

Abstract

Whereas the cleavage of alkenes by ozone typically generates peroxide intermediates that must be decomposed in an accompanying step, ozonolysis in the presence of pyridine directly generates ketones or aldehydes through a process that neither consumes pyridine nor generates any detectable peroxides. The reaction is hypothesized to involve nucleophile-promoted fragmentation of carbonyl oxides via formation of zwitterionic peroxyacetals.
The ozonolysis of alkenes, a widely used and environmentally sustainable oxidative transformation, is nearly always accompanied by a reaction to decompose the ozonides or other peroxide intermediates.1 However, the proclivity of ozonides towards exothermic and self-accelerating decomposition reactions, combined with their low rate of reaction with many reducing agents, can create serious hazards.2,3,4 An attractive alternative to a traditional stepwise approach would involve in situ capture and decomposition of the carbonyl oxide intermediates. We recently described two approaches to “reductive” ozonolyses based upon trapping of carbonyl oxides by amine N-oxides or water.5a,b However, the first of these requires basic conditions while the latter generates hydrogen peroxide as a stoichometric byproduct. We became interested in a handful of reports describing the direct formation of carbonyl groups for ozonolyses conducted in the presence of pyridine.6,7 This mechanistically unexplained process has received little synthetic attention.8,9 We now report that ozonolysis in the presence of pyridine involves an unprecedented organocatalyzed decomposition of carbonyl oxides via the formation and fragmentation of zwitterionic peroxyacetals. The overall process offers a fast, general, and high-yielding route to aldehydes and/or ketones.
Our initial investigations, illustrated in Table 1, directly compared ozonolysis in pyridine against a traditional two-step protocol.10,11 For example, ozonolysis of the acetate of 9-decenol, followed by reduction of the intermediate ozonide with Ph3P, furnished the aldehyde in 78% yield. The same product was available in 83% – in only 2-3 minutes and without any reductive workup - if ozonolysis was conducted in the presence of pyridine. Performing the reaction in the presence of substoichometric pyridine resulted in the isolation of significant amounts of ozonide, and the best yields of aldehyde or ketone were generally obtained in the presence of two or more equivalents of pyridine. In control reactions, we demonstrated that isolated ozonides were unreactive towards pyridine under the reaction conditions.
Table 1.
O3/pyridine versus a standard two-step procedure.
| substrate | cond | product | yield(s) |
|---|---|---|---|
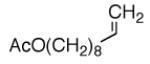 |
A | 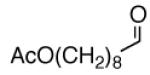 |
78% |
| B | 83% | ||
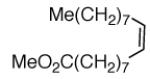 |
A | Me(CH2)7CHO + MeO2C(CH2)7CHO |
90, 75% |
| B | 82, 78% | ||
 |
A | 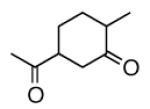 |
80% |
| B | 81% | ||
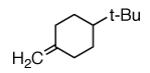 |
A | 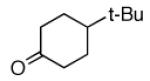 |
79% |
| B | 85% | ||
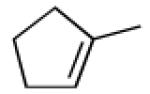 |
A | 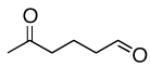 |
70% |
| B | 77% | ||
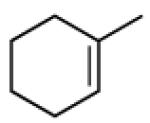 |
A | 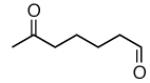 |
87% |
| B | 93% |
A) O3, CH2Cl2, −78 °C; Ph3P, 24 h; B) O3, 2-3 equiv pyridine, CH2Cl2, −78 °C, 2-3 min; C) As per “B” but 1 equiv pyridine
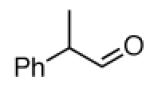
We next investigated ozonolysis of enol ethers (Table 2); the carbonyl oxide/ester pair derived from cycloreversion of the primary ozonide does not readily undergo cycloaddition to ozonides. As a result, enol ethers typically generate monomeric products only in the presence of an added alcohol or aldehyde able to capture the carbonyl oxide.12 However, ozonolysis of enol ethers 1a - 4a in the presence of pyridine furnished good yields of the carbonyl products (1b - 4b); neither carbonyl products nor ozonides were obtained in the absence of pyridine (not shown).
Table 2.
Reductive ozonolysis of enol ethersa
| substrate | product | yield | ||
|---|---|---|---|---|
| 1a | 1b | 74% | ||
| 2a | 2b | 86% | ||
| 3a | 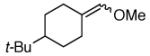 |
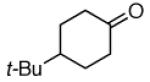 |
3b | 83% |
| 4a | 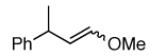 |
 |
4b | 70% |
Conditions: O3/O2, −78 °C, 2-3 equiv pyridine in CH2Cl2
The influence of pyridine electronic and steric factors was further investigated using enol ether 3a (Table 3). Similar yields of ketone 3b were obtained in the presence of electron-rich and electron-poor pyridines.13 However, the presence of steric bulk adjacent to the pyridine nitrogen suppresses formation of carbonyl product. Similar results were obtained with alkene substrates (not shown); for example, terminal alkenes furnish aldehydes in the presence of pyridine or 2,6-lutidine but not 2,6-di-t-butylpyridine. The replacement of pyridine with other heterocyclic bases (thiophene, imidazole, 1-methylimidazole) led to much lower yields of reduction products and this theme was not pursued.
Table 3.
Probing steric and electronic influences
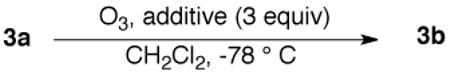
| additive | ketone (%)a |
|---|---|
| pyridine | 83 |
| 3-nitropyridine | 53 |
| N,N’-dimethylaminopyridine | 65 |
| 2,6-lutidine | traces |
| 2,6-di-t-butylpyridine | traces |
Isolated yield
The synthetic utility of the reductive ozonolysis can be seen in the ability to directly apply the crude reaction to trapping of a product aldehyde by a stoichometric amount of an organometallic reagent, a transformation typically performed on the purified products of ozonolysis (eq 1).
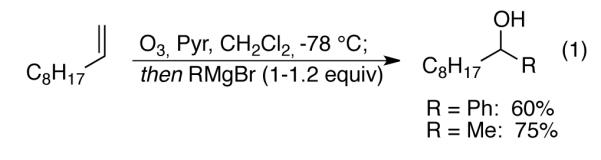 |
As illustrated in Scheme 1, reductive ozonolysis could in principle involve reaction of pyridine with either the primary ozonide (POZ), the carbonyl oxide (CO), or the secondary ozonide (SOZ).12 Unhindered alcohols are known to be effective carbonyl oxide trapping reagents,12,14 and we investigated reaction of several substrates in the presence of pyridine and added methanol. In each case, we observed hydroperoxyacetals (path a), demonstrating that the reductions involve either the CO or a downstream species.12 Control reactions demonstrated that the carbonyl products did not arise from pyridine-promoted E1cB fragmentation of the SOZ (path b).15 These observations implied the intermediacy of the CO,1a,12 but did little to explain the mechanism. The absence of hydrogen peroxide in crude reaction mixtures ruled out trapping of the CO by traces of solubilized water (not shown),5b while NMR monitoring of a reaction conducted in CD2Cl2 observed only carbonyl products and recovered pyridine, excluding a redox process (path c).8
Scheme 1.
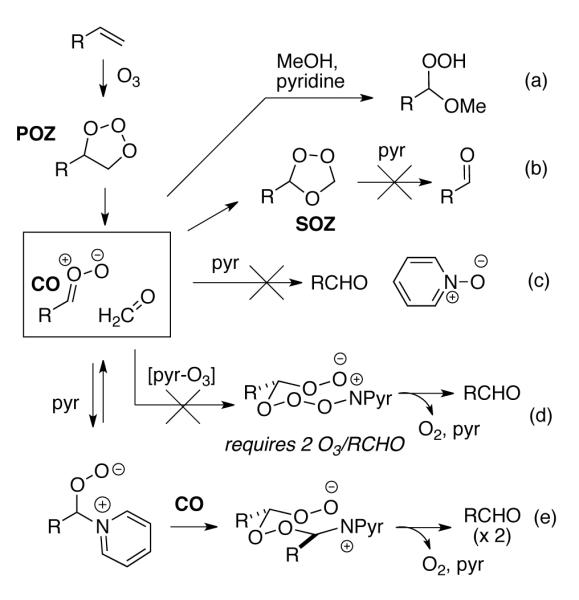
Mechanistic possibilities
Given the inabilty to explain the observed products via traditional reaction pathways, we next considered whether attack of a pyridine-ozone complex6a,9 on the CO could generate zwitterionic peroxyacetals able to fragment to pyridine, oxygen, and a carbonyl (path d). However, this mechanism requires two equivalents of O3 per molecule of carbonyl product, something not supported by experimental observations.16 A more viable mechanism involves addition of pyridine to the CO to generate a zwitterion which can react with another molecule of carbonyl oxide (path e).17 The resulting zwitterionic bisperoxyacetal would be highly activated towards fragmentation to generate a molecule of O2, two carbonyl groups, and pyridine.
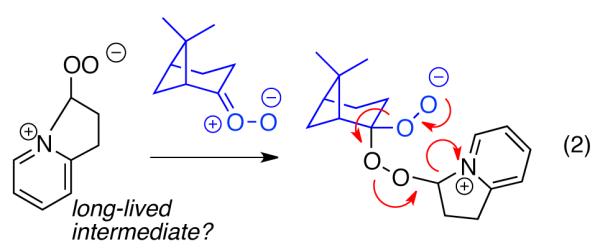
The proposed mechanism is consistent with the observed steric influences on the reduction process, and suggested that reductive ozonolysis of bulky substrates might be enhanced by an unhindered “helper” CO which could trap pyridine to generate the nucleophilic zwitterion. As illustrated in Table 4, this hypothesis was tested on β-pinene. This hindered substrate predominantly generates ozonides or polymeric peroxides even in the presence of pyridine.18 However, performance of ozonolysis in the presence of pyridine and ethyl vinyl ether, the latter a source of formaldehyde O-oxide, produced an improved yield of the ketone. More intriguingly, ozonolysis of pinene in the presence of an unsaturated pyridine designed to generate a pyridine-stabilized carbonyl oxide (eq. 2) resulted in a dramatically improved yield of ketone.
Table 4.
Influence of added carbonyl oxide sources
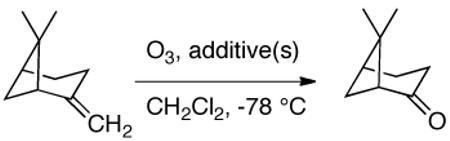
| additive A | additive B | ketone (%)a |
|---|---|---|
| - | - | traces |
| - | pyridine (3 equiv) | 12 |
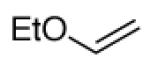 |
pyridine (3 equiv) | 30 |
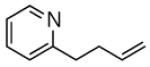 |
- | 65 |
Isolated yield
 |
In conclusion, we demonstrate a high-yielding and convenient procedure for the direct ozonolytic generation of anhydrous solutions of aldehydes and ketones. The reaction provides the first example of organocatalysis in ozonolysis, and suggests the existence of yet unglimpsed avenues of carbonyl oxide reactivity.
Supplementary Material
Note on Safety.
Although the process described above largely precludes formation of peroxides or ozonides, ozonolyses should always be conducted with an awareness of the potential for spontaneous and exothermic decompositions.4a In particular, experimenters should verify the absence of significant amounts of peroxides before concentrating crude reaction mixtures.19
Acknowledgment
Research was conducted with funding from NSF (CHE -0749916, CHE-1057982) in facilities remodeled with support from NIH (RR016544). We thank Profs. Stephen DiMagno and James Takacs (University of Nebraska-Lincoln) for useful suggestions.
Footnotes
Supporting Information Available. Experimental protocols, spectral characterization and 1H NMR spectra for all products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1)(a).Bailey PS. Ozonation in Organic Chemistry: Vol 1. Academic Press; New York: 1978. (Olefinic Compounds) [Google Scholar]; (b) McGuire J, Bond G, Haslam PJ. Handbook of Chiral Chemicals. 2nd ed Taylor & Francis; Boca Raton: 2006. Ozonolysis in the Production of Chiral Fine Chemicals; pp. 165–184. [Google Scholar]; (c) Van Ornum SG, Champeau RM, Pariza R. Chem. Rev. 2006;106:2990. doi: 10.1021/cr040682z. [DOI] [PubMed] [Google Scholar]
- (2).Kula J. Chem. Health Safety. 1999;6:21. [Google Scholar]
- (3)(a).Gordon PM. Chem. Eng. News. 1990;68:2.; Lavallee P, Bouthillier G. J. Org. Chem. 1986;51:1362–5.; see note 27 and references within; Ferreira JTB. Chem. Eng. News. 1990;68:4.
- (4).See: Hida T, Kikuchi J, Kakinuma M, Nogusa H. Org. Proc. Res. Dev. 2010;14:1485.; for discussions of membrane- or flow-through reactors, see: O’Brien M, Baxendale IR, Ley SV. Org. Lett. 2010;12:1596. doi: 10.1021/ol100322t.; Irfan M, Glasnov TN, Kappe CO. Org. Lett. 2010;13:984. doi: 10.1021/ol102984h.
- (5)(a).Schwartz C, Raible J, Mott K, Dussault PH. Org. Lett. 2006;8:3199. doi: 10.1021/ol061001k.; Tetrahedron. 2006;62:10747.; Schiaffo CE, Dussault PH. J. Org. Chem. 2008;73:4688. doi: 10.1021/jo800323x.; see also: Molander GA, Cooper DJ. J. Org. Chem. 2007;72:3558. doi: 10.1021/jo070130r.
- (6).Slomp G, Johnson JL. J. Am. Chem. Soc. 1958;80:915. [Google Scholar]
- (7).Lutidine has been employed as an additive for ozonolyses (Wang Y-G, Takeyama R, Kobayashi Y. Angew Chem. Int. Ed. 2006;45:3320. doi: 10.1002/anie.200600458.) but the substrates in this study would be expected to generate aldehydes regardless of conditions or additives.
- (8).Griesbaum K. Chem. Commun. 1966:920. [Google Scholar]
- (9).Pyridine has been applied as an additive to enhance the chemoselectivity of ozonolysis within polyunsaturated systems. See ref. 6 and: Trost BM, Machacek MR, Tsui HC. J. Am. Chem. Soc. 2005;127:7014. doi: 10.1021/ja050340q.
- (10).The alkene substrate (1-3 mmol) and dry pyridine (3-9 mmol) were dissolved in dry CH2Cl2 (15-20 ml) in a flame-dried flask under N2. The solution was cooled to−78 °C and a stream of O3/O2 was introduced through a disposable pipet for a period varying with the amount of alkene (typically ~ 1 min/mmol). Once judged to be complete (TLC, in situ IR, or time), the reaction was sparged with O2 and then N2. The crude reaction mixture was diluted with CH2Cl2 and sat. aq. NaHCO3 and the separated aqueous layer was extracted with additional CH2Cl2. The combined organic layers were dried over Na2SO4 and filtered through a cotton plug. The residue obtained upon concentration was purified via flash chromatography with ethyl acetate/hexanes to furnish the aldehyde or ketone. Comparable yields were obtained on a 10 mmol scale or when the sparged reaction mixture was directly concentrated and submitted to chromatography.
- (11).The best yields of aldehydes are obtained if reactions were stopped immediately upon consumption of alkene. Although direct monitoring (NMR) revealed formation of only small amounts (~ 5%) of carboxylic acids under typical reaction conditions, allowing reactions to proceed longer than necessary can result in significant overoxidation. Control reactions demonstrated that ozonolysis of mixtures of purified aldehydes and pyridine resulted in the slow formation of carboxylic acids.
- (12).Bunnelle WH. Chem. Rev. 1991;91:335. [Google Scholar]
- (13).Addition of ozone to CH2Cl2 solutions of pyridine and alkene often resulted in a yellowish tint which dissipated once the alkene substrate was consumed; the color varied with substrate and with different pyridines. We have been unable to isolate or to detect the colored intermediates (TLC, in situ IR, or 1H NMR on reactions conducted in CD2Cl2), which we assume are small amounts of pyridine-derived byproducts possessing a high extinction coefficient and which are consumed by excess ozone. Further investigations of this phenomenon are in progress.
- (14).Yamamoto Y, Niki E, Kamiya Y. Bull. Chem. Soc. Jpn. 1982;55:2677. [Google Scholar]
- (15).In contrast, trialkylamines readily decompose terminal ozonides to an aldehyde and formate. See Hon YS, Wu KC. Tetrahedron. 2003;59:493., and references within.
- (16).The effective delivery of ozone to a given substrate under a given set of conditions is easily calibrated; our experience has been that the rate of consumption of a given substrate is very similar in the presence or absence of pyridine. An example is provided in the Supporting Information.
- (17).Barton M, Ebdon JR, Foster AB, Rimmer S. Org. Biomol. Chem. 2005;3:1323. doi: 10.1039/b419174a. [DOI] [PubMed] [Google Scholar]
- (18).The ozonolytic cleavage of β-pinene to nopinone is notorious for the formation of explosive ozonides and peroxides which are decomposed only slowly by mild reductants. See ref. 3.
- (19).Smith LL, Hill FL. J. Chrom. 1972;66:101. doi: 10.1016/s0021-9673(01)82933-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.