMCs from patients with XHIM with anti-CD3 alone or in the presence of either CP-870,893 or rCD40L. After 36 hours, we measured IFN- γ secretion into the culture fluid by ELISA. As shown in Figure 1C and in keeping with previously published findings, XHM patient T cells produce markedly reduced amounts of IFN- γ in comparison to reference controls. CP-870,893 restored IFN- γ production in the five patients with XHIM at levels comparable to stimulation with rCD40L.
XHM patients have defective class switch recombination (CSR), resulting in hypogammaglobulinemia and reduced serum IgA and IgE levels. Importantly, this defect is due to a lack of functional CD40L, and is therefore extrinsic to the B cells. To assess the ability of CP-870,893 to substitute for CD40L in inducing CSR, we treated XHM patient B cells with CP-870,893 or rCD40L, together with IL-4 and IL-10. As expected, naïve XHM B cells did not produce IgE, but underwent CSR to IgE after treatment with rCD40L, IL-4, and IL-10 (Figure 1D). Although B cells in PBMCs treated with the CD40 agonist antibody plus cytokines underwent class switch recombination, the level of IgE production was lower when compared to stimulation with rCD40L, IL-4, and IL-10. These results indicate that CP-870,893 is able to signal through CD40 and can substitute for CD40L to a significant degree in vitro. Based on these findings, we investigated whether CP-870,893 would show the same efficacy in vivo and restore immune function to XHM patients with Cryptosporidium infection.
3.2. Patient Characteristics
The first patient is a 16-year-old male who developed cholangiopathy and obstructive jaundice secondary to chronic infection with Cryptosporidium. Baseline magnetic resonance cholangiopancreatography (MRCP) was unremarkable without intra and extra-hepatic biliary tract abnormalities. With continued infection, he presented with a picture of obstructive jaundice with an acute rise in total bilirubin from 0.4 mg/dL to 6.1 mg/dL and an alkaline phosphatase from 350 U/L to 603 U/L. Repeat imaging revealed significant intra and extra-hepatic biliary dilation. An endoscopic retrograde cholangiopancreatography (ERCP) was performed with sphincterotomy, cholangioscopy (SpyGlass®), and biopsies, which revealed an inflammatory biliary stricture in the main bile duct, and intermittent areas of stenosis and dilation in the right hepatic duct consistent with a secondary cholangitis picture from chronic infection. Post-sphincterotomy, the patient's obstructive symptoms improved along with bilirubin and alkaline phosphatase levels. However, his transaminase levels remained slightly above the upper limits of normal. The patient was then started on CP-870,893 at 0.05 mg/kg once weekly for 2 weeks, followed by dose escalation to 0.10 mg/kg once weekly for 3 weeks. On continued hepatic monitoring, the patient had an increase in his transaminase levels to >2.5-fold above baseline, as well as elevation of his bilirubin level from 0.5 to 3.5 mg/dL. At this point the study drug was discontinued. The patient was then started on ursodeoxycholic acid with return of his liver enzymes to baseline. Other treatment options were considered, including allogeneic bone marrow transplantation (BMT) and a combined liver/BMT. However, because of the poor clinical outcomes of the above procedures in the setting of Cryptosporidium infection, the patient opted for palliative care.
The second patient is a 19-year-old male who received 44 weeks of treatment with CP-870,893 for biliary Cryptosporidium infection. At baseline, the patient had normal transaminases with mildly elevated alkaline phosphatase at 160 U/L. The patient tolerated infusions of CP 870,893 once weekly starting at 0.05 mg/kg and escalating from 0.10 mg/kg to 0.20 mg/kg and finally to 0.30 mg/kg (Figure 2). With the dose escalation of CP-870-893 from 0.2 mg/kg to 0.3 mg/kg, the patient developed transient transaminitis less than two times the upper limit of normal following the drug infusion. However, these changes were not sustained, and returned to baseline prior to the next scheduled infusion. The patient also experienced intermittent infusion associated pain in the right upper abdomen, fever, and one episode of sever headache. These clinical symptoms were effectively controlled with pre-treatment analgesics. With weekly infusion of CP-870,893 at 0.30 mg/kg, the patient developed positive delayed hypersensitivity skin tests to Candida, whereas these tests had been negative at the start and after 10 weeks of therapy. The Candida antigen was applied subcutaneously after completing CP-870,893 infusions. The maximum diameter of induration was observed at 24 hours, and resolved completely by 72 hours. Qualitative stool tests at the 0.30 mg/kg dose of CP-870,893 were reported as positive for “rare Cryptosporidium species,” while they were read as “heavy” at the start of treatment. At 0.30 mg/kg, the patient did not mount IgG responses to rabies vaccine challenge, serum immunoglobulin levels were unchanged, and IgE as well as IgA levels remained below the level of detection. Following the once per week dosing schedule of CP-870,893 described above, the dosing frequency of the drug was increased in an attempt to eradicate the Cryptosporidium infection. The patient received a 10-week course of CP-870,893 at a dose of 0.2 mg/kg twice weekly that was followed by an additional 10-week course of infusion at 0.2 mg/kg three times per week for a total of 44 weeks of treatment. During the final 6 weeks of treatment with CP-870, 893, the patient's stool consistently tested negative for Cryptosporidium by microscopy. These samples subsequently tested positive by PCR, thus demonstrating that CP-870,893 therapy suppressed but did not eliminate oocysts shedding. Concurrent with the three times per week treatment with 0.2 mg/kg CP-870,893, the patient was found to have a sustained transaminitis and increased hepatic stiffness by non-invasive transient elastography measurements (Fibroscan®). Alkaline phosphatase levels also increased from 399-461 U/L during the once a week dosing interval to 520-692 U/L at 0.2 mg/kg three times a week. Concurrently, gamma-glutamyl transferase (GGT) levels increased from 282-369 U/L to 406-566 IU/L, while ALT (<107 U/L), AST (158 U/L), and bilirubin (<1.0 mg/dL) were unchanged. Due to the increasing levels of alkaline phosphatase and GGT, a liver biopsy was performed, which revealed peri-portal tract inflammation with fibrosis and bile stasis. There was also evidence of bile duct injury and a decline in the number of bile ducts. Given these findings, CP-870,893 was then discontinued. A repeat liver biopsy performed 3 months after discontinuation revealed very sparse portal inflammatory infiltrate, which was improved compared to the prior biopsy. Fibrosis and ductular reaction was also significantly reduced compared with the previous biopsy. By transient elastography (Fibroscan®), hepatic stiffness was also significantly improved and returned to baseline 3 months after discontinuation of CP-870,893. He has since been maintained on palliative treatment with nitazoxanide.
Figure 2.
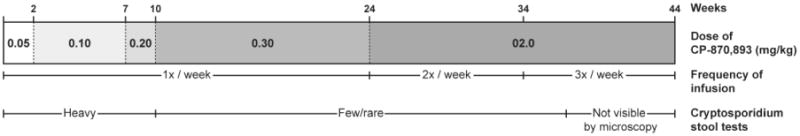
Clinical treatment design. The patient was treated weekly infusions of CD870,893 as follows: 0.5 mg/kg for 2 weeks, 0.10 mg/kg for 6 weeks, 0.2 mg/kg for 2 weeks, and 0.3 mg/kg for 14 weeks. Following this 24-week period, the patient was infused with CD870, 893 at 0.2 mg/kg two times per week for 10 weeks, after which the frequency at 0.2 mg/kg dose was increased to three times per week for an additional 10 weeks for a total of 44 weeks of treatment. Serial stool examination showed fewer Cryptosporidium cysts when CP970,893 was administered at higher or more frequent doses.
3.3. Peripheral leukocyte counts decline following infusion of XHM patients with CP-870,893
CP-870,893 was previously shown to deplete peripheral blood B cells, monocytes, and T cells following infusion. The effect on B cell numbers was severe but relatively short-lived and B cell counts showed near complete return to baseline by the following week. Depletion of monocytes and T cells was more modest and was not observed in either patient with weekly dosing. Here, we used flow cytometry to measure B cell, T cell, and monocyte numbers at baseline, 24-36 hours following CP-870,893 infusion, and at the end of the study. Immunophenotyping analysis was limited to the second patient because treatment of the other patient with CP-870,893 was suspended early due to progressive biliary disease. We observed a marked decrease in the number of CD20+ B cells during treatment with CP-870,893 at all dosing intervals with modest recovery of B cell number 1 week after the last dose of CP-870,893 (Figure 3). Changes in monocyte number were more variable but notable when CP-870,893 was infused more than once per week. A decrease in the number of CD4+ and CD8+ T cells was also observed with CP-870,893 treatment. Interestingly, the lowest absolute number of CD4+ cells was observed when CP-870,893 was infused three times per week, and both CD4+ and CD8+ T cell number returned to baseline 1 week after therapy. These results are consistent with previous studies that showed changes in blood cell composition following CP-870,893 infusion. As the dose or frequency of CP-870,893 infusions increased, the numbers of peripheral CD4+, CD8+, and CD19+ cells were all observed to decrease but subsequently recovered following cessation of therapy.
Figure 3.
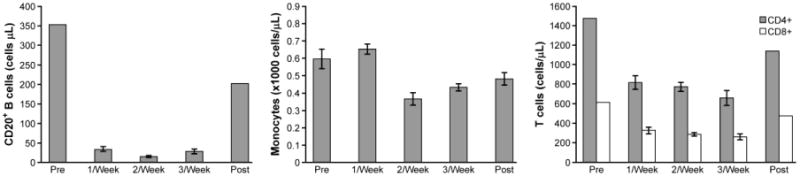
Measurement of circulating leukocytes after infusion of CP-870,893. A. B cell count; B absolute monocyte count; C. T cell count. Data are grouped in a given dose level and reported as mean values with standard deviation.
3.4. CP-870,893 treatment induces patient T cells to produce TNF-α or IFN-γ
We show that T cells from XHM patients are deficient in the production of TH1 effector cytokines following activation, and that this impairment in T cell function can be at least partially restored by the ex vivo addition of CP-870,893 (Figure 1). To determine whether in vivo administration of CP-870,893 can drive naive XHM T cells to produce TH1 cytokines, we cultured patient and control PBMCs with anti-CD3 for 36 hours and then measured TNF-α or IFN-γ secretions into the culture fluid by specific ELISA. Measurements were made at baseline and at monthly intervals during CP-870,893 treatment. As shown in Figure 4A, prior to treatment with CP-870,893 the patients' T cells exhibited markedly reduced levels of TNF-α and IFN-γ secretion in response to these stimuli. After initiation of CP-870,893 treatment the T cells displayed an increase in the capacity to produce both TNF-α and IFN-γ, although below the reference range obtained from normal donors. We next assessed the ability of CP-870,893 treatment to prime CD4+ T cell subsets by measuring intracellular TNF-α and IFN-γ or IL-4 by flow cytometry after stimulation with Staphylococcus enterotoxin B (SEB) (Figure 4B). The number of TNF-α, IFN-γ, or IL-4 producing cells, as a percentage of total CD4+ CD69+ cells, was lower at baseline in XHM patients than in healthy donors, and was unchanged following CP-870,893 treatment. The same was also observed for CD8+ cells. These results demonstrate that while frequent dosing of CP-870,893 treatment in vivo primed patient T cells to produce effector cytokines, the magnitude of these responses was modest and well below the levels secreted by activated T cells from healthy donors.
Figure 4.
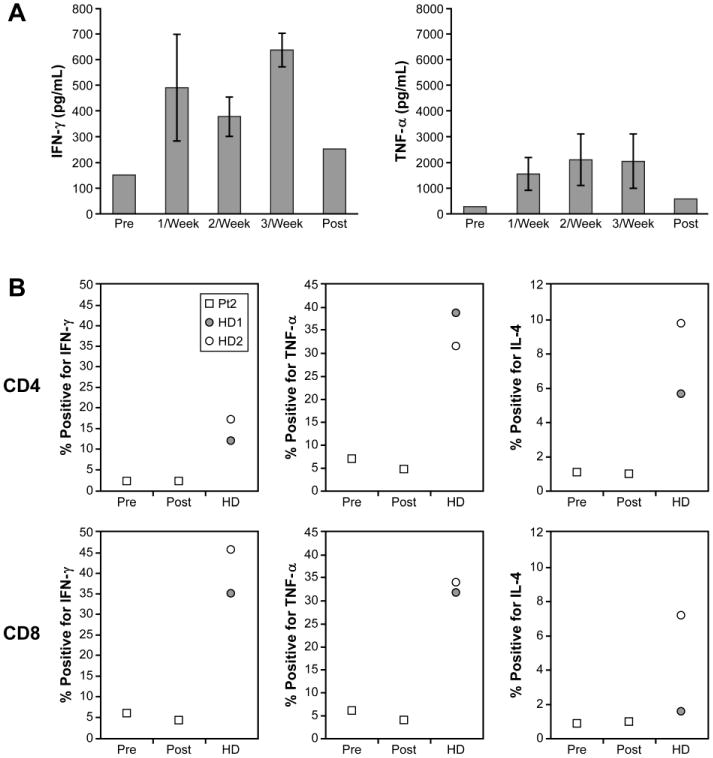
Cytokine production by T cells after treatment with CP-870,893. A. PMBCs (2 × 106) from patients with XHM and normal controls were stimulated with anti-CD3. Cytokine measurements were performed by ELISA at 36 hours and are reported as mean values with standard deviation. Healthy adult controls (n= 20) were used to determine the reference range because pediatric controls were not available. Reference range values for TNF-α production: 2,343-23,968. Reference range for IFN-γ: 1,242-17,703. B. Lymphocytes from healthy donors (HD1 and HD2), and XHM patients (Pt2) pre- and post-treatment, were stimulated in vitro with staphylococcal enterotoxin B (SEB), then permeabilized and stained for intracellular IFN-γ, TNF-α, or IL-4. The results are shown as the percentage of positive-staining CD4+ cells among total CD4+ CD69+ cells, or as the percentage of positive-staining CD8+ cells among total CD8+ CD69+ cells (for CD8).
3.5. CP-870,893 treatment is associated with increased intrahepatic infiltration of T cells and histiocytes
It was recently reported that CP-870,893 CD40 in combination with gemcitabine induced clinical responses in patients with surgically incurable pancreatic ductal carcinoma. The patients received weekly gemcitabine and a single dose of CP-870,893 every 28 days. In those patients that achieved a partial remission, histological analysis of primary tumors and liver metastasis revealed necrosis and an infiltrate dominated by macrophages with an absence of lymphocytes. We therefore investigated the impact of more frequent and higher dose CP-870,893 on the leukocytes infiltrating the liver of one of our study patients with biliary Cryptosporidum infection (Figure 5). Interestingly, CD3+ T cells and CD 68+ histiocytes were detected in representative portal spaces during CP-870,893 treatment at 0.2 mg/kg three times per week, but the numbers of T cells and histiocytes were clearly reduced once the therapy was withdrawn. Further studies will be necessary to address whether the CD3+ T cells infiltrating the liver during CP-870,893 treatment are specific for Cryptosporidium or represent the recruitment of bystander T lymphocytes to the liver from protracted CP-870,893 treatment. Regardless of the precise mechanism, frequent and high doses of CP-870,893 are not sufficient to eradicate Cryptosporidum infection in XHM.
Figure 5.
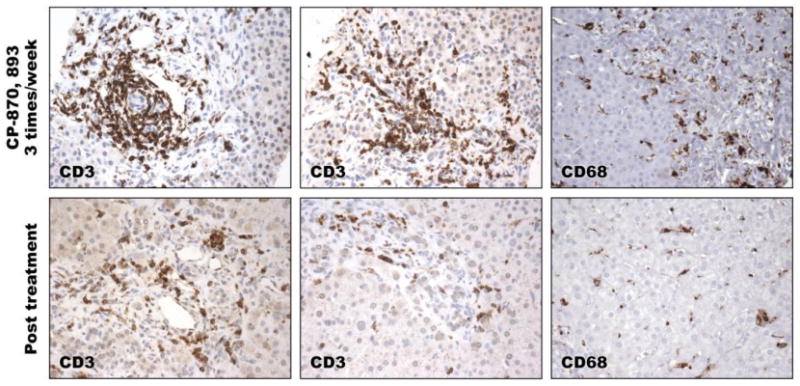
Immunohistochemical staining of CD3+ T cells and CD68+ monocytes in representative liver biopsy specimens during CP870, 893 therapy (top panel), and in the absence of therapy (bottom panel). Magnfication 400X.
3.6. CD40 is rapidly internalized after binding to CP-870,893
Although CP-870,893 was a potent agonist in vitro, its ability to reconstitute the immune systems of the XHM patients in the clinic was limited. While CP-870,893 is known to have a half-life of 8 hours in patients, the mechanism of its rapid clearance is not known. We hypothesized that CP-870,893 may induce receptor internalization after binding CD40 on the cell surface. To test this, we used flow cytometry to monitor CD40 expression on B cells and monocytes before and after infusion of CP-870,893 at 0.30 mg/kg using FITC conjugated CP-870,893 and FITC-conjugated anti-CD40 mAb LOB7/6. These antibodies were first tested by flow cytometry in cross competition studies using peripheral blood B cells to confirm that each monoclonal antibody interacts with a distinct extracellular epitope of CD40 (data not shown). Prior to infusion, patient monocytes and B cells expressed normal levels of cell-surface CD40, as detected using FITC-conjugated CP-870,893 or a commercially available FITC-conjugated LOB7/6 (Figure 6A). At 6 hours post-infusion, CD40 expression on patient monocytes was barely detectable. At 12 hours post-infusion, CD40 expression on monocytes began to increase and was even more pronounced at 24 hours. CD20+ peripheral B cells were largely undetectable with chronic administration of CP-870,893. However, the few B cells present showed reduced CD40 expression at 6 hours after CP-870,893 with gradual recovery of CD40 expression at 24 hours.
Figure 6.
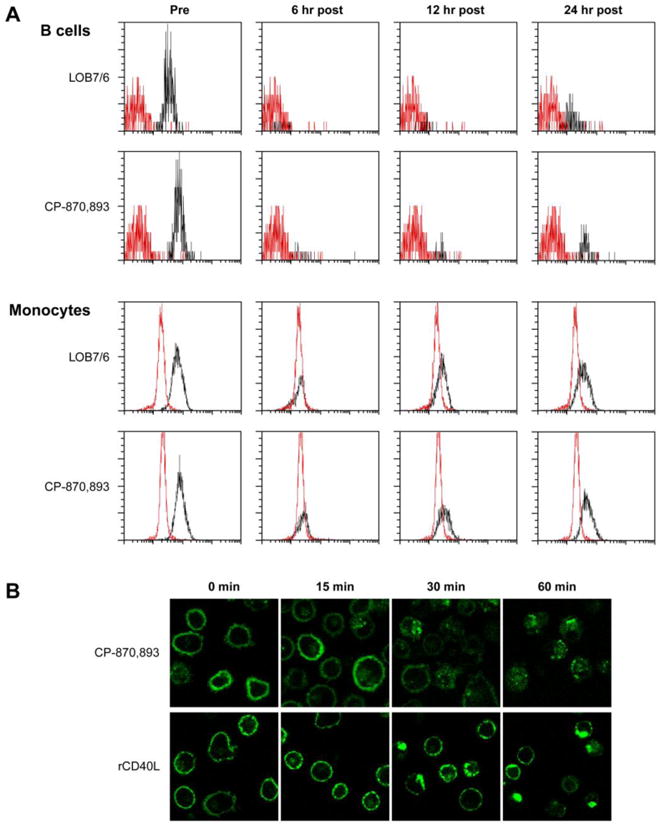
Time-dependent internalization of CP-870,893. A. Profile of surface CD40 expression on peripheral blood B cells and monocytes before and after CP-870,893 infusion. Cells were stained with FITC labeled anti-CD40 clone 5C3, CP-870,893, or isotype control (red histogram). B. Monocyte-derived dendritic cells were labeled with CP-870,893 FITC or a non-agonistic FITC-conjugated anti-CD40 antibody (clone 5C3) followed by incubation with recombinant CD40L trimer. Images were taken at 0, 15, 30, and 60 minutes.
To further test our hypothesis that CP-870,893 induces receptor internalization after binding CD40 on APCs, we MoDCs from healthy donors with FITC-conjugated CP-870,893 and examined CD40 localization by confocal microscopy (Figure 6B). Incubation of MoDCs with FITC-conjugated CP-870,893 at 4°C resulted in distinct cell membrane labeling. Upon warming of these cells to 37°C, internalization of CP-870,893 was observed and found to be time-dependent. By 60 minutes, sparse labeling extended throughout the cytoplasm. To better understand the fate of CD40 receptors after binding to its physiologic ligand, we first incubated MoDCs with FITC labeled 5C3 at 4°C and, after washing, subsequently incubated the cells with recombinant CD40 ligand trimer. Surface cell membrane labeling of CD40 was noted at 4°C. Small clusters of CD40 formed on the cell surface 15 minutes after warming to 37°C. At 30 and 60 minutes of stimulation, these clusters enlarged to form cap-like structures. Taken together, these findings indicate that in contrast to CP-870,893, CD40 ligand induces CD40 clustering on the cell surface and not receptor internalization.
4. Discussion
X-linked hyper-IgM syndrome is a rare primary immune deficiency disorder caused by mutations in the gene encoding CD40L (CD154)1,2. Reflecting the importance of CD40L in the biology of B cells and antigen-presenting cells, XHM patients suffer from defects in both humoral and cell-mediated immunity. Central to the disorder, activated CD4+ T cells fail to express functional CD40L and are thus unable to activate CD40-expressing B cells and other APCs4,5. Because stimulation of B cells through CD40 is an important signal for induction of the antibody-diversifying mechanisms of somatic hypermutation and class switch recombination, XHM patients are limited in their ability to generate high-affinity antibodies with different effector functions4,5,32. CD40 signaling also activates APCs to produce proinflammatory cytokines such as IL-8, TNF-α, and IL-12, and costimulatory molecules such as B710,11,33. XHM APCs are less capable of facilitating T cell priming, and XHM patients are thus particularly susceptible to malignancy and Cryptosporidiosis, conditions that are normally kept in check by a Th1 immune response12,13. This added deficiency of cell-mediated immunity explains why fewer than 20% of XHM patients survive beyond age 25 despite treatment with immunoglobulin replacement therapy and appropriate antibiotic therapy for breakthrough infections—a statistic that highlights the need for better-targeted and more specific therapies for XHM13.
One prospective new treatment is CP-870,893, a fully human IgG2 agonist antibody to CD40 that has already been tested in early clinical trials for patients with solid tumors25. Moreover, CP-870,893 showed therapeutic efficacy in patients with metastatic pancreatic ductal carcinoma and in a mouse model of PDA22. In both humans and mice, tumor regression resulted from infiltration of CD40-activated macrophages into the tumor stroma22. In this report, we present preclinical data and the results of a pilot study conducted on two patients with XHM and biliary Cryptosporidium.
CP-870,893 was shown to have potent biological activity in both preclinical experiments and in the two XHM patients. Importantly, CP-870,893 was effective in activating both B cells and T cells in vitro, restoring class switch recombination in XHM B cells, and IFN-γ production by XHM T cells. The latter was likely due to CD40 induced secretion of TNF-α and IL-12 by MoDCs. CP-870,893 infusions were well tolerated without signs of severe cytokine release syndrome and caused a rapid disappearance of peripheral B cells from the blood. As others have suggested, the disappearance and rapid reappearance of peripheral B cells is most likely due to redistribution, and CP-870,893 is known to upregulate chemokines and cell adhesion molecules on B cells25. Decreases in the number of CD4+ and CD8+ T cells were also observed with CP-870,893 treatment. The lowest absolute number of CD4+ cells was observed when CP-870,893 was infused three times per week, and both CD4+ and CD8+ T cell number returned to baseline 1 week after therapy.
CP-870,893 had limited effect on immune reconstitution in vivo studies. Patient B cells in the peripheral blood remained invariably naïve, serum IgE and IgA were notably absent, and there was no improvement in the ability to generate IgG responses to specific antigens. Frequent administration of the drug induced patient T cells to produce TNF-α, and IFN-γ and this correlated with decreased Cryptosporidium cyst burden.
Although IgG2 antibodies typically have a half-life of 3 weeks, previous reports indicate that the half-life of CP-870,893 is 8 hours. This indicated that the failure of the patients to experience significant immune reconstitution may be due to rapid clearance of the drug, and we hypothesized that binding of CP-870,893 may cause rapid internalization of the CD40 receptor. In support of this, we observed cytoplasmic translocation of labeled CP-870,893 from the cell surface of monocyte-derived dendritic cells after 1 hour of incubation. This is in contrast to the constant presence of a saturating amount of antibody in vitro, and most likely explains the discrepancy between the potency of CP-870,893 in preclinical studies and its lack of efficacy in vivo. Although CP-870,893 may have limited utility in the treatment of patient with XHM, the demonstration that CP-870,893 causes rapid internalization of the CD40 receptor opens up a potential novel strategy for the treatment of CD40+ malignancies in which CP-870,893 is used as a vehicle to deliver recombinant toxin or radiolabels. In this form, CP-870,893 conjugates would specifically recognize CD40+ cells and efficiently mediate cell entry of therapeutic agents.
We recently published a clinical study where we investigated recombinant CD40L (rCD40L) in the treatment of XHM34. The improvements in T cell immune function with rCD40L administration were more prominent that those observed with CP-870,893. rCD40L therapy was also associated with changes in lymph node size and architecture based on comparison of biopsies taken before and after therapy. The question as to why rCD40L led to greater improvements in T cell responses awaits further investigation. However, it has been shown that each drug binds to different epitopes on the CD40 receptor23 and our confocal studies demonstrate that rCD40L induces CD40 clustering on the cell surface that was not observed with CP-870,893. rCD40L also has a longer half-life of 24-36 hours after in vivo administration35. Such differences between the two drugs may be associated with differences in expression of co- stimulatory molecules that are important for T cell priming.
In closing, it is important to note that the expression of CD40L is tightly regulated on the surface of CD4+ T cells and constitutive expression of CD40L in mice led to the development of lymphomas that involved multiple organs36,37. XHM patients enrolled in future clinical trials that use CD40 agonists will need careful monitoring. In addition, both patients were forced to discontinue treatment with CP-870,893 due to liver function test abnormalities. Patient 2 while receiving infusion of CP-870,893 at 0.2 mg/kg three times per week was found to have a sustained transaminitis as well as increased hepatic stiffness by non-invasive transient elastography measurements. A liver biopsy revealed peri-portal tract inflammation with fibrosis as well as bile stasis, and there was also evidence of bile duct injury with a decline in the number of bile ducts. These changes in the liver reversed with discontinuation of therapy. While pre-existing biliary Cryptosporidium may have contributed to hepatic injury in this patient, it is important to note grade 3 or 4 elevations in liver transaminases were observed in cancer patients receiving recombinant CD40 ligand35. Given the observed hepatic toxicity associated with CD40 agonists, future studies should include interim evaluations and clear stopping rules for toxicity.
Highlights.
Use of CP-870,893, a CD40 agonist monoclonal antibody in hyper IgM Syndrome.
CP-870,893 infusions were well tolerated and showed significant activity in vivo.
Frequent dosing primed T cells and suppressed oocyst shedding in the stool.
The CD40 receptor was rapidly internalized following binding with CP-870,893.
Acknowledgments
This work was supported, in whole or in part, by the intramural program of the National Institutes of Allergy and Infectious Diseases, NIH.
We thank the families studied for their invaluable contribution to this project; W. Strober for helpful discussions; Harry Malech for careful review of the manuscript; and R. Huhn for facilitating access to CP870, 893.
The project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400, and supported by the intramural program of NIAID/NIH.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Notarangelo LD, Duse M, Ugazio AG. Immunodeficiency with hyper-IgM (HIM) Immunodefic Rev. 1992;3:101–121. [PubMed] [Google Scholar]
- 2.Korthauer U, et al. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:539–541. doi: 10.1038/361539a0. [DOI] [PubMed] [Google Scholar]
- 3.Aruffo A, et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell. 1993;72:291–300. doi: 10.1016/0092-8674(93)90668-g. [DOI] [PubMed] [Google Scholar]
- 4.Elgueta R, et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DiSanto JP, Bonnefoy JY, Gauchat JF, Fischer A, de Saint Basile G. CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature. 1993;361:541–543. doi: 10.1038/361541a0. [DOI] [PubMed] [Google Scholar]
- 6.Bhushan A, Covey LR. CD40:CD40L interactions in X-linked and non-X-linked hyper-IgM syndromes. Immunol Res. 2001;24:311–324. doi: 10.1385/IR:24:3:311. [DOI] [PubMed] [Google Scholar]
- 7.Winkelstein JA, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) 2003;82:373–384. doi: 10.1097/01.md.0000100046.06009.b0. [DOI] [PubMed] [Google Scholar]
- 8.Lougaris V, Badolato R, Ferrari S, Plebani A. Hyper immunoglobulin M syndrome due to CD40 deficiency: clinical, molecular, and immunological features. Immunol Rev. 2005;203:48–66. doi: 10.1111/j.0105-2896.2005.00229.x. [DOI] [PubMed] [Google Scholar]
- 9.Castigli E, et al. CD40 ligand/CD40 deficiency. Int Arch Allergy Immunol. 1995;107:37–39. doi: 10.1159/000236923. [DOI] [PubMed] [Google Scholar]
- 10.Caux C, et al. Activation of human dendritic cells through CD40 cross-linking. J Exp Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cella M, et al. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aghamohammadi A, et al. Clinical and laboratory findings in hyper-IgM syndrome with novel CD40L and AICDA mutations. J Clin Immunol. 2009;29:769–776. doi: 10.1007/s10875-009-9315-7. [DOI] [PubMed] [Google Scholar]
- 13.Levy J, et al. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997;131:47–54. doi: 10.1016/s0022-3476(97)70123-9. [DOI] [PubMed] [Google Scholar]
- 14.Hayward AR, et al. Cholangiopathy and tumors of the pancreas, liver, and biliary tree in boys with X-linked immunodeficiency with hyper-IgM. J Immunol. 1997;158:977–983. [PubMed] [Google Scholar]
- 15.Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature. 1995;378:617–620. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- 16.Bayrakci B, et al. The efficacy of immunoglobulin replacement therapy in the long-term follow-up of the B-cell deficiencies (XLA, HIM, CVID) Turk J Pediatr. 2005;47:239–246. [PubMed] [Google Scholar]
- 17.Schneider LC. X-linked hyper IgM syndrome. Clin Rev Allergy Immunol. 2000;19:205–215. doi: 10.1385/CRIAI:19:2:205. [DOI] [PubMed] [Google Scholar]
- 18.Pantenburg B, et al. Human CD8(+) T cells clear Cryptosporidium parvum from infected intestinal epithelial cells. Am J Trop Med Hyg. 2010;82:600–607. doi: 10.4269/ajtmh.2010.09-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Wilson JM. CD40 ligand-dependent T cell activation: requirement of B7-CD28 signaling through CD40. Science. 1996;273:1862–1864. doi: 10.1126/science.273.5283.1862. [DOI] [PubMed] [Google Scholar]
- 20.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 21.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 22.Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gladue RP, et al. The CD40 agonist antibody CP-870,893 enhances dendritic cell and B-cell activity and promotes anti-tumor efficacy in SCID-hu mice. Cancer Immunol Immunother. 2011;60:1009–1017. doi: 10.1007/s00262-011-1014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carpenter EL, Mick R, Ruter J, Vonderheide RH. Activation of human B cells by the agonist CD40 antibody CP-870,893 and augmentation with simultaneous toll-like receptor 9 stimulation. J Transl Med. 2009;7:93. doi: 10.1186/1479-5876-7-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruter J, Antonia SJ, Burris HA, Huhn RD, Vonderheide RH. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10:983–993. doi: 10.4161/cbt.10.10.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuss IJ, Kanof ME, Smith PD, Zola H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr Protoc Immunol. 2009;Chapter 7(Unit7 1) doi: 10.1002/0471142735.im0701s85. [DOI] [PubMed] [Google Scholar]
- 27.Rich RR, Mollick JA, Cook RG. Superantigens: interaction of staphylococcal enterotoxins with MHC class II molecules. Trans Am Clin Climatol Assoc. 1990;101:195–204. discussion 204-196. [PMC free article] [PubMed] [Google Scholar]
- 28.Foster B, Prussin C, Liu F, Whitmire JK, Whitton JL. Detection of intracellular cytokines by flow cytometry. Curr Protoc Immunol. 2007;Chapter 6(Unit6 24) doi: 10.1002/0471142735.im0624s78. [DOI] [PubMed] [Google Scholar]
- 29.Jain A, et al. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2:223–228. doi: 10.1038/85277. [DOI] [PubMed] [Google Scholar]
- 30.Xiao L, et al. Phylogenetic analysis of Cryptosporidium parasites based on the small-subunit rRNA gene locus. Appl Environ Microbiol. 1999;65:1578–1583. doi: 10.1128/aem.65.4.1578-1583.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain A, et al. Defects of T-cell effector function and post-thymic maturation in X-linked hyper-IgM syndrome. J Clin Invest. 1999;103:1151–1158. doi: 10.1172/JCI5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ochs HD, Hollenbaugh D, Aruffo A. The role of CD40L (gp39)/CD40 in T/B cell interaction and primary immunodeficiency. Semin Immunol. 1994;6:337–341. 1042–1994. doi: 10.1006/smim.1994.1042. [DOI] [PubMed] [Google Scholar]
- 33.Koch F, et al. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med. 1996;184:741–746. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain A, et al. Partial immune reconstitution of X-linked hyper IgM syndrome with recombinant CD40 ligand. Blood. 2011;118:3811–3817. doi: 10.1182/blood-2011-04-351254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vonderheide RH, et al. Phase I study of recombinant human CD40 ligand in cancer patients. J Clin Oncol. 2001;19:3280–3287. doi: 10.1200/JCO.2001.19.13.3280. [DOI] [PubMed] [Google Scholar]
- 36.Brown MP, et al. Thymic lymphoproliferative disease after successful correction of CD40 ligand deficiency by gene transfer in mice. Nat Med. 1998;4:1253–1260. doi: 10.1038/3233. [DOI] [PubMed] [Google Scholar]
- 37.Sacco MG, et al. Lymphoid abnormalities in CD40 ligand transgenic mice suggest the need for tight regulation in gene therapy approaches to hyper immunoglobulin M (IgM) syndrome. Cancer Gene Ther. 2000;7:1299–1306. doi: 10.1038/sj.cgt.7700232. [DOI] [PubMed] [Google Scholar]