

Summary
Novel inhibitors are needed to counteract the rapid emergence of drug-resistant HIV variants. HIV-1 reverse transcriptase (RT) has both DNA polymerase and RNase H (RNH) enzymatic activities, but approved drugs that inhibit RT target the polymerase. Inhibitors that act against new targets, like RNH, would be effective against all of the current drug-resistant variants. Here, we present 2.80 Å and 2.04 Å resolution crystal structures of an RNH inhibitor, β-thujaplicinol, bound at the RNH active site of both HIV-1 RT and an isolated RNH domain. β-thujaplicinol chelates two divalent metal ions at the RNH active site. We provide biochemical evidence that β-thujaplicinol is a slow-binding RNH inhibitor with non-competitive kinetics and suggest that it forms a tropylium ion that interacts favorably with RT and the RNA:DNA substrate.
Introduction
HIV-1 reverse transcriptase (RT) is a key target of anti-AIDS drugs. RT converts the single-stranded viral genomic RNA into double-stranded DNA that is subsequently integrated into the genome of the host cell. RT has two enzymatic activities that cooperate to carry out this synthesis: (1) A DNA polymerase that can use either RNA or DNA as a template, and (2) RNase H (RNH). Like many other DNA polymerases, RT requires both a template and a primer; the primer for the first, or “minus”, strand DNA is tRNA lys3. Synthesis of the minus strand DNA generates an RNA:DNA duplex that is a substrate for RNH; RNH degrades the viral RNA, leaving a purine-rich segment of the viral RNA (the polypurine tract, or PPT) which serves as the primer for the second, or “plus” strand of viral DNA. After plus strand DNA synthesis copies the first 18 nucleotides of the minus strand, RNH removes the tRNA primer. The polymerase and RNH activities are both required for viral replication; the RNH activity of RT cannot be replaced by endogenous cellular RNases H[1, 2].
RT is comprised of a 66 kDa (p66) and a 51 kDa (p51) subunit (Figure 1), which derive, by cleavage by the viral protease, from the Gag-Pol polyprotein precursor. The first 440 residues of p66 and p51 are identical. In both subunits, these residues comprise the four polymerase subdomains: thumb, palm, fingers, and connection[3, 4]. The RNH domain is formed by the C-terminal residues 427-560 of p66[3-6]. The individual subdomains in p51 and p66 have similar structures but are arranged differently. Amino acid residues directly responsible for both enzymatic activities reside entirely within the p66 subunit, while the p51 subunit is believed to play a more structural role. The p66 subunit can be in an “open” conformation, in which the thumb rotates away from the fingers to form a large cleft that accommodates double-stranded nucleic acid substrates. Conversely, in the absence of nucleic acid, the p66 subunit assumes a “closed” conformation, in which the thumb rotates toward the fingers to fill much of this cleft[7].
Figure 1. Overview of RT structure.
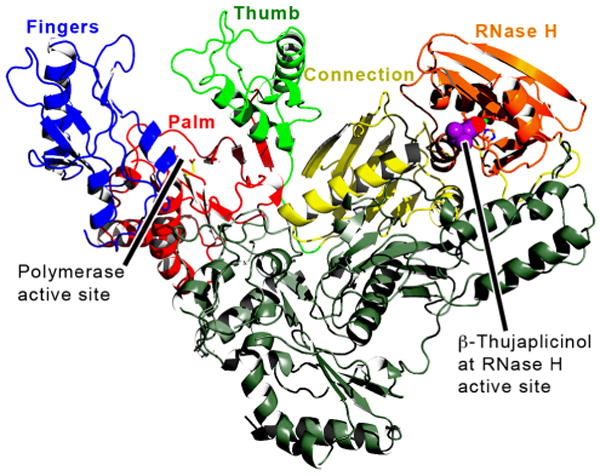
An RT ribbon diagram of the RT/β-thujaplicinol structure is shown. The subdomains of the p66 subunit (including the RNase H domain) are: fingers, blue; palm, red; thumb, green; connection, yellow; RNH, orange; and the p51 subunit, gray. β-thujaplicinol is shown space-filled in magenta and red.
The RT inhibitors used to treat HIV-1 infections are typically given to patients as part of a cocktail of therapeutic agents in a treatment strategy known as highly active antiretroviral therapy (HAART). However, The efficacy of these therapies is limited by the emergence of drug-resistant variants of the virus (reviewed in ref. [8]). To address this problem, new inhibitors must be developed that can block the replication of the existing drug-resistant viruses. This means that new inhibitors that act against the same targets as the existing drugs must be relatively effective against the extant resistant viruses, or the new inhibitors can inhibit essential viral functions that are not blocked by existing drugs. To date, all of the RT inhibitors that have been approved for clinical use target the polymerase activity of RT, not its RNH activity. Given that RNH activity is essential for viral replication[1, 2], RNH inhibitors (RNHIs) have considerable potential as anti-AIDS therapeutics.
One problem in developing an RNHI active site inhibitor is the absence of a deep pocket into which the inhibitors can bind[9]. However, it should be possible to use the active site metal ions as anchor points for inhibitor binding. A diketo acid inhibitor[10] was shown to bind in a metal-dependent manner to the RNH domain of RT. The authors postulated that this RNHI has a metal ion-dependent inhibition mechanism that is similar to that of related HIV integrase inhibitors[11]. N-hydroxyimide inhibitors were designed to chelate the active site magnesium ions of the RNH domain, based on a specific interaction with the two metal ions[12]. These inhibitors bind directly to the RNH domain; there is no binding in the absence of metal ions. Le Grice[13] and coworkers have presented compelling evidence that β-thujaplicinol (2,7-dihydroxy-4-(propan-2-yl)-cyclohepta-2,4,6-trien-1-one), a natural product from Western red cedar[14], inhibits RNH activity. They proposed that β-thujaplicinol chelates the active site metal ions at the RNH active site[13]. A structure has previously been reported showing an RNHI, DHBNH, bound near the RT polymerase active site, more than 50 Å from the RNase H active site[15], and a structure for an active, isolated HIV-RNH domain in a complex with an N-hydroxyimide inhibitor has been briefly described[16], but no coordinates have yet been published for an RNHI in complex with the RNH domain. We present here a structure of HIV-1 RT at 2.80 Å resolution and a structure of the isolated RNH domain of HIV-1 RT (isoRNH) at 2.04 Å resolution, each in a complex with β-thujaplicinol. These structures reveal the details of the interactions of the RNHI and RNH, including the coordination of the two catalytic metal ions.
Results
Binding and Inhibitory Properties of β-Thujaplicinol
It has previously been reported that β-thujaplicinol inhibits the RNH activity of HIV-1 with an IC50 of 0.21 μM in a manner that is not competitive with the nucleic acid substrate[13]. We find that β-thujaplicinol is a time-dependent inhibitor of the RNH that displays non-competitive kinetics (Figure 2). The inhibitor does not affect the polymerase activity of RT (data not shown). β-thujaplicinol is also an inhibitor of the isolated RNase H domain. This inhibition is non-competitive in steady state experiments (data not shown). To evaluate the mechanism of the time dependence, kobs was determined for RT by fitting the progress curves as described in the Methods section. The relationship between kobs and β-thujaplicinol concentration was linear (Figure 2b), suggesting simple reversible inhibition. Kiapp was estimated from Figure 2b to be 0.14 μM. Progress curves generated at multiple substrate concentrations revealed no significant dependence of kobs on substrate concentration, consistent with non-competitive inhibition (Figure 2c, d). Direct binding studies performed using both surface plasmon resonance (SPR) and intrinsic protein fluorescence revealed that β-thujaplicinol has relatively weak binding affinity for the free enzyme (KDSPR = 5.7 μM, KDfluor = 1 μM; Table 1, Figure 3). Binding studies were also performed in the presence of a non-hydrolyzable double-stranded RNA substrate analog using intrinsic protein fluorescence. The presence of a substrate analog significantly decreased the KD value (KDfluor = 0.185 μM; Table 1, Figure 3), which is consistent with the Kiapp generated by kinetic studies. These data suggest that the presence of a bound RNA:DNA substrate is necessary to achieve maximal binding of β-thujaplicinol to RT. The time-dependence of inhibition may be due to the fact that assembly of the enzyme-inhibitor-substrate ternary complex is slow relative to enzyme turnover. The increased binding affinity of β-thujaplicinol in the presence of a substrate could be the result of a positive interaction between the substrate and the bound inhibitor. Alternatively, there could be a decrease in the dissociation rate constant for the inhibitor as a result of steric occlusion following substrate binding. SPR studies confirmed that there was no detectable inhibitor binding (IC50 ≫ 100 μM, data not shown) in the absence of divalent metal ions.
Figure 2. Inhibition of HIV-1 RT RNH activity by β-thujaplicinol.

a) Progress curves describing cleavage by RNH in the presence of ■ 0, ▲ 30, ▼ 45, ♦ 60, ● 110, + 200, ו 400, × 600 nM β-thujaplicinol. Progress curves were fit as described in materials and methods. b) Kobs vs. β-thujaplicinol concentration. Data plotted are the average of three measurements ± SD. c) Progress curves describing cleavage by RNH in the presence of 100 nM β-thujaplicinol and various concentrations of substrate (□10, ∆20, ∇30, ◊40, ○60, ■80, ▲100, ▼120, ♦140, ●1ff70, x210, +300 nM). d) Plot of Kobs as a function of substrate concentration. Data are the average of duplicates ± SD. All data shown are representative examples of greater than three independent experiments.
Table 1. Equilibrium Binding Constants for binding of β-thujaplicinol to HIV-1 RT.
| Binding Condition | KDfluor (μM) | KDSPR (μM) | Kiapp (μM) |
|---|---|---|---|
| Enzyme, no metal | - | >100 | - |
| Enzyme + MgCl2 | 0.734±0.09 | 6±1 | - |
| Enzyme + MgCl2 + ds RNA | 0.185±0.008 | - | - |
| Enzyme + MgCl2 + RNA:DNA hybrid | - | - | 0.14±0.03 |
KDfluor are equilibrium binding constants determined by quenching the intrinsic protein fluorescence of RT. See Figure 3.
KDSPR are equilibrium binding constants determined by surface plasmon resonance. See Figure 3.
Kiapp is the Ki value determined kinetically. See Figure 2.
All numbers represent the average of at least 2 independent experiments.
Figure 3. Binding of β-thujaplicinol to HIV-1 RT.

(a) Surface plasmon resonance sensorgrams of increasing concentrations of β-thujaplicinol interacting with RT in the absence of substrate. (b) Binding isotherm from (a). (c) Intrinsic protein fluorescence quenching as a result of incubation with increasing concentrations of β-thujaplicinol in the presence of a dsRNA substrate analog. (d) Example binding isotherm describing rfu max as a function of β-thujaplicinol concentration in the presence of a non-hydrolyzable substrate analog consisting of dsRNA. Data shown are representative examples of at least two independent examples.
Overall Protein Conformations
The RT69A HIV-1 RT[17] that was used to generate the complex with β-thujaplicinol crystallized with the symmetry of space group C2 (Table 2) with one protein molecule per asymmetric unit, and the protein was in a closed conformation typical of unliganded (no bound nucleic acid or inhibitor) RT. The isolated RNH domain crystallized in space group P31, with two protein molecules per asymmetric unit. The structures of the RNH domain in the intact RT and the isolated RNH domain corresponded closely to previously published RT structures. Although the last helix of the p66 subunit (helix E', secondary structure labeled as in ref. [4]) was partially unwound in some of the previously reported structures, we found helix E' intact and well-ordered in both of the structures described in this report.
Table 2. Data Collection and Refinement Statistics.
| Unit Cell and Space Group | |||||||
|---|---|---|---|---|---|---|---|
| RT | 164.1 | 71.3 | 108.5 Å | 90.0° | 105.1° | 90.0° | C2 |
| isoRNH | 51.1 | 41.1 | 112.4° | 90.0° | 90.0° | 120.0° | P31 |
| RT | isoRNH | ||||||
| PDB ID Code | 3IG1 | 3K2P | |||||
| Data Collection | |||||||
| Resolution Range (Å) | 35 - 2.80 | 50 - 2.04 | |||||
| Rsym (%) | 6.1 | 4.8 | |||||
| Average I/σ | 15.3 | 23.7 | |||||
| Completeness (%): | 94.3 | 99.8 | |||||
| Unique Reflections/Multiplicity | 28769/2.7 | 20314/3.4 | |||||
| Refinement | |||||||
| Sigma Cutoff | 0.0 | 0.0 | |||||
| Resolution Range Used (Å) | 33.8 - 2.80 | 44.3 - 2.04 | |||||
| Completeness in Range (%) | 91.9 | 99.1 | |||||
| R-factor/Rfree(%) | 23.8/25.5 | 21.1/25.4 | |||||
| Cross-validated Coordinate Error (Å)[52-54]: | 0.45 | 0.179 | |||||
| No. of Protein/Solvent Atoms | 7875/74 | 2045/219 | |||||
| No. of Inhibitor/Cation Atoms | 13/2 | 13/2 | |||||
| Average B-factors (Å2) | |||||||
| Protein/Solvent | 89.3/58.6 | 34.0/45.0 | |||||
| Inhibitor/cations | 104.2/65.7 | 38.19/31.88 | |||||
| RMS Bond Lengths (Å)/Angles (°)[55] | 0.008/1.70 | 0.022/1.99 | |||||
| Ramachandran Regions | |||||||
| Most favored | 95.1% | 93.4% | |||||
| Additional Allowed | 4.8% | 6.1% | |||||
| Generous or disallowed | 0.1% | 0.4% | |||||
Our two structures, which were determined and refined independently, from different starting models, showed good overall agreement. When all main chain atoms of each of the isolated RNH copies in the asymmetric unit were superimposed on those of RT, we calculated a root mean square difference of 0.54 to 0.55 Å between corresponding main chain atoms and 1.63 to 1.66 Å between side chain atoms. The agreement between the atoms of active site residues (Asp443, Glu478, Asp498, His539, and Asp549) was even better, with an rms difference of 0.29 to 0.36 Å between main chain atoms, 1.01 to 1.02 Å between side chain atoms, 0.56 to 0.58 Å between inhibitor atoms, and 0.17 to 0.27 Å between corresponding Mn2+ atoms.
β-Thujaplicinol Binds at the RNH Active Site
The binding of the inhibitor is essentially identical in both the RT and isolated RNH structures (Figures 4 and 5). Both structures exhibited strong difference Fourier (Fo-Fc) electron density peaks for two manganese ions at the RNH active site (Figure 4), separated by a distance of about 3.8 Å. We designate these manganese ions “A” and “B” following the convention for a two-cation mechanism which is discussed below. Glu478 and Asp 498 coordinate metal ion B, and Asp549 coordinates metal ion A (Figure 6). The side chain of Asp443 is between the Mn2+ ions and coordinates both metals. In both structures, β-thujaplicinol coordinates the two Mn2+ ions and forms ionic or hydrogen-bond interactions with the side chains of residues Glu478, Asp498, His539, and Asp549 (Figure 5 and 6). All three oxygen atoms of the tropolone ring of β-thujaplicinol coordinate the manganese ions. The middle oxygen (at the 1-position) of the tropolone ring coordinates both manganese ions. Metal ion A is coordinated by four ligands in both of the structures, including the 2-hydroxyl on the tropolone ring. Metal ion B, however, is coordinated by six or seven ligands (in vivo, where the cation is magnesium, six-atom-coordination is expected). The isolated RNH structure shows that there is an ionic or hydrogen-bond contact between one of the inhibitor hydroxyls and the side chain of Arg557. Arg557 is not present in the full-length RT crystallized here. In the isolated RNH structure, Arg557 also forms a salt bridge with Asp549. Ala538 has a 4 Å non-polar interaction with the tropolone ring.
Figure 4. Electron density at the binding site.

For (a) RT/β-thujaplicinol and (b) the isolated RNH domain in complex with β-thujaplicinol, three electron density maps are shown in “wall-eyed” stereo: a 2Fo-Fc difference map (cyan), contoured at 1 sigma; a simulated-annealing Fo-Fc omit map in which β-thujaplicinol and both manganese cations were omitted (magenta), contoured at 3.5 sigma in (a) and 2.5 sigma in (b); and a simulated-annealing Fo-Fc omit map in which only the cations were omitted (dark green), contoured at 9.5 sigma.
Figure 5. Superposition of the binding sites in the two RNH structures.

Residues Val442 to Asp443, Glu 478, and Asp549 were superimposed for the coordinates for RT (carbons in yellow) and the isolated RNH domain (carbons in black). The coordinates of the two structures, which were refined independently, agree remarkably well with each other. Manganese cations are labeled A and B according to the convention used in the previously proposed two-cation mechanism[28, 29].
Figure 6. Contact distances between protein, cations, and inhibitor.

Electrostatic (red) and several hydrophobic (gray) interactions are indicated with distances in Å. Solid lines designate coordination of manganese cations A and B, whereas dashed lines indicate direct protein-inhibitor interactions. Inset: A diagram of the tropylium ion charge distribution and its application to β-thujaplicinol.
Discussion
Tropylium Chemistry and Mn2+ Coordination
A tropylium ion is a seven-membered aromatic ring, containing three conjugated double bonds and an overall +1 charge (for reviews, see refs [18-20], Figure 6 Inset). If one of the carbon atoms is changed to a carbonyl, the ring is called a tropolone, and the ring retains some aromatic character[21]. β-Thujaplicinol is a member of the thujaplicin family of compounds, which are characterized by a tropolone ring and an acidic hydroxyl (for review, see ref. [22]). Based on the properties of tropolones, β-thujaplicinol might be expected to carry at least a partial positive charge on its tropolone ring and have a corresponding partial negative charge shared by resonance involving its carbonyl and hydroxyls (Figure 6 Inset).
RNH Enzymatic Mechanism
For over a decade, the enzymatic mechanism of HIV RNH and related RNases has been the subject of debate, with several hypotheses being advanced. All of the proposed mechanisms suggest that cleavage of substrate likely occurs by general base catalysis in which an activated water molecule, acting as a Lewis base, makes a nucleophilic attack on a phosphate of the RNA strand. This scissile phosphate becomes a good leaving group when the product is reprotonated by general acid catalysis. In E. coli RNase HI, it has been suggested that His124 (RT His539) serves as the general base[23]. The hypotheses diverge on whether one cation[24-27] or two cations[28, 29] are required for catalysis, and it has also been proposed that the second cation (when present) serves to attenuate catalysis[30, 31]. In the two-cation mechanism, Metal A helps to position the catalytic water for nucleophilic attack and Metal B stabilizes the pentacovalent transition state of reaction[28, 29]. Recently published structures of B. halodurans RNase H/RNA:DNA[32] and human RNase H1/RNA:DNA[33], both with an active site point mutation to inactivate the enzyme, show the substrate bound to the active site in position appropriate for cleavage along with two cations, lending support to the two-cation mechanism.
Mechanism of Inhibition
The presence of a carbonyl and at least one hydroxyl on the tropolone ring of the thujaplicins gives this class of compounds the ability to chelate metal cations, and α-thujaplicin weakly inhibits (IC50 50 μM) HIV RNase H, suggesting that this compound coordinates one of the RNH active site cations[13]. The structures reported in the present study show that the additional hydroxyl of β-thujaplicinol at the 7-position of the tropolone ring (Figure 6) allows this compound to coordinate both divalent cations.
To investigate the mechanism of inhibition, a structure for human RNase H1 (PDB code 2QKK, ref. [33]) was superimposed on our current RT/β-thujaplicinol structure (superposition was based on p66 residues Val442 to Gly444 focusing on Asp443, which coordinates both cations). The human RNase H1 is in a complex with an RNA:DNA substrate appropriately positioned for cleavage; catalysis is prevented by a point mutation, Asp210Asn, which is at a position equivalent to RT residue Asp498[33]. The active sites of the two structures superimpose well, including the two RNase H1 Ca2+ ions in the structure of the human enzyme that occupy virtually the same positions as the Mn2+ ions of the two RNH structures reported here (Figure 7). This superposition suggests that β-thujaplicinol usurps the position of both the scissile phosphate and the water believed to serve as the nucleophile during catalysis.
Figure 7. Comparison of substrate and inhibitor binding.

The RNH active sites of human Ca2+-RNase H1 from a structure of a complex with RNA:DNA (magenta protein carbon atoms and cyan nucleic acid carbon atoms, Ca2+ atoms magenta)[33] and HIV-1 RT from the complex with β-thujaplicinol are superimposed. The RT Mn2+ atoms are shown in black and labelled A and B as in Figure 5. The separation distance between each Mn2+ atom and the corresponding Ca2+ atom is 0.6 Å for Mn2+ atom A and 1 Å for Mn2+ atom B. Some of the RT RNH active site side-chains are shown with yellow carbon atoms and labelled to indicate the quality of the superposition (RT residue=corresponding human RNase H1 residue: Asp443=Asp145; Glu478=Glu186; Asp498=Asp210; Asp549=Asp274; His539=His264). The nucleophile (red) is a water molecule in the human Ca2+-RNase H1/RNA:DNA structure. This superposition suggests that β-thujaplicinol (translucent gray and pink atoms) occupies the position that would normally be occupied by the scissile phosphate of the RNA template strand and the attacking nucleophilic water molecule
The kinetic analysis reported here (Figure 2) supports the view that β-thujaplicinol is a non-competitive inhibitor, suggesting that the RNA:DNA substrate and β-thujaplicinol can be bound to the enzyme together, forming an Enzyme·Substrate·Inhibitor (E·S·I) complex. A recently published paper describes the inhibition of RT by β-thujaplicinol under pre-steady state conditions (Bielhartz et al). In this experiment the authors pre-form the Enzyme·Substrate (E·S) complex and show that β-thujaplicinol is unable to inhibit RNase H cleavage. In a separate experiment, the authors demonstrate that β-thujaplicinol is not an inhibitor of polymerase dependent cleavage. Both experiments provide compelling evidence that when bound, the RNA:DNA substrate blocks access to the RNase H active site. However, these experiments don't provide information as to whether the substrate is able to bind to the Enzyme·Inhibitor (E·I) complex. An inhibitor which is able to bind only the free enzyme, and not the E·S complex can still demonstrate non-competitive kinetics if the substrate is able to bind the E·I complex thereby forming the E·S·I complex. Non-competitive kinetics have been shown in other enzyme systems[34, 35] where a small molecule is known to bind at the active site, but is insufficient to displace the macromolecular substrate. In these cases, the inhibitor is able to block access to the active site but does not block the ability of the substrate to bind to the enzyme through interactions at regions beyond the active site. Given that the RNA:DNA substrate interacts with RT over a large region extending from the polymerase active site to the RNH active site[36], it is reasonable to propose that the RNA:DNA substrate can be bound to RT/β-thujaplicinol even as the inhibitor competes with the RNA strand for access to the RNH active site.
We have previously shown that an RNA:DNA substrate can bind to RT without engaging the RNH active site. In a structure of RT complexed with an RNA:DNA substrate containing a polypurine tract (PDB code: 1HYS)[6], the polypurine tract of the RNA strand interacts with the RNase H domain but is not cleaved because the scissile phosphate is about 4 Å away from the active site[6]. To model what might happen when RT/β-thujaplicinol binds an RNA:DNA substrate, the aforementioned RT/RNA:DNA structure (PDB code: 1HYS) was superposed on our RT/β-thujaplicinol structure (the superposition was based on residues Val442 to Asp443, Glu 478, and Asp549). This superposition suggests that the two non-bridging oxygen atoms of the scissile phosphate could form ionic interactions with the positively charged tropolone ring of β-thujaplicinol (Figure 8). As stated above, binding studies using both SPR and intrinsic protein fluorescence showed a significant increase in the affinity of RT for β-thujaplicinol when a double-stranded nucleic acid substrate analog was present (Table 1). The increase in affinity of β-thujaplicinol for RT in the presence of substrate could be the result of direct favorable interactions between the inhibitor and substrate in the ternary complex. Alternatively, the increased affinity could be caused by a decrease in the off-rate if the bound substrate interfered with the release of the inhibitor from the complex. In this case, the on-rate for the inhibitor would also be reduced in the enzyme-substrate complex relative to the free enzyme complex. β-thujaplicinol would be expected to bind preferentially to the free enzyme. Subsequent binding of substrate would then trap the E·S·I complex. This could explain the time-dependent nature of the inhibition if dissociation of the E·S complex is slow relative to both the formation of the E·I complex and turnover of the E·S complex. Interestingly, inhibition of the isolated RNase H domain by β-thujaplicinol is not time dependent. In this case, the binding affinity of the RNA:DNA substrate is significantly weaker and turnover of the E·S complex significantly slower. If association and dissociation of β-thujaplicinol are hindered by the presence of nucleic acid substrate as suggested by these studies, this might partly explain why potent in vitro inhibition of RNH activity has not yet been translated into potent in vivo antiviral activity. The processive nature of the RT polymerase could limit the presence of free RT available for binding by active site RNHIs. However, polymerase-independent activities of RNH such as primer removal steps should still be sensitive to active site directed RNHIs. Beilhartz et al. have shown that β-thujaplicinol inhibits polymerase-independent “secondary cuts” in the RNA strand caused by the RNH[37]. Additional structural and kinetic studies are necessary to fully understand the interactions of the inhibitor with the RT/RNA:DNA complex.
Figure 8. Modeling inhibitor-substrate interactions.
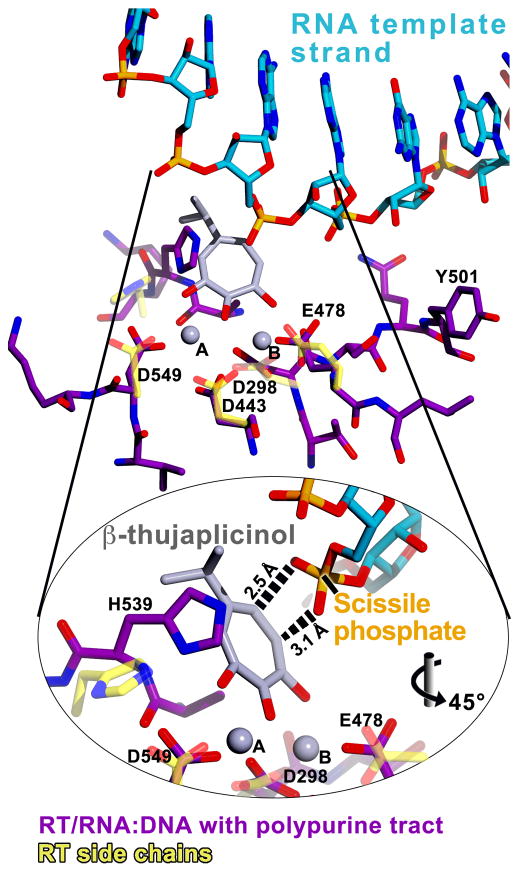
Shown is a superposition of the active sites of two crystal structures: (1) RT/β-thujaplicinol (same vantage point as in Figure 7); (2) RT/RNA:DNA with no bound inhibitor (magenta protein carbon atoms and cyan nucleic acid carbon atoms)[6]. The RT Mn2+ atoms are shown in gray and labelled A and B as in Figures 5 and 7. Inset: Close-up view of the RNH active site rotated 45° as indicated to show the distance between the inhibitor and the scissile phosphate of the substrate. Some of the RT/β-thujaplicinol active site side-chains are shown with yellow carbon atoms to indicate the quality of the superposition. The polypurine tract of the RNA:DNA substrate positions the scissile phosphate about 4 Å away from the RNH active site, providing a space for β-thujaplicinol to make the same interactions with RT that are present in our current structures. According to the superposition, the positively charged tropolone ring of β-thujaplicinol would be positioned to interact favorably with the scissile phosphate of the substrate (dashed lines).
A recent model suggests that the tropolone ring of β-thujaplicinol forms π-stacking interactions with the side chain of Tyr501, in addition to coordinating one of the active site divalent cations[37]. Further, it was found that substituting benzoyl-L-phenylalanine, an artificial amino acid, in place of Tyr501 conferred resistance to inhibition by β-thujaplicinol. These results were explained by proposing that the artificial side chain rotated away from β-thujaplicinol to form more favorable interactions with other RT residues, depriving β-thujaplicinol of π-stacking interactions. Our structural results do not support this picture. Instead, they indicate that β-thujaplicinol coordinates both divalent cations at the RNH active site and is unavailable for interactions with Tyr501. Resistance by benzoyl-L-phenylalanine may be explained by assuming that the bulky side chain interferes sterically with the binding of β-thujaplicinol but can be displaced by the natural substrate.
Inhibitors containing diketo acid moieties interfere with the enzymatic activities of both HIV-1 RNH and HIV integrase[10, 11]. In both cases, it has been proposed that these inhibitors act by coordinating two divalent cations at the active site[11]. Other integrase inhibitors have subsequently been developed based on this hypothesis[38], but no crystal structure is currently available which shows the interactions between integrase and these inhibitors. Because the active site of RNH is structurally similar to those of integrases and transposases[39], the structures presented here may be helpful in the development of structural models of the interaction of integrase with its inhibitors.
Conclusions
We have presented a 2.80 Å resolution structure of HIV-1 RT and a 2.04 Å structure of the isolated RNH domain, both with β-thujaplicinol bound in virtually identical modes at the RNH active site. Although a conclusive picture of protein-inhibitor-substrate interactions will require a structure of RT in complex with both an RNA:DNA substrate and β-thujaplicinol, our current structural results are consistent with a mechanism of inhibition in which β-thujaplicinol forms a stable inhibitory complex with protein and substrate that directly prevents the attacking water and the scissile phosphate from being properly positioned for catalysis. These studies provide a detailed visualization of a metal-chelating inhibitor at the active site of RNH and could help the development of therapeutic agents that bind to the RNH active site, blocking viral replication.
Our biochemical results agree with previously published data[13] that β-thujaplicinol displays non-competitive inhibitory kinetics. We propose that the nucleic acid substrate is able to bind the E·I complex and for a stable E·S·I complex. The formation of this ternary complex results in non-competitive kinetics in the steady state. Furthermore, we propose that the substrate binding enhances inhibitor binding either by forming direct positive interactions with the inhibitor or by sterically blocking inhibitor dissociation. Our modeling studies suggest the substrate binds in a manner where it lays over the top of the inhibitor while it's bound at the active site. This model would predict that a pre-formed E·S complex would be resistant to inhibition by an active site directed inhibitor, consistent with recently published pre-steady state inhibition studies and polymerase dependent RNAse H inhibitions studies[37]. We cannot exclude the possibility that β-thujaplicinol has two binding modes, one at higher affinity that gives rise to non-competitve kinetics, and one at the RNH active site which is far weaker and does not affect the biochemical kinetics that we observe. However, there is no evidence to date that suggests multiple binding modes.
Designing an RNHI that can be used as an anti-AIDS drug presents a challenge, both because the structure of the active site of HIV-1 RNase H is very flat and because it is quite similar to the active site of human RNase H (Figure 7). A therapeutically useful RNHI will have to inhibit HIV RNase H activity potently and specifically, without inhibiting the corresponding cellular enzymes. The RNHI β-thujaplicinol inhibits HIV-1 RT by binding to catalytically essential residues of the RNH active site and chelating the essential metal ions. The IC50 of β-thujaplicinol for the RNH of HIV-1 RT is 30-fold greater than that of human RNase H1, almost 300-fold greater than that of E. coli RNase H[13], and over 10-fold greater than that of HIV-1 integrase (data not shown). The fact that compounds have been developed that interact with the active site of HIV-1 integrase with a much greater specificity suggests that additional improvements can be made in the development of RNH inhibitors. The current structures should provide guidance that can be used to introduce additional substituents onto β-thujaplicinol that should enhance discrimination between the retroviral and the human enzymes.
Experimental Procedures
Expression and Purification of HIV-1 RT for structural studies
An HIV-1 RT variant (designated RT69A) was used for X-ray diffraction studies. In this variant the p66 subunit contained mutations F160S and C280S, and was truncated at residue 555[17]. The p51 subunit contained a HRV14 3C protease cleavable N-terminal hexahistidine tag, a C280S mutation, and was truncated at residue 428. RT69A was expressed and purified as previously described[17]. Briefly, 1 mM IPTG was used to induce BL21-CodonPlus®-RIL (Stratagene) containing a plasmid encoding both subunits of RT69A at an OD600 of 0.9 and the culture was incubated for three hours at 37°C. The cells were pelleted and lysed by sonication. Protein was purified by Ni-NTA according to manufacturers' recommendations (Qiagen) with the following modifications: each buffer contained 600 mM NaCl, no lysozyme was added, and a 1.2 M NaCl wash was added. Eluted protein was incubated with HRV14 3C protease overnight at 4°C. A Mono Q purification step was performed as described[40]. The RT69A protein was concentrated to 20 mg ml-1 in “RT storage buffer” (10 mM Tris pH 8.0, 75 mM NaCl) and stored at -80°C.
Expression and purification of HIV-1 RT for biochemical studies
Full length wild-type HIV-1 RT p66, from strain BH10, was expressed in E. coli BL21(DE3) and induced with 0.1 mM IPTG overnight at 25°C[41]. A cell pellet corresponding to 3 L of culture was resuspended in 30 mM Tris pH 8.0, 10% glycerol, 250 mM NaCl, 1 mM EDTA and 2 mM DTT and disrupted by microfluidization (Microfluidics, model M-110 Y). After adjustment of the NaCl concentration to 1 M, the lysate was sonicated for 1 min 45 s. The lysate was clarified by ultracentrifugation (Beckman L8-M) at 125,171 × g for 45 minutes and dialyzed overnight against buffer A (25 mM Tris 8.0, 2 mM DTT). The dialyzed RT was loaded onto a 110 ml heparin-Sepharose CL-6B column (GE Healthcare) equilibrated in buffer A. The protein was loaded overnight at a flow rate of 0.5 ml/min. The column was washed and RT was eluted with a linear gradient using 8 column volumes of buffer A plus 0.8 M NaCl. Fractions were pooled based on SDS-PAGE and biochemical assays. At this point the RT had been converted to the p66/p51 heterodimer by E. coli proteases. The heparin pool was dialyzed overnight in buffer B (25 mM HEPES pH 7.0, 25 mM NaCl, 2 mM DTT) and was loaded onto a 20 ml Source 15Q column connected in series to a 10 ml SP-sepharose HP column (GE Healthcare) equilibrated in buffer B. HIV-1 RT was eluted from SP sepharose with a 30 column volume gradient of buffer B plus 0.5 M NaCl. SP fractions were pooled based on SDS-PAGE and dialyzed overnight against a buffer containing 50 mM Tris pH 8.0 and 2 mM DTT. Dialyzed SP fractions were concentrated and loaded onto a HiPrep 26/60 Sephacryl S-200 HR column (GE Healthcare). Fractions were pooled, aliquoted, and stored at -80°C.
Expression and purification of isolated RNH domain
The isolated RNH domain (amino acids 427-560, HIV-1 BH10) was cloned into a modified pET vector that coded for an N-terminal 6×-His tag followed by a TEV protease recognition sequence. Expression was induced by 0.1M IPTG in E. coli BL21(DE3) cells at 16°C overnight[41], and the isolated RNH was purified as follows: A cell pellet corresponding to 2 L of culture was suspended in buffer C (25 mM Tris–HCl, pH 8.0, 25 mM NaCl, 20 mM imidazole, and 0.25 mM TCEP) and was disrupted and clarified as described above for the purification of RT for Biochemical assays. The supernatant fraction was collected and loaded onto a 5 ml ProBond nickel affinity column (Invitrogen) equilibrated in Buffer C. The column was washed with 30 column volumes of buffer C plus 1 M NaCl, followed by a second 30 column volume wash with buffer C. The isolated RNH domain was step-eluted with 30 column volumes of buffer D (25 mM Tris–HCl, pH 8.0, 25 mM NaCl, 400 mM imidazole, and 0.25 mM TCEP). Eluted isolated RNH domain was dialyzed with recombinant TEV protease at room temperature overnight against buffer containing 25 mM Tris–HCl, pH 8.0, 50 mM NaCl, 20 mM imidazole, and 0.25 mM TCEP. The dialyzed solution containing the isolated RNH domain was further incubated the following morning in a 37°C water bath for 8 hours. The solution containing the cleaved isolated RNH domain was adjusted to 0.5 M NaCl and uncleaved material was removed with a second passage over a 5 ml nickel affinity column. The isolated RNH domain was collected in the flow through, concentrated, and loaded to a gel filtration (HiLoad 26/60 Superdex-75) column equilibrated in (25 mM Tris–HCl, pH 7.6, 50 mM NaCl, and 0.25 mM TCEP. The isolated RNH domain eluted in two peaks corresponding to monomeric and dimeric forms of the protein. Monomer fractions were pooled, concentrated to ∼20 mg/ml, and stored at -80°C.
Isolated RNase H domain used for kinetic experiments contained a C-terminal histidine tag to facilitate enzymatic activity. This protein was made as described[42]
Crystallization and Data Collection
HIV-1 RT was co-crystallized with β-thujaplicinol by vapor diffusion in microseeded hanging drops containing 1.3 μl each of 20 mg ml-1 protein in RT buffer (10 mM Tris pH 8.0, 75 mM NaCl) and reservoir solution (50 mM Bicine pH 8.2, 45 mM ammonium sulfate, 15 mM manganese sulfate, 10 mM spermine, 5 mM taurine, 2% (v/v) PEG 400, 10% (w/v) PEG 8000) at 4°C. Crystals were transferred to a stabilization solution containing 14% (w/v) PEG 8000 in the above mother liquor, and 5% PEG 200 instead of PEG 400. In the presence of 0.86 mM β-thujaplicinol, the PEG 200 concentration was raised stepwise in 5% increments by solution replacement at one to three minute intervals per step to a final concentration of 26% (w/v) PEG 200. The crystals were subsequently flash-cooled and stored in liquid N2. X-ray data were collected at 100K and a wavelength of 1.1 Å at the National Synchrotron Light Source at Brookhaven National Laboratories, Beamline X25. The data were processed using DENZO/SCALEPACK[43, 44].
Crystals of the isolated RNH domain were grown by vapor diffusion in hanging drops containing 1.0 μl each of 21.6 mg ml-1 protein (in 25 mM Tris pH 7.6, 50 mM NaCl, 0.25 mM TCEP) and well solution (200 mM formate pH 7.0, 10% (w/v) PEG 3350, 10 mM MnCl2) at 21°C. Drops were soaked with 0.04 μl of 200 mM β-Thujaplicinol in DMSO for 4.5 hrs. The crystal was cryo-protected by a brief immersion in well solution containing 20% (v/v) glycerol followed by flash-cooling and storage in liquid N2. Data were collected on a RuH2R generator with a MAR345 detector. The data were integrated and scaled using DENZO/SCALEPACK[43, 44].
Structure Determination and Refinement
Phases for the HIV-1 RT/β-thujaplicinol data were determined by molecular replacement with the CCP4 program PHASER[45], using an unliganded RT structure (PDB accession number 1DLO)[7] as an initial search model. Stepwise model building and refinement were conducted using the “O” graphics package[46], the Coot graphics package (version 0.3.1)[47], and CNS 1.1[48] with a bulk solvent correction. Water molecules were built in manually. The geometry of the inhibitor and refinement of the RNH active site were improved by energy minimization using the Impact and PrimeX facilities of the Schrödinger software package (Schrödinger, LLC).
The structure of the isolated RNH domain was solved by molecular replacement with CCP4 PHASER[45] using an unliganded isolated RNH domain structure (PDB accession number 1HRH)[9] as a search model. The model was refined using CCP4 Refmac[49], with ligand restraints generated by Sketcher in CCP4. Root mean square differences between corresponding atoms of the isolated RNH domain and RT were calculated in CNS.
Enzymatic Activity Assays
The RNH activity of HIV-1 RT was determined essentially as described[50] with minor modifications. Briefly, 1 nM HIV-1 RT was incubated with 150 nM 18-mer RNA:DNA hybrid FRET substrate in 1× FRET buffer (50 mM Tris pH 8.0, 60 mM KCl, 5 mM MgCl2, 0.02% Tween-20). The enzyme-mediated increase in fluorescence was monitored at 520 nm using a Tecan Safire plate reader. Product formation was determined at various concentrations of β-thujaplicinol under initial velocity conditions. The progress curves were fit to Equation 1, solving for kobs, vi and vs using GraphPad Prism (GraphPad Software Inc.). Estimates of Kiapp were generated by (1) plotting kobs versus β-thujaplicinol] concentration and fitting the data to Equation 2 and (2) plotting vs vs. [β-thujaplicinol] and fitting to the standard hyperbolic IC50 equation (Equation 3).
| Equation 1 |
where vi is initial velocity, vs is steady state velocity, and kobs is the rate constant for the conversion from the initial rate to the steady state rate.
| Equation 2 |
where k4 is the dissociation rate constant, kobs is determined from equation 1, and [I] is the inhibitor concentration.
| Equation 3 |
The enzymatic activity of the isolated RNase H domain was determined as described for HIV-1 RT with the following changes. The enzyme concentration used was 500 μM, the 18-mer FRET substrate concentration was varied between 1-20 μM (Km = 7 μM), and the reaction was run in buffer containing 50 mM Tris pH 7.5, 20 mM KCl, 0.02% Tween-20, and 8 mM MnCl2.
Direct Binding Assay Using Intrinsic Protein Fluorescence
β-thujaplicinol binding to HIV-1 RT was detected by monitoring the quenching of intrinsic tryptophan fluorescence as a function of inhibitor concentration. The fluorescence emission spectra was monitored using a Perkin Elmer LS-50B fluorimeter by exciting the protein (0.8 uM in 1 ml) in 1× FRET buffer minus the Tween-20 +/- double-stranded 18-mer RNA substrate analog, at 295 nm. The dsRNA used is identical to the hybrid substrate used for the activity assay except that it contains only RNA. HIV-1 RT is unable to cleave this dsRNA analog in the presence of MgCl2 (data not shown). Multiple small aliquots of β-thujaplicinol in 10% (v/v) DMSO (25 μl total per experiment) were titrated into the protein sample and the quench in protein fluorescence was determined. Multiple scans were performed at various incubation times to assure protein stability and equilibrium conditions during titrations. Protein fluorescence measurements in the presence of inhibitor were corrected for protein dilution. DMSO titrations showed no quenching of the fluorescence signal at concentrations achieved in the β-thujaplicinol titrations (< 1%). Inner filter effects were corrected for by performing β-thujaplicinol titrations into free N-Acetyl-L-tryptophanamide[51]. The quench observed upon titration into free N-Acetyl-L-tryptophanamide was subtracted from the quench observed upon titration into HIV-1 RT. The IC50 for binding was estimated by plotting the corrected relative fluorescence value as a function of inhibitor concentration.
Direct binding Assay Using Surface Plasmon Resonance
Small molecule binding studies were carried out using a Biacore 3000 instrument (GE Healthcare) at 20°C in a 60 mM KCl, 50 mM Tris.HCl pH 8.0, 8 mM MgCl2, 0.005% P20 detergent, 2.0 mM DTT, 1% (v/v) DMSO running buffer. Samples of the isolated RNH domain or full length HIV-1 RT were immobilized on a Biacore CM5 chip by standard EDC/NHS amine coupling chemistry, using a 0.4 μM solution of the protein, in 10 mM sodium acetate buffer pH 6.0 at 25°C. Immobilization was carried out in the presence of a saturating concentration of β-thujaplicinol to minimize perturbation of the active site upon coupling to the chip surface. In a typical experiment compound injections were made at a flow rate of 50 μl per minute using the Biacore 'kinject' mode, which is designed to minimize sample dispersion during injection (GE Healthcare). Compound injections were made in duplicate using a two-fold dilution series from a 100 μM starting concentration. Data analysis was performed using the Scrubber2 software package (BioLogic Software, Pty., Australia). Compound injections were referenced to both a blank surface and by a buffer blank. Processed data were fit to an equilibrium model for single set of binding sites to obtain the observed equilibrium dissociation constant KD. SPR-based β-thujaplicinol binding studies could not be performed in the presence of double-stranded RNA substrate analog. In order to ensure continuous exposure to the enzyme, the substrate analog must be present in the running buffer and compound samples. This would have required impractical quantities of substrate analog. Binding studies done in the absence of magnesium were performed identically except MgCl2 was omitted from the buffer and 0.1 mM EDTA was added. Control experiments were run immediately before and after the magnesium omit studies to verify active protein on the chip.
Acknowledgments
This research was supported by Pfizer and NIH grant AI 27690. We are grateful to synchrotron staff members at Brookhaven National Light Source, Meirong Xu and Darin Vanderpool for work on the fluorescence binding experiments, Zdenek Hostomsky for encouragement, and other members of our laboratories for their assistance and helpful discussions. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. This publication has been funded in part with Federal funds from the National Cancer Institute, NIH, under Contract No. NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- HAART
- PPT
- RNH
- RNHI
- RT
- SPR
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tisdale M, Schulze T, Larder BA, Moelling K. Mutations Within the RNase H Domain of HIV-1 RT Abolish Virus Infectivity. J Gen Virol. 1991;72:59–66. doi: 10.1099/0022-1317-72-1-59. [DOI] [PubMed] [Google Scholar]
- 2.Schatz O, Cromme F, Naas T, Lindermann D, Gruninger-Leitch F, Mous J, Le Grice SFJ. Inactivativation of the RNaseH domain of HIV-1 Reverse Transcriptase Blocks Viral Infectivity. In: Papas T, editor. Oncogenesis and AIDS. Houston, TX: Portfolio Publishing Company; 1990. pp. 55–68. [Google Scholar]
- 3.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal Structure at 3.5 Å Resolution of HIV-1 Reverse Transcriptase Complexed with an Inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 4.Jacobo-Molina A, Ding J, Nanni RG, Clark AD, Jr, Ju X, Tantillo C, Williams RL, Kamer G, Ferris AL, Clark P, Hizi A, Hughes SH, Arnold E. Crystal Structure of Human Immunodeficiency Virus Type 1 Reverse Transcriptase Complexed with Double-stranded DNA at 3.0 Å Resolution Shows Bent DNA. Proc Natl Acad Sci USA. 1993;90:6320–6324. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a Covalently Trapped Catalytic Complex of HIV-1 Reverse Transcriptase: Implications for Drug Resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 6.Sarafianos SG, Das K, Tantillo C, Clark AD, Jr, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold E. Crystal Structure of HIV-1 Reverse Transcriptase in Complex with a Polypurine Tract RNA:DNA. EMBO J. 2001;20:1449–1461. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsiou Y, Ding J, Das K, Clark AD, Jr, Hughes SH, Arnold E. Structure of Unliganded HIV-1 Reverse Transcriptase at 2.7 Å Resolution: Implications of Conformational Changes for Polymerization and Inhibition Mechanisms. Structure. 1996;4:853–860. doi: 10.1016/s0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- 8.Sarafianos SG, Das K, Hughes SH, Arnold E. Taking Aim at a Moving Target: Designing Drugs to Inhibit Drug-Resistant HIV-1 Reverse Transcriptases. Curr Opin Struct Biol. 2004;14:716–730. doi: 10.1016/j.sbi.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Davies JF, 2nd, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. Crystal Structure of the Ribonuclease H Domain of HIV-1 Reverse Transcriptase. Science. 1991;252:88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- 10.Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen R, Olsen DB, Carroll SS, Embrey M, Wai JS, Miller MD, Cole JL, Hazuda DJ. Inhibition of HIV-1 Ribonuclease H by a Novel Diketo Acid, 4-[5-(benzoylamino)thien-2-Yl]-2,4-dioxobutanoic Acid. J Biol Chem. 2003;278:2777–2780. doi: 10.1074/jbc.C200621200. [DOI] [PubMed] [Google Scholar]
- 11.Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai JS, Young S, Vacca J, Hazuda DJ. Diketo Acid Inhibitor Mechanism and HIV-1 Integrase: Implications For Metal Binding in the Active Site of Phosphotransferase Enzymes. Proc Natl Acad Sci U S A. 2002;99:6661–6666. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klumpp K, Hang JQ, Rajendran S, Yang Y, Derosier A, Wong Kai In P, Overton H, Parkes KE, Cammack N, Martin JA. Two-Metal Ion Mechanism of RNA Cleavage by HIV RNase H and Mechanism-Based Design of Selective HIV RNase H Inhibitors. Nucleic Acids Res. 2003;31:6852–6859. doi: 10.1093/nar/gkg881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. Selective Inhibition of HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity by Hydroxylated Tropolones. Nucleic Acids Res. 2005;33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner JAF, Baron GM, MacLean H. Occurrence of 2,7-dihydroxy-4-isopropyl-2,4,6-cycloheptatrien-1-one (7-hydroxy-4-iso-propyltropolone) in Western Red Cedar (Thuja plicata Donn) Can J Chem. 1957;35:1039–1048. [Google Scholar]
- 15.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD, Jr, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. HIV-1 Reverse Transcriptase Structure With RNase H Inhibitor Dihydroxy Benzoyl Naphthyl Hydrazone Bound at a Novel Site. ACS Chem Biol. 2006;1:702–712. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klumpp K, Mirzadegan T. Recent Progress in the Design of Small Molecule Inhibitors of HIV RNase H. Curr Pharm Des. 2006;12:1909–1922. doi: 10.2174/138161206776873653. [DOI] [PubMed] [Google Scholar]
- 17.Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD, Jr, Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E. Crystal Engineering of HIV-1 Reverse Transcriptase for Structure-based Drug Design. Nucleic Acids Res. 2008;36:5083–5092. doi: 10.1093/nar/gkn464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietra F. Seven-membered Conjugated Carbo- and Heterocyclic Compounds and their Homoconjugated Analogs and Metal Complexes. Synthesis, Biosynthesis, Structure, and Reactivity. Chem Rev. 1973;73:293–364. [Google Scholar]
- 19.Bertelli DJ. Ground State Structures of Heptafulvene •-Systems. In: Nozoe T, Breslow R, Itô S, Hafner K, Murata I, editors. Top Nonbenzenoid Aromat Chem. Vol. 1. New York: John Wiley & Sons, Inc.; 1973. pp. 29–46. [Google Scholar]
- 20.Kolomnikova GD, Parnes ZN. Advances in the Chemistry of the Tropylium Ion. Russ Chem Rev. 1967;36:735–753. [Google Scholar]
- 21.Dewar MJS. Structure of Stipitatic Acid. Nature. 1945;155:50–51. [Google Scholar]
- 22.Pauson PL. Tropones and Tropolones. Chem Rev. 1955;55:9–136. [Google Scholar]
- 23.Oda Y, Yoshida M, Kanaya S. Role of Histidine 124 in the Catalytic Function of Ribonuclease HI From Escherichia Coli. J Biol Chem. 1993;268:88–92. [PubMed] [Google Scholar]
- 24.Nakamura H, Oda Y, Iwai S, Inoue H, Ohtsuka E, Kanaya S, Kimura S, Katsuda C, Katayanagi K, Morikawa K, et al. How Does RNase H Recognize a DNA RNA Hybrid? Proc Natl Acad Sci U S A. 1991;88:11535–11539. doi: 10.1073/pnas.88.24.11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oda Y, Nakamura H, Kanaya S, Ikehara M. Binding of Metal Ions to E. Coli RNase HI Observed by 1H-15N Heteronuclear 2D NMR. J Biomol NMR. 1991;1:247–255. doi: 10.1007/BF01875518. [DOI] [PubMed] [Google Scholar]
- 26.Katayanagi K, Okumura M, Morikawa K. Crystal Structure of Escherichia Coli Rnase HI in Complex with Mg2+ at 2.8 A Resolution: Proof for a Single Mg(2+)-Binding Site. Proteins. 1993;17:337–346. doi: 10.1002/prot.340170402. [DOI] [PubMed] [Google Scholar]
- 27.Kanaya S, Oobatake M, Liu Y. Thermal Stability of Escherichia Coli Ribonuclease HI and its Active Site Mutants in the Presence and Absence of the Mg2+ Ion. Proposal of a Novel Catalytic Role for Glu48. J Biol Chem. 1996;271:32729–32736. doi: 10.1074/jbc.271.51.32729. [DOI] [PubMed] [Google Scholar]
- 28.Yang W, Hendrickson WA, Crouch RJ, Satow Y. Structure of Ribonuclease H Phased at 2 A Resolution by MAD Analysis of the Selenomethionyl Protein. Science. 1990;249:1398–1405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]
- 29.Steitz TA, Steitz JA. A General Two-Metal-Ion Mechanism for Catalytic RNA. Proc Natl Acad Sci U S A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keck JL, Goedken ER, Marqusee S. Activation/Attenuation Model for RNase H. a One-Metal Mechanism with Second-Metal Inhibition. J Biol Chem. 1998;273:34128–34133. doi: 10.1074/jbc.273.51.34128. [DOI] [PubMed] [Google Scholar]
- 31.Goedken ER, Marqusee S. Co-Crystal of Escherichia coli RNase HI with Mn2+ Ions Reveals Two Divalent Metals Bound in the Active Site. J Biol Chem. 2001;276:7266–7271. doi: 10.1074/jbc.M009626200. [DOI] [PubMed] [Google Scholar]
- 32.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal Structures of RNase H Bound to an RNA/DNA Hybrid: Substrate Specificity and Metal-Dependent Catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 33.Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. Structure of Human RNase H1 Complexed with an RNA/DNA Hybrid: Insight into HIV Reverse Transcription. Mol Cell. 2007;28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 34.Krishnaswamy S. Exosite-driven Substrate Specificity and Function in Coagulation. J Thromb Haemost. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 35.Wittwer AJ, Hills RL, Keith RH, Munie GE, Arner EC, Anglin CP, Malfait AM, Tortorella MD. Substrate-dependent Inhibition Kinetics of an Active Site-directed Inhibitor of ADAMTS-4 (Aggrecanase 1) Biochem. 2007;46:6393–6401. doi: 10.1021/bi7000642. [DOI] [PubMed] [Google Scholar]
- 36.Ding J, Hughes SH, Arnold E. Protein-nucleic Acid Interactions and DNA Conformation in a Complex of Human Immunodeficiency Virus Type 1 Reverse Transcriptase with a Double-stranded DNA Template-primer. Biopolymers. 1997;44:125–138. doi: 10.1002/(SICI)1097-0282(1997)44:2<125::AID-BIP2>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 37.Beilhartz GL, Wendeler M, Baichoo N, Rausch J, Le Grice S, Gotte M. HIV-1 Reverse Transcriptase Can Simultaneously Engage its DNA/RNA Substrate at Both DNA Polymerase and RNase H Active Sites: Implications for RNase H Inhibition. J Mol Biol. 2009;388:462–474. doi: 10.1016/j.jmb.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garvey EP, Johns BA, Gartland MJ, Foster SA, Miller WH, Ferris RG, Hazen RJ, Underwood MR, Boros EE, Thompson JB, Weatherhead JG, Koble CS, Allen SH, Schaller LT, Sherrill RG, Yoshinaga T, Kobayashi M, Wakasa-Morimoto C, Miki S, Nakahara K, Noshi T, Sato A, Fujiwara T. The Naphthyridinone GSK364735 is a Novel, Potent Human Immunodeficiency Virus Type 1 Integrase Inhibitor and Antiretroviral. Antimicrob Agents Chemother. 2008;52:901–908. doi: 10.1128/AAC.01218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richardson JM, Dawson A, O'Hagan N, Taylor P, Finnegan DJ, Walkinshaw MD. Mechanism of Mos1 Transposition: Insights from Structural Analysis. Embo J. 2006;25:1324–1334. doi: 10.1038/sj.emboj.7601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark AD, Jr, Jacobo-Molina A, Clark P, Hughes SH, Arnold E. Crystallization of Human Immunodeficiency Virus Type 1 Reverse Transcriptase With and Without Nucleic Acid Substrates, Inhibitors and an Antibody Fab Fragment. Methods in Enzymol. 1995;262:171–185. doi: 10.1016/0076-6879(95)62017-6. [DOI] [PubMed] [Google Scholar]
- 41.Brodsky O, Cronin CN. Economical Parallel Protein Expression Screening and Scale-up in Escherichia coli. J Struct Funct Genomics. 2006;7:101–108. doi: 10.1007/s10969-006-9013-0. [DOI] [PubMed] [Google Scholar]
- 42.Hang JQ, Rajendran S, Yang Y, Li Y, In PW, Overton H, Parkes KE, Cammack N, Martin JA, Klumpp K. Activity of the Isolated HIV RNase H Domain and Specific Inhibition by N-Hydroxyimides. Biochem Biophys Res Commun. 2004;317:321–329. doi: 10.1016/j.bbrc.2004.03.061. [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. DENZO and SCALEPACK. In: Rossmann MG, Arnold E, editors. Crystallography of Biological Macromolecules. F. Boston: Kluwer Academic Publishers; 2001. pp. 226–235. [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. In: Charles J, Carter W, Sweet RM, editors. Macromolecular Crystallography Part A. Vol. 276. New York: Academic Press Inc.; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 45.Read RJ. Pushing the Boundaries of Molecular Replacement with Maximum Likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 46.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved Experimental Procedures for Building Protein Models in Electron-density Maps and the Location of Errors in These Models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 49.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 50.Parniak MA, Min KL, Budihas SR, Le Grice SF, Beutler JA. A Fluorescence-Based High-Throughput Screening Assay for Inhibitors of Human Immunodeficiency Virus-1 Reverse Transcriptase-Associated Ribonuclease H Activity. Anal Biochem. 2003;322:33–39. doi: 10.1016/j.ab.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 51.Cogan U, Kopelman M, Mokady S, Shinitzky M. Binding Affinities of Retinol and Related Compounds to Retinol Binding Proteins. Eur J Biochem. 1976;65:71–78. doi: 10.1111/j.1432-1033.1976.tb10390.x. [DOI] [PubMed] [Google Scholar]
- 52.Kleywegt GJ, Brünger AT. Checking Your Imagination: Applications of the Free R Value. Structure. 1996;4:897–904. doi: 10.1016/s0969-2126(96)00097-4. [DOI] [PubMed] [Google Scholar]
- 53.Kleywegt GJ, Bergfors T, Senn H, LeMotte P, Gsell B, Shudo K, Jones TA. Crystal Structures of Cellular retinoic Acid Binding Proteins I and II in Complex with All-trans-retinoic Acid and a Synthetic Retinoid. Structure. 1994;2:1241–1258. doi: 10.1016/s0969-2126(94)00125-1. [DOI] [PubMed] [Google Scholar]
- 54.Luzzati V. Traitement Statistique des Erreurs dans la Determination des Structure Cristallines. Acta Cryst. 1952;5:802–810. [Google Scholar]
- 55.Engh RA, Huber R. Accurate Bond and Angle Parameters for X-ray Protein Structure Refinement. Acta Cryst. 1991;A47:392–400. [Google Scholar]