

Abstract
It is well known that 6-nitroquipazine exhibits about 150-fold higher affinity for the serotonin transporter (SERT) than quipazine and recently we showed quipazine buspirone analogues with high to moderate SERT affinity. Now we have designed and synthesized several 6-nitroquipazine buspirone derivatives. Unexpectedly, their SERT binding affinities were moderate, and much lower than that of the previously studied quipazine buspirone analogues. To explain these findings, docking studies of both groups of compounds into two different homology models of human SERT was performed using a flexible target-ligand docking approach (4D-docking). The crystal structures of leucine transporter from Aquifex aeolicus in complex with leucine and with tryptophan were used as templates for the SERT models in closed and outward-facing conformations, respectively. We found that the latter conformation represents the most reliable model for binding of buspirone analogues. Docking into that model showed that the nitrated compounds acquire a rod like shape in the binding pocket with polar groups (nitro- and imido-) at the ends of the rod. 6-Nitro substituents gave steric clashes with amino acids located at the extracellular loop 4, which may explain their lower affinity than corresponding quipazine buspirone analogues. The results from the present study may suggest chemical design strategies to improve the SERT modulators.
Keywords: serotonin transporter, serotonin reuptake inhibitors, nitroquipazine buspirone analogues, flexible docking, molecular modeling
Introduction
The serotonin transporter (SERT), together with the dopamine and noradrenaline transporters, belongs to the neurotransmitter:sodium symporter family (NSS) [1]. SERT acts as a co-transporter of sodium and chloride ions, and serotonin molecules [2–4]. The human SERT consists of 630 amino acids with 12 transmembrane domains and cytoplasmic N- and C-terminals [5–6]. According to site directed mutagenesis Tyr95 and Asp98, located in the first transmembrane helix (TMH), are crucial for serotonin binding [7–8].
SERT is the target for several antidepressant drugs including the selective serotonin reuptake inhibitors (SSRIs) [9]. SSRIs are today the most widely used agents for the treatment of depression and many other related disorders [10–11]. The SSRIs act by blocking serotonin reuptake thus increasing the concentration of serotonin at the synapse, enhancing the serotonergic neuronal transmission. A variety of SSRIs such as fluoxetine and paroxetine have been developed for the treatment of depression [14, 15]. However, because a large number of neurological and psychiatric diseases are associated with transporter defects it is a considerable interest in identification of novel and improved SERT modulators. The disadvantages of the available SSRIs include multiple side-effects, such as anxiety, impotency, and sleep disorders, as well as a delayed onset of action: it may take about 6–8 weeks for the effects of the drugs to develop [12–13]. Thus, safer and more effective therapeutic agents are needed.
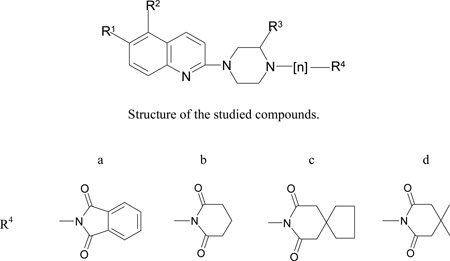
In our former papers we described synthesis and biological evaluation of a series of quipazine buspirone analogues with moderate affinity for SERT (Table 1, compounds 12 to 19) [16–20]. 6-Nitroquipazine (1, Table 1) is known as one of the most potent and selective inhibitors of SERT in vitro [21–22] and in vivo [23–24]. Its nitro group plays an important role in retaining sub-nanomolar affinity to SERT (Ki = 0.17 nM) [26], since removing the group diminishes the affinity substantially (quipazine Ki = 30 nM [25]). Since 6-nitroquipazine (1) exhibits approximately 150 times higher affinity to SERT than quipazine (2) we decided to prepare and evaluate the affinity of several buspirone-like 6-nitroquipazine derivatives with similar substituents as our previous quipazine buspirone analogues (Table 1). Several papers describe structure-affinity relationship for aromatic and piperazine ring substituted 6-nitroquipazines [27–29] but only two describing single N-4 substituted 6-nitroquipazine derivatives [30–31].
Table 1.
The molecular structure and affinities to SERT (IC50, nM) and 5-HT1A receptor affinities (Ki, nM) of the studied compounds. In the brackets the inhibition constant of [3H]5-HT at human tissue is included.
| Compound | R1 | R2 | R3 | n | R4 | SERT IC50 ± SEM, nM |
5-HT1A Ki ± SEM, nM |
|---|---|---|---|---|---|---|---|
| 1 | NO2 | H | H | - | H | ND (Ki=0.17nM) | ND |
| 2 | H | H | H | - | H | ND (Ki=30 nM) | 230 |
| 3 | NO2 | CH3 | H | 4 | b | 267.8 ± 26.3 | 711.0 ± 21.0 |
| 4 | NO2 | H | H | 4 | b | 921.1 ± 8.2 | 10600 ± 3500 |
| 5 | NO2 | CH3 | H | 4 | c | 1350 ± 211 | 776.5 ± 117.5 |
| 6 | NO2 | H | H | 4 | c | 25900 ± 1100 | 1100 ± 400 |
| 7 | NO2 | CH3 | H | 4 | d | 806.8 ± 85.2 | 221.0 ± 34.6 |
| 8 | NO2 | H | H | 4 | d | 616.3 ± 1.1 | 2100 ± 400 |
| 9 | NO2 | H | H | 4 | a | 4130 ± 900 | 59000 ± 23100 |
| 10 | H | H | Cl | 4 | d | 2800 ± 200 | 1400 ± 100 |
| 11 | C(O)NHCH3 | H | H | 4 | c | 18800 ± 700 | 3200 ± 800 |
| 12 | H | CH3 | H | 4 | a | 33.4 ± 17.0 (8)* | 13[17] |
| 13 | H | H | H | 4 | a | 95.5 ± 37.2 | 39[17] |
| 14 | H | CH3 | H | 3 | a | (80)* | 275[19] |
| 15 | H | CH3 | H | 4 | b | (50)* | 37 ± 6[18] |
| 16 | H | CH3 | H | 4 | c | 75.0 ± 16.3 | 24 ± 3[18] |
| 17 | H | H | H | 4 | c | 111.6 ±27.2 | 39 ± 7[16] |
| 18 | H | CH3 | H | 4 | d | 170.7 ±68.5 | 15 ± 3[18] |
| 19 | H | H | H | 4 | d | 146.4 ±19.4 | 11 ± 2[16] |
data obtained from Servier laboratories
Molecular modelling studies of ligand interactions with SERT were performed in order to investigate the structural differences in SERT binding between the compounds.
The leucine transporter (LeuT) is a bacterial homologue of SERT which belongs to Na+/Cl− -dependent transporter family [32]. In our previous paper [33] a SERT model was built using the X-ray crystal structure of the leucine transporter (LeuT) from Aquifex aeolicus [32] in complex with leucine as template for the homology modelling. This LeuT structure is believed to represent the substrate occluded conformation, and was interpreted as a conformational intermediate between inward and outward-facing conformations. In 2008, the structure of LeuT in complex with tryptophan in an outward-facing (open-to-out) conformation was published [34]. The recently developed homology model of SERT in the open-to-out conformation [37] was used in the present study. The compounds (Table 1) were docked to both SERT models and analyzed using molecular modelling methods. The predicted models were analyzed for their ability to explain the weak binding of the synthesized 6-nitroquipazine buspirone analogues and suggest different chemical strategies for improvement of SERT modulators.
Materials and methods
Synthesis
The synthesis of compounds 12–19 was described previously [16–19]. The synthesis of compounds 3–11 was as follows:
Compounds 3–9
Compounds 3–9 were prepared by the condensation of 6-nitroquipazine (1) or 3’-methyl-6-nitroquipazine (20) with bromobutylimide (A) (Fig. 1), whereas starting (1) and (20) were prepared as described in [26, 27], respectively.
Fig. 1.

General scheme of the preparation of 6-nitroquipazine buspirone analogues 3–9.
Fragments of the general structure A were synthesized by the condensation of imides (21–24) with 1,4-dibromobutane to give bromobutyloimides (25–28) (Fig. 2).
Fig. 2.

Synthesis of bromobutylimides (A) 17–20.
Compounds 10 and 11
The 5-Chloroquipazine derivative (10) was prepared by the condensation of 5-chloroquipazine (29) with imide 26 (Fig. 3), while 6-acetamido derivative (11) was obtained from the nitro derivative 6 by reduction with tin/hydrochloric acid followed by acetylation (Fig. 4).
Fig. 3.

Synthesis of 5-chloro derivative 10.
Fig. 4.

Synthesis of 6-amido derivative 11.
SERT binding experiments
The assay was performed according to the method of Owens et al. [35] with slight modifications. Rat cerebral cortex was homogenized in 30 volumes of ice-cold 50 mM Tris-HCl containing 150 mM NaCl and 5mM KCl, pH=7.7 at 25°C and centrifuged at 20,000×g for 20 min. The supernatant was decanted and the pellet was resuspended in 30 volumes of buffer and centrifuged again. The resulting pellet was resuspended in the same quantity of the buffer and centrifuged for the third time in the same conditions. [3H]-Citalopram (spec. act. 50 Ci/mmol, NEN Chemicals) was used for labelling 5-HT-transporter. 240 µl of the tissue suspension, 30 µl of 1 µM imipramine (displacer), 30 µl of 1nM [3H]-Citalopram and 100 µl of the analyzed compound were incubated at 22°C for 1 h. The concentrations of the analyzed compounds ranged from 10−10 to 10−4 M.
Incubations were terminated by vacuum filtration over Whatman GF/B filters and washed two times with 100 µl of ice-cold buffer. Radioactivity was measured in a WALLAC 1409 DSA – liquid scintillation counter. All assays were done in duplicates.
5-HT1A receptor binding experiments
[3H]-8-Hydroxy-2-(di-n-propylamino)-tetralin ([3H]-8-OH-DPAT, spec. act. 106 Ci/mmol, NEN Chemicals) was used for labelling 5-HT1A receptors. The membrane preparation and the assay procedure were carried out according to the published procedure [36] with slight modifications. Briefly, the cerebral cortex tissue was homogenized in 20 vol. of 50 mM Tris-HCl buffer (pH 7.7 at 25°C) using Ultra-Turrax® T 25, and was then centrifuged at 32000 g for 10 min. The supernatant fraction was discarded, and pellet was resuspended in the same volume of Tris-HCl buffer and was then centrifuged. Before the third centrifugation, the samples were incubated at 37°C for 10 min. The final pellet was resuspended in Tris-HCl buffer containing 10 µM pargyline, 4 mM CaCl2 and 0,1% ascorbic acid. One millilitre of the tissue suspension (9 mg of wet weight), 100 µl of 10 µM serotonin (for unspecific binding), 100 µl of [3H]-8-OH-DPAT and 100 µl of analyzed compound were incubated at 37°C for 15 min. The incubation was followed by a rapid vacuum filtration through Whatman GF/B glass filters, and was when washed 3 times with 5 ml of a cold buffer (50 mM Tris-HCl, pH 7.7) using a Brandel cell harvester. The final [3H]-8-OH-DPAT concentration was 1 nM, and the concentrations of the analyzed compounds ranged from 10−10 to 10−4 M.
Molecular modelling
SERT models
Two SERT models representing different conformational states were used in the present study. The templates for homology modelling of SERT were crystal structures of the leucine transporter from Aquifex aeolicus: (i) in complex with leucine (PDB-id: 2A65) [32] and (ii) in complex with tryptophan (PDB-id: 3F3A) [34]. The first SERT model, closed at both sites of the membrane, represents a substrate-bound (occluded) conformation (Fig. 5a), whereas the second, corresponds to the open-to-out conformation (Fig. 5b).
Fig. 5.

Serotonin transporter (SERT) models based on the X-ray structure of the LeuTAa from Aquifex aeolicus: (a) the substrate-occluded conformation, (b) the outward-facing conformation. The models are coloured from amino (red) to carboxy (blue) terminal. Binding pockets identified by using ICM PocketFinder: putative binding pocket is displayed in orange color, vestibular binding pocket, consisting of the two subpockets, is displayed in a blue and pink color. The large binding pocket of SERT in the outward-facing conformation is displayed in red color.
The generation of SERT homology models has been described in detail in two other papers (occluded [33], open-to-out [37]). A comprehensive alignment of NSS family members from the literature [38] was used to guide the alignment of the SERT (UniProt [38] accession number P31645) and LeuT sequences. The homology models were generated using the BuildModel macro available in ICM (Internal Coordinate Mechanics) [39]. Energy refinement of SERT models was performed using the RefineModel macro of ICM, which gradually relaxes the model using the combination of the ICM force field [41] and harmonic positional restraints with the strength adjusted to counter the initial conformational stress. The RefineModel refinement was followed by regularisation, i.e., fitting the amino acids with the ideal covalent geometry. The loops were included in both SERT models and were present during docking.
Docking
The ligands were docked both using a regular rigid receptor-flexible ligand docking approach [39] and a new ensemble docking approach (described in detail in another paper [37]). SERT conformational ensembles were prepared in two steps: (i) detection of a ligand binding pocket using ICM PocketFinder (tolerance level 3) [67], and (ii) generation of different SERT binding pocket conformations by biased probability Monte Carlo (BPMC [41]) sampling of the binding site amino acid side chains in the presence of repulsive grid density representing a generic ligand. Each ligand was docked simultaneously into all SERT conformations using the four-dimensional (4D) flexible ligand docking [42]. During the 4D docking, the receptor conformations represent the fourth dimension [42]. The 3D grids of each conformation are stored as a single data structure, and serve as a variable in the global optimization. This allows ligand docking to the multiple binding pocket conformations in a single docking simulation.
Prior to docking the ligand sampling in vacuo was performed. A set of ligands conformers generated was placed into the binding site in four principal orientations and used as starting points for Monte Carlo global energy optimization.
The binding scores of the docked compounds were predicted using the VLS scoring function implemented in ICM [43, 44]. The VLS scoring function consists of the internal force field energy of the ligand and the ligand-receptor interaction energy. The ligand-receptor interaction energy includes several weighted terms: van der Waals, a hydrophobicity term based on the solvent accessible surface buried upon binding, an electrostatic solvation term calculated using a boundary-element solution of the Poisson equation, hydrogen- bond interactions and an entropic term proportional to the number of flexible torsions in the ligand.
Detection of binding pockets, side chain sampling and docking
Three putative binding pockets were identified in the occluded SERT model: one pocket in an area corresponding to the binding pocket of the substrate leucine in the LeuT structure [32] that is believed to represent the substrate binding pocket of SERT, and two pockets in the vestibular region of the model (Fig 5a). Superimposition of the SERT homology model with the LeuT crystal structures cocrystallised with SERT inhibitors (PDB id: 3GWW, 3GWV, 3GWU, 2Q6H, 2Q72, 2QB4, 2QEI and 2QJU) showed that the ligands were located in an area of LeuT corresponding to the two vestibular binding pockets of the SERT model. Thus, based on the similarities with LeuT a binding pocket in SERT may consist of amino acids located in the two vestibular binding pockets of SERT. Further, the superimposition also revealed that parts of the cocrystallised inhibitors were in a region corresponding to the region occupied by the amino acids at the tip of EL4 of our SERT model.
The two vestibular binding pockets were separated by parts of an insert into EL4 only, and thus are located in a structurally uncertain region of the model. In order to check if the buspirone analogues could bind SERT in pocket similar to TCA/SSRI in LeuT we decided to fuse these pockets into one and then see of the present compounds could bind. In order to generate one fused vestibular binding pocket to be used as input for BPMC side chain sampling [41], two identical occluded SERT homology models were superimposed, and the tip of EL4 (amino acids 398–402) was removed from one of the models. ICM PocketFinder [67] was then used to identify a fused binding pocket in the altered model (lacking the tip of EL4) before the model was removed, while the pocket was kept. The unchanged model with the fused binding pocket (generated from the other model lacking the EL4 tip) was used as an input for the conformational ensemble generation routine. During second step of the protocol, the side chains of amino acids Leu99, Trp103, Arg104, Tyr107, Tyr175, Tyr176, Ile179, Trp182, Tyr232, Val236, Gln238, Ile251, Asp328, Phe335, Lys399, Asp400, Ser404, Leu406, Ile408, Leu481, Tyr487, Val489, Lys490, Glu494, Arg564 and Tyr568 were sampled using BPMC [41] in the presence of a repulsive grid density representing a generic ligand. Out of 45 generated binding pocket conformations, five were selected and used for 4D docking to the fused vestibular binding pocket.
Similarly, five conformations of the putative substrate binding site were generated. The following side chains were sampled using BPMC [41]: Tyr95, Asp98, Ile172, Tyr176, Phe335, Ser336, Phe341, Val343 and Ser438.
In the previous study [37] the studied compounds bound mainly to 5 conformations of the SERT model, and thus, these conformations were used for docking in the present study.
In the model of SERT in an open-to-out conformation, one larger binding pocket was detected consisting of amino acids in both the putative substrate binding pocket, and two vestibular pockets of the substrate occluded conformation (Fig 5b). The side chains of amino acids Tyr95, Asp98, Leu99, Trp103, Arg104, Tyr107, Tyr175, Tyr176, Ile179, Phe335, Ser336, Leu337, Phe341, Val343, Lys399, Asp400, Pro403, Leu405, Leu406, Phe407, Ser438, Thr439, Glu493 and Thr497 were sampled using BPMC [41] during the second step of the protocol. As previously described, this resulted in the generation of 47 SERT binding pocket conformations [37], out of which the three, with the lowest energy were used for ensemble docking (Fig S1, supplementary materials).
Preliminary docking revealed (see results) that further modification of SERT models was necessary to dock such large compounds. Thus, two complexes with the lowest score, but with oppositely oriented ligands (9 and 13), were selected for refinements using induced fit (Fig S1, supplementary materials). During the refinement, the side-chain torsion angles of the amino acids within 4.0 Ĺ of the ligands were changed in each random step using the BPMC procedure [41].
All ligands, including quipazine and 6-nitroquipazine, were docked to the two different SERT conformations obtained after induced fit refinement by using usual rigid protein-flexible ligand approach (four parallel dockings performed).
Modelling of ligands
The examined ligands (Table 1) were protonated at the nitrogen atom attached to the butyl fragment. Ligands 12, 14, 15, 16, 18 of the quipazine analogues and 3, 5, 7 of the nitroquipazine analogues additionally possessed a methyl group in an alpha position with respect to the protonated nitrogen atom. Structures of the ligands were constructed based on the X-ray structure of buspirone [16] using the ICM program [39].
Results
Binding affinities
Our studies showed that 6-nitroquipazine buspirone analogues have lower affinity than the quipazine derivatives (Table 1). Comparison of binding affinities of six pairs of methylated and non-methylated quipazine analogues (12–13, 16–17, 18–19) and nitroquipazine analogues (3–4, 5–6 and 7–8) showed that introduction of the methyl group gave relatively higher binding affinities and complex stabilization (for the four pairs: 4→3, 6→5, 13→12, and 17→16) or no change (or minor loss) of binding affinity and complex destabilization (for two of the pairs: 8→7, 19→18) for SERT (Table 1). Compound 14 with shorter n-propyl chain exhibited slightly lower affinity than the n-butyl analogue 12.
Compound 11, with an amide substituent at C6 position of the quinoline moiety, showed higher affinity for SERT than compound 6. The 5-chloroquipazine analogue 10 showed lowest affinity of all derivatives possessing imide fragment (d) (7, 8, 18, 19, Table 1).
SERT models
Three ligand binding pockets were identified in the SERT model representing the closed SERT conformation: the putative substrate binding pocket and two vestibular binding pockets that were fused into one before the docking (Fig. 5a). The putative substrate binding pocket consisted of amino acids within TMHs: 1, 3, 6 and 8, while amino acids in TMHs: 1, 3, 6, 10, 11 and EL4 constituted the fused vestibular binding pocket. Both binding regions were separated by a gate formed by Tyr176 in TMH3 and Phe335 in TMH6.
In the model representing the outward-facing SERT conformation the putative substrate binding pocket and the vestibular pocket created one larger binding pocket (Fig. 5b) that included amino acids from TMHs 1, 3, 6, 8, 10 and EL4.
Docking study
Docking of the compounds into the putative substrate binding site of the occluded SERT model indicated that the ligands were located in the pore formed between TMHs 1, 3, 6, 8 (Fig 6a). However, only quipazine and nitroquipazine fitted well into the binding pocket and did not exhibit substantial unfavourable steric interactions (Fig 7a–b, Fig S2-supplementary materials). Steric and electrostatic hindrance was observed between compounds 3–11 and amino acids forming the binding pocket. The nitroquipazine derivatives 3–9 had their NO2 group located next to the charged amino acid Asp400 that may cause electrostatic repulsion. The NO2 group was also located close to Ile179 and Trp103 resulting in steric clashes with SERT. Steric interactions were also observed between the Cl substituent of the 5-chloroquipazine and Tyr175 of SERT. The ligands 3–19 were close to Tyr176 and Phe335 and steric interactions were observed. Amino acids lining the putative binding pocket for quipazine and nitroquipazine were: Tyr95, Ala96, Asp98, Gly100 in TMH 1; Ala169, Phe170, Ile172, Ala173, Tyr176, Asn177 in TMH 3; Phe335, Ser336, Leu337, Gly338, Phe341 in TMH 6; Ser438, Thr439, Ala441, Gly442, Leu443, Leu444 in TMH 8.
Fig. 6.

The docking of nitroquipazine derivatives to the a) putative binding site, b) vestibular binding site of the SERT model in the closed conformation.
Fig. 7.

The docking of a) 6-nitroquipazine and b) quipazine to SERT model in closed conformation to the putative binding site. The aminoacids Tyr176 and Phe355 form an aromatic lid that prevents from binding of long arylopiperazine analogues to SERT in closed conformation.
Docking into the vestibular binding pocket, that corresponds to tricyclic antidepressant binding site in LeuT, showed that the compounds were too large for that binding site, and instead, they bound to the extracellular facing cavity surface region consisting of transmembrane helices: 1, 6, 10, 11 and extracellular loops EL4, EL5, EL6 (Fig. 6b, Fig S2- supplementary materials). The residues involved in the ligand binding to the surface regions were: Arg104, Tyr107, Gln111 in TMH1; Ile327, Asp328, Ala331 in TMH6; Lys490, Glu493, Glu494 in TMH10, Phe556 in TMH11 and Lys399, Asp400, Ala401 in EL4, Tyr487 in EL5, Arg564, Tyr568 in EL6. The ligands were located so their imide group was in close contact to charged amino acids: Asp328, Glu493, Glu494.
The preliminary ligands docking to the outward-facing SERT model (three the lowest energy conformations, Fig S1- supplementary materials) showed that only quipazine, nitroquipazine, 9, 13, 14, 16, and 18 formed complexes with negative scores. Interestingly, quinoline moiety of 9 pointed toward EL4, whereas 13 was located with quinoline fragment directed to the bottom (intracellular side) of SERT model. Since other complexes were characterized by positive (unfavorable) scores and ligands 6 and 11 did not bind to any of the SERT conformations, a subsequent refinement step involving induced fit of complexes with 9 and 13 was performed. Re-docking into the two resulted SERT conformations showed that most of compounds followed binding mode of 9 (with the quinoline moiety next to EL4 and imide moiety at the bottom of the binding site; Fig S3- supplementary materials).
In that position the ligands were in contact with residues (Fig 8a–b): Tyr95, Ala96, Asp98, Gly100, Asn101, Trp103, Arg104, Tyr107 in TMH 1; Ile172, Tyr175, Tyr176, Asn177, Ile179 in TMH 3; Phe335, Ser336, Leu337, Gly338, Phe341 in TMH 6; Ser438 in TMH 8, Glu493, Thr497 in TMH 10 and Ala399, Asp400, Pro403, Ser404, Leu405, Leu406, Phe407 in EL4. Only compound 5 was found to prefer a binding mode opposite to the others. All ligands adopted a folded conformations in the binding site. The methyl substituent in the piperazine ring was located equatorially in almost all the methylated compounds except for compounds 5, 7, 18 where the CH3 group was in axial position. The substituent group of the buspirone analogues was placed next to the amino acids: Ala399, Pro403, Leu405, Leu406 in EL4 and Trp103, Arg104, Tyr107 in TMH1.
Fig. 8.

The docking of nitrated and non-nitrated buspirone analogues to SERT model in outward-facing conformation: a) ligand 12 b) ligand 9. The ligand were docked in similar manner with quinoline moiety located at the extracellular vestibule of SERT next to EL4 and imide moiety directed to the central cavity of SERT.
Discussion
The function of SERT is to transport 5-HT from the synaptic cleft back into the presynaptic cell. Several studies indicate that widespread co-operative conformational changes including sliding and tilting motions of the TMHs may occur during ion and serotonin transport [45, 46]. SERT may therefore involve several conformational changes during its transport cycle, both in TMHs and in loop segments, and it is not obvious which transporter conformation inhibitors prefer. Models representing different conformational states of SERT were therefore considered for docking in the present study (Fig. 5). The SERT model, constructed by homology to the LeuT crystal structures in complex with leucine, should correspond to a substrate-bound occluded conformation (Fig. 5a), while the model based on the X-ray structure of LeuT in complex with tryptophan should correspond to open-to-out conformation (Fig. 5b). The experimental data of ligand binding affinity (Table 1) together with molecular modelling studies provide the possibility of exploring the binding area of SERT.
When constructing homology models of transporter proteins in general there are pitfalls in regard to several of the main steps in the homology modeling procedure. The accuracy of homology models depends on the sequence identity and functional similarity between the template and target proteins, the amino acid sequence alignments between the template and the targets and the resolution at which the template protein crystal structure was solved [47, 48]. For membrane transporters in general there are few templates available, and the resolution of these templates is generally low. Furthermore, the homology between the target transporter and the template may also be low. Docking into homology based models of transporter proteins is therefore also challenging. In the present study the best docking results were obtained with open-to-out SERT model using the same docking protocol and open-to-out SERT model conformations as in our previous study [37]. In that study docking of a test set of actives and decoys indicated that the open-to-out model and the present docking protocol were able to score the majority of the active compounds above decoys.
Since the substitution at C6 position of the quinoline ring of compound 2 with a nitro group yields compound 1 with a much higher SERT affinity [25, 26]. However, the NO2 substituted buspirone derivatives show moderate SERT affinity, and lower than unsubstituted analogues. Further, a methyl group connected to the piperazine ring seems favourable, but not for compounds 7 and 18 (Table 1). The chloro- (10) and amide- (11) analogues posses lower affinity for SERT than did their counterparts without these groups (compounds 6, 17 and 8, 19, respectively).
The experimental binding studies of the 6-nitroquipazine buspirone analogues 3–9 showed also the low binding affinity of these compounds toward 5-HT1A receptor (Table 1). The 5-HT1A receptor activity of the quipazine analogues was reported previously [16–19]. The affinity for other CNS targets has not been tested.
Docking to the closed conformation of SERT (Fig. 6 and 7) indicated that the binding pocket lacks space to accommodate the long chain arylopiperazine derivatives, resulting in steric hindrance between the compounds and amino acids of the putative binding pocket (Fig. 7). Among the amino acids responsible for that were Tyr176 in TMH3 and Phe335 in TMH6 which together form an aromatic lid normally occluding the substrate binding site from the extracellular vestibule in SERT model in the closed conformation. The distance between Tyr176 and Phe335 is approximately 2.4 Ĺ in the closed conformation, while in the open-to-out conformation the distance is 4.4 Ĺ. Therefore, SERT in outward-facing conformation can accommodate in the binding site compounds with different size from smaller, such as quipazine, to larger, such as the buspirone analogues (Fig. 8). In addition, in the open-to-out conformation the distance between side chains of Arg104 in TMH1 and Glu493 in TMH10 is larger than in the model of SERT in the closed state, where these amino acids form a salt bridge. However, steric conflicts were observed for the buspirone analogues with nitro-, acetamide- or chloro- substituents located next to amino acids in TMH 1, 6 and EL4.
Ligand docking to the vestibular binding pocket of the closed SERT conformation indicated that the compounds were located in the region consisting of the extracellular loops: EL4, EL5, EL6. These loops are flexible, especially EL4 that is known to be important for the transport mechanism [60]. Thus, the docking of the ligands into the extracellular facing cavity of SERT indicated only weak interactions with the binding site. However, it cannot be ruled out that other compounds can bind to this pocket with substantial strength.
Our results showed that all nitrated and non-nitrated quipazine buspirone analogues were bound in the large binding pocket of the open-to-out SERT model. This pocket is formed by TMHs 1, 3, 6, 8, 10 and EL4 (Fig. 8a–b). Evidence from many laboratories confirm involvement of amino acids in TMH1 [49–52], TMH3 [49, 53–55], TMH6 [56–57] and TMH8 [58] for binding of compounds to SERT. Experimental accessibility studies of SERT indicate that also TMH10 might participate in the transition to a cytoplasmic-facing SERT conformation [59]. Mutagenesis studies involving amino acids in EL4 also indicated a role of this loop in the conformational changes associated with substrate transport [60].
Docking into the large binding pocket of the open-to-out SERT model indicated that the compounds preferred poses where the quinoline moiety was situated in the extracellular vestibule of SERT next to: Trp103, Arg104, Tyr107 in TMH1 and Ala398, Lys399, Asp400, Pro403, Ser404, Leu405, Phe407 in EL4 with the imide moiety deeper inside in the model close to: Ala96, Tyr95 in TMH1; Tyr175, Tyr176 in TMH3; Phe341 in TMH6 and Thr497 in TMH10 of SERT. Interactions between the nitrogen atom in the quinoline moiety and Arg104 in TMH1 were observed. The C-H……π interactions between the C-H group of the amino acids Tyr103 and Tyr107 in TMH1 and quinoline moiety of the ligand might be important for binding of quipazine like buspirone analogues. The ligands in SERT were located near Phe407 in EL4 that is the most conserved amino acid in all known sequences of the NSS family. The experimental data are suggesting that Phe407 may play an important structural role [60]. The involvement of aminoacids Tyr103 in TMH1 and Ile179 in TMH3 in a conformational change associated with SERT transport was also confirmed experimentally [66]. Hydrogen bonding interactions between the side chains of Tyr95 in TMH1, Tyr175 and Tyr176 in TMH3, Ser438 in TMH8 and imide group of the buspirone analogues without substituent at the quinoline moiety seemed to stabilize the SERT-compound complexes.
Introduction of the nitro substituent to the quinoline led to rod-shaped compounds with two large polar groups at both ends. The nitrated buspirone analogues had the NO2 group next to EL4. The presence of this polar group next to hydrophobic amino acids forming extracellular loop 4 can result in steric interactions between the compounds and SERT.
The amide substituted buspirone derivative 11 obtained low (positive) scoring values after docking to the SERT model in the open-to-out conformation which is in agreement with the experimental results (Table 1).
For compounds 3, 12, 14, 15, which possess a methyl group in the piperazine ring, weak C-H……π interactions between the hydrogen atoms of the methyl group and the aromatic moiety of Tyr175 in TMH3 or Phe335 (TMH 6) was observed (Fig 9a). These weak interactions may play an important role during ligand-receptor complex formation and stabilization [61–62]. However, for compounds 7 and 18, (as compared to 8 and 19, respectively) an increase of binding affinity was not observed (Table 1). It should be noted that these compounds possess two additional methyl groups in their imide ring. For the complexes of SERT with ligands 7 and 18 the methyl group was directed toward hydrophobic aminoacids: Ile179 in TMH3 and Leu99 in TMH1, and interactions between the methyl group and Phe355 in TMH6 were not observed (Fig. 9b). In addition, the methyl group was located in the axial position in ligands: 5, 7, 18, instead of the less hindered equatorial. It seems that Phe355 in TMH 6 can play important roles in ligand binding to SERT.
Fig. 9.

The interactions between methyl group at the piperazine ring of buspirone analogues with SERT model in the outward-facing conformation: (a) ligand 12 (b) ligand 18.
Conclusions
In the present paper, synthesis, binding studies and docking of a series of buspirone analogues (3–11) that target the SERT have been described. We also performed docking of a series of compounds from a previous study (12–19). The introduction of a nitro, amide or chloro substituent into the buspirone analogues resulted in reduced SERT binding affinity. The compounds were docked to two different SERT models that should represent a closed conformation and an outward-facing (open-to-out) conformation of SERT. The docking indicated that the SERT model in open-to-out conformation could accommodate all studied compounds in the binding pocket, and seems to represent the most reliable model for binding these compounds. The docking also showed that the introduction of a nitro, amide or chloro substituent into the buspirone analogues resulted in steric clashes with amino acids of SERT that may explain their reduced affinities for SERT.
Experimental protocols
General procedure for the preparation of N-(4-bromobutyl)imides 25–28.
To the solution of an imide of general structure B in dry toluene anhydrous K2CO3 (2.5 equiv), 18-crown-6 (1 mol %) and 1,4-dibromobutane (4 equiv) were added. The reaction mixture was stirred under reflux for 24 h under inert gas. Then, the reaction mixture was filtered off and the filter cake washed with dichloromethane. The combined filtrates were evaporated under reduced pressure and crude product was distilled under reduced pressure.
1-(4-Bromo-butyl)-piperidine-2,6-dione (25) from glutarimide (21) and 1,4-dibromobutane: bright yellow oil (89%). 1H NMR (CDCl3) δ (ppm) = 3.78 (t, 2H, J = 7.1 Hz), 3.41 (t, 2H, J = 6.5 Hz), 2.65 (t, 4H, J = 6.55 Hz), 1.78 – 1.88 (m, 4H), 1.61 – 1.73 (m, 2H); 13C NMR (CDCl3) δ (ppm) = 172.3, 38.37, 33.0, 32.7, 29.9, 26.6, 17.0. IR (CHCl3): v (cm−1) = 1725, 1671; Rf = 0.28 (30% AcOEt/hex); b.p. = 197°C / 3 tor. Spectroscopic data consistent with reported previously [63].
1-(4-bromobutyl)-4,4-dimethylpiperidine-2,6-dione (26) from 3,3-dimethylglutarimide (22) and 1,4-dibromobutane: bright yellow oil (83%). 1H NMR (CDCl3) δ (ppm) = 3.79 (t, 2H, J = 7.1 Hz), 3.41 (t, 2H, J = 6.5 Hz), 2.5 (s, 4H), 1.83–1.89 (m, 2H), 1.03–1.70 (m, 2H), 1.08 (s, 6H); 13C NMR (CDCl3) δ (ppm) = 171.8, 46.3, 38.35, 32.9, 30.08, 29.0, 27.6, 26.6; IR (CHCl3): v (cm−1) = 1724, 1671; Rf = 0.38 (30% AcOEt/hex); b.p. = 205°C / 3 tor. Spectroscopic data consistent with reported previously [63–64].
8-(4-bromobutyl)-8-azaspiro[4.5]decane-7,9-dione (27) from 3,3-tetramethyleneglutarimide (23) and 1,4-dibromobutane: bright yellow oil (75%). 1H NMR (CDCl3) δ (ppm) = 3.79 (t, 2H, J = 7.1Hz), 3.41 (t, 2H, J = 6.5 Hz), 2.59 (s, 4H), 1.81 – 1.85 (m, 2H), 1.65 – 1.78 (m, 6H), 1.47 – 1.53 (m, 4H); 13C NMR (CDCl3) δ (ppm) = 171.9, 44.5, 39.2, 38.14, 37.27, 32.8, 29.8, 26.4, 23.9; IR (CHCl3): v (cm−1) = 1724, 1668; Rf = 0.41 (30% AcOEt/hex), b.p.= 215°C / 3 tor. Spectroscopic data consistent with reported previously [63–64].
N-(4-bromobutyl)isoindoline-1,3-dione (28) from phthalimide (24) and dibromobutane: white crystals (83%). 1H NMR (CDCl3) δ (ppm) = 7.82 – 7.87 (m, 2H), 7.69 – 7.74 (m, 2H), 3.72 (t, 2H, J = 6.6 Hz), 3.44 (t, 2H, J = 6.3 Hz), 1.86 – 1.91 (m, 4H); 13C NMR (CDCl3) δ (ppm) = 168.2, 133.9, 131.9, 123.1, 36.8, 32.7, 29.7, 27.1; IR (CHCl3): v (cm−1) = 1618, 1604, 1507; Rf: 0.59 (30% AcOEt/hex), b.p. = 230°C / 3 Tor. Spectroscopic data consistent with reported previously [65].
General procedure of 6-nitroquipazine (1) and 3’-methyl-6-nitroquipazine (20) N-alkylation.
To the solution of 6-nitroquipazine (1) or 3’-methyl-6-nitroquipazine (20) in dry toluene were added anhydrous K2CO3 (4 equiv.), 18-crown-6 (0.01 equiv.) and 1-(4-bromobutyl)imide of general structure A (Scheme 2, 2 equiv.) were added and the mixture was stirred under refluxed for 5 days. Then the reaction mixture was filtrated and the filter cake washed with dichloromethane (DCM). The combined filtrates were evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel (0–3 % MeOH in DCM).
1-{4-[2-Methyl-4-(6-nitroquinolin-2-yl)piperazin-1-ylo]butyl}piperidine-2,6-dione (3). From 3’-methyl-6-nitroquipazine (20) and 1-(4-bromobutyl)piperidine-2,6-dione (25). Brown solid. 1H NMR (CDCl3) δ (ppm) = 8.49 (d, 1H, J = 2.54), 8.26 (dd, 1H, J1 = 2.62, J2 = 9.21), 7.92 (d, 1H, J = 9.29), 7.62 (d, 1H, J = 9.24), 7.03 (d, 1H, J = 9.37), 4.30-4.20 (m, 2H), 3.85-3.75 (m, 2H), 3.50-3.35 (m, 1H), 3.15-2.90 (m, 2H), 2.64 (t, 4H, J = 6.57), 2.45-2.25 (m, 4H), 2.00-1.90 (m, 2H), 2.60-2.40 (m, 4H), 1.13 (d, 3H, J = 6.18). 13C NMR (CDCl3) δ (ppm) = 172.3, 158.1, 141.5, 138.4, 126.8, 124.0, 123.4, 120.8, 110.6, 54.6, 52.8, 51.0, 44.7, 39.2, 32.7, 25.9, 23.3, 17.0, 15.6; Rf = 0.35 (10% MeOH/DCM).
3•HCl; C23H29N5O4•HCl m.p. = 203 °C–206 °C. Anal. calcd. for C23H29N5O4∙HCl 0.33 H2O: C, 57.32; H, 6.41; N, 14.53; Found: C, 57.34; H, 6.53; N, 14.27%.
1-{4-[4-(6-Nitro-quinolin-2-yl)-piperazin-1-yl]-butyl}-piperidine-2,6-dione (4) from 6-nitroquipazine (1) and 1-(4-bromobutyl)piperidine-2,6-dione (25). 60% dark brown semi-solid. 1H NMR (CDCl3) δ (ppm) = 8.52 (d, 1H, J = 2.6 Hz), 8.28 (dd, 1H, J1 = 2.5 Hz, J2 = 9.22 Hz), 7.95 (d, 1H, J = 9.27 Hz), 7.64 (d, 1H, J = 9.24 Hz), 7.05 (d, 1H, J = 9.29 Hz), 3.87 – 3.76 (m, 6H), 2.68 – 2.54 (m, 6H), 2.45 – 2.38 (m, 4H), 1.93 (t, 4H, J = 6.38), 1.61 – 1.53 (m, 2H). Rf: 0.45 (10% MeOH/DCM).
4•HCl: C22H27N5O4•HCl; m.p. = 180 °C–182 °C.
1H NMR (CDCl3) δ(ppm)=8.78 (d,1H,J=2.57Hz), 8.38 (d,1H, J = 9.3 Hz), 8.25 (dd, 1H, J1 = 2.63 Hz, J2 = 2.63 Hz), 7.67 (d, 1H, J = 9.25 Hz), 7.5 (d, 1H, J = 9.3 Hz), 4.78 (d, 2H, J = 5.24), 3.65–3.55 (m, 6H), 3.12–3.04 (m, 4H), 2.6 (t, 4H, J = 6.44), 1.89–1.64 (m, 4H), 1.58–1.43 (m, 2H);
Anal. calcd for C22H27N5O4•HCl•0.5 H2O: C, 56.11; H, 6.21; N, 14.87 %. Found: C, 55.94; H, 6.17; N, 14.77 %.
8-{4-[2-Methyl-4-(6-nitro-quinolin-2-yl)-piperazin-1-yl]-butyl}-8-aza-spiro[4.5]-decane-7,9-dione (5) from 3’-methyl-6-nitroquipazine (20) and 8-(4-bromobutyl)-8-azaspiro[4.5]decane-7,9-dione (27). Brown semi-solid 60%. 1H NMR(CDCl3) δ (ppm) = 8.52 (d, 1H, J = 2.56), 8.29 (dd, 1H, J1 = 2.54, J2 = 9.22), 7.95 (d, 1H, J = 9.33), 7.65 (d, 1H, J = 9.24), 7.05 (d, 1H, J = 9.35), 4.25 (d, 2H, J = 12.94), 3.79 (t, 2H, J = 6.62), 3.49-3.40 (m, 1H), 3.15-3.07 (m, 1H), 3.03-2.93 (m, 1H), 2.79-2.70 (m, 1H), 2.59 (s, 4H), 2.53-2.50 (m, 1H), 2.42-2.32 (m, 2H), 1.75-1.65 (m, 6H), 1.54-1.50 (m. 10H), 1.15 (d, 3H, J = 6.18); 13C NMR (CDCl3) δ (ppm) = 172.0, 158.1, 151.4, 141.7, 138.3, 126.8, 124.0, 123.3, 120.7, 110.6, 54.5, 52.8, 51.0, 50.2, 44.7, 39.3, 39.1, 37.3, 25.8, 24.0, 23.1, 15.6; Rf = 0.46 (10% MeOH/DCM).
5•HCl: C27H35N5O4•HCl; m.p. 192–195 °C.
Anal. calcd for C27H35N5O4•HCl•0.5 H2O: C, 60.16; H, 6.92; N, 12.99 %. Found: C, 60.15; H, 7.08; N, 12.99 %.
8-{4-[4-(6-Nitro-quinolin-2-yl)-piperazin-1-yl]-butyl}-8-aza-spiro[4.5]decane-7,9-dione (6) from 6-nitroquipazine (1) and 8-(4-bromobutyl)-8-azaspiro[4.5]decane-7,9-dione (27). Dark brown oil 67%.1H NMR (CDCl3) δ (ppm) = 8.53 (d, 1H, J = 2.5 Hz), 8.28 (dd, 1H, J1 = 2.53 Hz, J2 = 9.21 Hz), 7.95 (d, 1H, J = 9.31 Hz), 7.65 (d, 1H, J = 9.2 Hz), 7.05 (d, 1H, J = 9.4 Hz), 3.88 – 3.76 (m, 6H), 2.59 – 2.54 (m, 8H), 2.42 (t, 2H, J = 6.8), 1.75 – 1.68 (m, 4H), 1.63 – 1.47 (m, 6H); 13C NMR (CDCl3) δ (ppm) = 172.1, 158.3, 151.4, 141.6, 138.5, 126.9, 124.1, 123.4, 120.9, 110.7, 58.0, 52.9, 44.7, 44.5, 39.6, 39.2, 37.4, 29.9, 25.8, 24.1; Rf: 0.54 (10% MeOH/DCM).
6•HCl: C26H33N5O4•HCl, m.p. = 193–194°C.
1H NMR (CDCl3) δ (ppm) = 8.81 (d, 1H, J = 2.46 Hz), 8.42 (d, 1H, J = 9.32 Hz), 8.31 (dd, 1H, J1 = 2.7 Hz, J2 = 2.78 Hz), 7.77 (d, 1H, J = 9.22 Hz), 7.54 (d, 1H, J = 9.33 Hz), 4.78 (d, 2H, J = 13.8), 3.69–3.55 (m, 6H), 3.09 (s, 4H), 2.62 (s, 4H), 1.8–1.65 (m, 2H), 1.62–1.59 (m, 4H), 1.48–1.41 (m, 6H);
Anal. calcd for C26H33N5O4•HCl•0.5 H2O: C, 59.48; H, 6.72; N, 13.34 %. Found: C, 59.49; H, 6.93; N, 13.25 %.
4,4-Dimethyl-1-{4-[2-methyl-4-(6-nitro-quinolin-2-yl)-piperazin-1-yl]-butyl}-piperidine-2,6-dione (7) from 3’-methyl-6-nitroquipazine (20) and 1-(4-bromobutyl)-4,4-dimethylpiperidine-2,6-dione (26). Brown semi oil 52 %. 1H NMR(CDCl3) δ (ppm) = 8.51 (d, 1H, J = 2.59), 8.28 (dd, 1H, J1 = 2.65, J2 = 9.26), 7.94 (d, 1H, J = 9.30), 7.64 (d, 1H, J = 9.27), 7.05 (d, 1H, J = 9.34), 4.26 (d, 2H, J = 12.94), 3.80 (t, 2H, J = 6.70), 3.49-3.37 (m, 1H), 3.14-3.08 (m, 1H), 3.03-2.92 (m, 1H), 2.80-2.74 (m, 1H), 2.56-2.50 (m, 1H), 2.50 (s, 4H), 2.44-2.32 (m, 2H), 1.54-1.25 (m, 4H), 1.14 (d, 3H, J = 6.22), 1.08 (s, 6H); 13C NMR (CDCl3) δ (ppm) = 171.8, 158.2, 151.5, 141.6, 138.5, 126.9, 124.1, 123.5, 120.8, 110.7, 54.6, 52.9, 51.1, 50.3, 46.3, 44.7, 39.2, 29.0, 27.6, 25.9, 23.3, 15.6; Rf = 0.45 (10% MeOH/DCM).
7•HCl: C25 H33 N5O4•HCl; m.p. 177–181°C.
Anal. Calcd for C25H33N5O4•HCl•0.5 H2O: C, 58.53; H, 6.88; N, 13.65 %. Found: C, 58.36; H, 7.03; N, 13.66 %.
4,4-Dimethyl-1-{4-[4-(6-nitro-quinolin-2-yl)-piperazin-1-yl]-butyl}-piperidine-2,6-dione (8) from 6-nitroquipazine (1) and 1-(4-bromobutyl)-4,4-dimethylpiperidine-2,6-dione (26): 60 % dark brown oil. 1H NMR (CDCl3) δ (ppm) = 8.52 (d, 1H, J = 2.6 Hz), 8.28 (dd, 1H, J1 = 2.6 Hz, J2 = 9.25 Hz), 7.95 (d, 1H, J = 9.33 Hz), 7.65 (d, 1H, J = 9.24 Hz), 7.05 (d, 1H, J = 9.28 Hz), 3.88 – 3.76 (m, 6H), 2.57 (t, 6H, J = 4.9 Hz), 2.50 (s, 4H), 2.42 (t, 2H, J = 6.8 Hz), 1.56 (t, 4H, J = 3.55 Hz), 1.08 (s, 6H); 13C NMR (CDCl3) δ (ppm) = 171.8, 158.2, 151.3, 141.6, 138.4, 126.8, 124.0, 123.4, 120.8, 110.7, 57.9, 52.8, 46.2, 44.4, 38.9, 29.7, 28.9, 27.4, 25.7; Rf: 0.38 (10% MeOH/DCM).
8•HCl: C24H31N5O4• HCl, m.p. = 173 °C–174 °C.
1H NMR (CDCl3) δ (ppm) = 8.79 (d, 1H, J = 2.65 Hz), 8.4 (d, 1H, J = 9.36 Hz), 8.29 (dd, 1H, J1 = 2.6 Hz, J2 = 2.71 Hz), 7.78 (d, 1H, J = 9.27 Hz), 7.51 (d, 1H, J = 9.4 Hz), 4.76 (d, 2H, J = 13.77), 3.7–3.5 (m, 7H), 3.1–3.05 (m, 4H), 2.54 (s, 4H), 1.85–1.7 (m, 2H), 1.49–1.45 (m, 2H), 0.98 (s, 6H).
Anal. calcd for C24H31N5O4•HCl•0.5 H2O: C, 57.77; H, 6.67; N, 14.03 %. Found: C, 57.67; H, 6.74; N, 13.95 %.
2-{4-[4-(6-Nitro-quinolin-2-yl)-piperazin-1-yl]-butyl}-isoindole-1,3-dione (9) from 6-nitroquipazine (1)) and 2-(4-bromobutyl)-1H-isoindole-1,3(2H)-dione (28). Dark brown crystals 61 %. 1H NMR (CDCl3) δ (ppm) = 8.52 (d, 1H, J = 2.55 Hz), 8.28 (dd, 1H, J1 = 2.67 Hz, J2 = 9.26 Hz), 7.95 (d, 1H, J = 9.32 Hz), 7.87–7.82 (m, 2H), 7.76–7.69 (m, 2H), 7.64 (d, 1H, J = 9.28), 7.05 (d, 1H, J = 9.35), 3.85 (t, 4H, J = 5.02), 3.73 (t, 2H, J = 6.89), 2.57 (t, 2H, J = 4.99), 2.45 (t, 2H, J = 7.25), 1.80–1.59 (m, 4H); Rf: 0.41 (10% MeOH/DCM).
9•HCl: C25H25N5O4•HCl; m.p. = 219 °C–220 °C.
1H NMR (CDCl3) δ (ppm) = 8.77 (d, 1H, J = 2.66 Hz), 8.38 (d, 1H, J = 9.35 Hz), 8.27 (dd, 1H, J1 = 2.59 Hz, J2 = 2.72 Hz), 7.66 (d, 1H, J = 9.23 Hz), 7.49 (d, 1H, J = 9.3 Hz), 4.71 (d, 2H, J = 13.8 Hz), 3.64-3.57 (m, 6H), 3.27–3.12 (m, 4H), 1.77–1.65 (m, 4H).
Anal. calcd for C25H25N5O4•HCl•0.5 H2O: C, 59.46; H, 5.39; N, 13.87 %. Found: C, 60.26; H, 5.49; N, 13.85 %.
Preparation of compounds 10 and 11.
1-{4-[4-(5-Chloroquinolin-2-yl)piperazin-1-yl]butyl}-4,4-di-methylopiperydine-2,6-dione (10). To the solution of 5-chloroquipazine (29) (1.223 g, 4.9 mmol) in 40 mL of anhydrous toluene 2.74 g, (2 equiv.) of bromoimide (26), 2.76 g (4 equiv.) of anhydrous potassium carbonate and 0.19 g of 18-crown-6 were added. The reactions mixture was heated under reflux for 4 days. The resulted suspension was filtered off and the residue was evaporated. Combined solid residues were purified with the aid of column chromatography on silica gel (0–3% MeOH/DCM) to give brown oil of 10 (0.940, 43%). 1H NMR (CDCl3) δ (ppm) = 8.22 (d, 1H, J = 9.36 Hz), 7.56 (d, 1H, J = 7.62 Hz), 7.63 (d, 1H, J = 8.95 Hz), 7.38 (t, 1H, J = 8.41 Hz), 7.38 (dd, 1H, J1 = 7.60 Hz, J2 = 8.40 Hz), 6.98 (d, 1H, J = 9.40 Hz), 3.77 (t, 2H, J = 6.76 Hz), 3.73 (t, 4H, J = 4.88 Hz), 2.52 (t, 4H, J = 5.00 Hz), 2.46 (s, 4H), 1.55-1.52 (m, 4H), 1.04 (s, 6H). 13C NMR (CDCl3) δ (ppm) = 171.7, 157.3, 148.7, 133.9, 130.9, 129.1, 125.6, 122.1, 120.7, 110.0, 77.0, 58.1, 52.9, 46.3, 44.8, 39.2, 29.0, 27.5, 25.9, 24.2; Rf = 0.25 (5% MeOH/DCM).
10•HCl: C22H33N5O2•HCl.
Anal. calcd. for C22H33N5O2•HCl•3 H2O: C, 55.92; H, 7.04; N, 10.87%. Found: C, 55.92; H, 6.85; N, 10.96%.
8-{4-[4-(6-Aminoquinolin-2-yl)piperazin-1-yl]butyl}-8-azaspiro-[4.5]decane-7,9-dione (30). 1-{4-[4-(6-Nitrochinolin-2-ylo)-piperazyn-1-ylo]butylo}-8-azaspiro[4.5]decano-7,9-dion (6) (0.325 g, 0.7 mmol) was dissolved in concentrated hydrochloric acid (8 mL) and 0,52 g (4 equiv.) of tin (II) chloride was added. The reaction mixture was heated at 60°C for 3 h., poured out on crushed ice, alkalized to pH 12 with 12 M NaOH do pH=12 and extracted with DCM (3×50 mL). Combined organic layers were dried over anhydrous MgSO4 and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (0–6% MeOH/DCM) to give 0.18 g (58%) of brown solid which was directly used to the next reaction. 1H NMR (CDCl3) δ (ppm) = 7.71 (d, 1H, J = 9.11 Hz), 7.57 (d, 1H, J = 8.85 Hz), 7.03 (dd, 1H, J1 = 2.60 Hz, J2 = 8.79 Hz), 6.92 (d, 1H. J = 9.13 Hz), 6.82 (d, 1H, J = 2.54 Hz), 3.79 (t, 2H, J = 6.69 Hz) 3.68 (t, 4H, J = 4.66 Hz), 2.62-2.56 (m, 8H), 2.45-2.40 (m, 2H), 1.75-1.67 (m, 4H), 1.59-1.47 (m, 8H). IR (CHCl3): ν (cm−1) = 3446,3374, 29571724, 1668, 1606, 1507; Rf = 0.42 (10% MeOH/DCM).
N-(2-{4-[4-(7,9-Dioxo-8-azaspiro[4.5]dec-8-yl)butyl]piperazin-1-yl}quinolin-6-yl)-acetamide (11). To the solution of compound 30 (0.17 g, 0.4 mmol) in glacial acetic acid (5 mL), acetic anhydride (0.36 mL, 10 equiv.) was added and the mixture was stirred at room temperature for 1 hr. The reaction mixture was poured onto ice, alkalised with 12 M NaOH to pH = 12 and extracted with DCM (3–50 mL). The combined organic layers were dried over anhydrous MgSO4 i evaporated to dryness. The resulted crude product was purified by column chromatography (0–6% MeOH/DCM) to give white-gray solid residue (0.193 g, 93%). 1H NMR (CDCl3) δ (ppm) = 8.06 (d, 1H, J = 2.23 Hz), 7.84 (d, 1H, J = 9.19 Hz), 7.63 (d, 1H, J = 8.95 Hz), 7.49 (s, 1H), 7.38 (dd, 1H, J1 = 2.36 Hz, J2 = 8.85 Hz), 6.96 (d, 1H, J = 9.18 Hz), 3.79-3.70 (m, 6H), 2.59-2.54 (m, 8H), 2.42-2.40(m, 2H), 2.20 (s, 3H), 1.75-1.67 (m, 4H), 1.54-1.50 (m, 8H). 13C NMR (CDCl3) δ (ppm) = 129.4, 128.0, 127.8, 127.5, 127.3, 127.1, 125.9, 56.1, 51.2, 48.7, 46.4, 20.0. IR (CHCl3): ν (cm−1) = 3437, 2956, 1724, 1668, 1609, 1533; Rf = 0.35 (10% MeOH/DCM).
11•HCl: C22H33N5O2•HCl.
Anal. calcd for C22H33N5O2•3 HCl•2.5 H2O: C, 52.06; H, 7.02; N, 10.84%. Found: C, 52.07; H, 7.20; N, 10.67%.
Supplementary Material
Acknowledgements
This study was partly supported by a grant PNRF-103-AI-1/07 from Norway through the Norwegian Financial Mechanism and grant NIH-R01-GM071872.
Abbreviations
- LeuTAa
leucine transporter
- NSS
neurotransmitter:sodium symporter family
- SERT
serotonin transporter
- TMH
transmembrane α-helix
- ICM
4D docking
References
- 1.Kanner BI, Zomot E. Sodium-coupled neurotransmitter transporters. Chem. Rev. 2008;108:1654–1668. doi: 10.1021/cr078246a. [DOI] [PubMed] [Google Scholar]
- 2.Schlosss P, Williams DC. The serotonin transporter: a primary target for antidepressant drugs. J. Psychopharmacol. 1998;12:115–121. doi: 10.1177/026988119801200201. [DOI] [PubMed] [Google Scholar]
- 3.Masson J, Sagne C, Hamon H, El Mestikawy S. Neurotransmitter transporters in the central nervous system. Pharmacol. Rev. 1999;51:439–464. [PubMed] [Google Scholar]
- 4.Reizer J, Reizer A A, Saier HH., Jr A functional superfamily of sodium/solute symporters. Biochim. Biophys. Acta. 1994;1197:133–166. doi: 10.1016/0304-4157(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 5.Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, Gomapathy Y, Blakely RD. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc. Natl. Acad. Sci. USA. 1993;90:2542–2546. doi: 10.1073/pnas.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitayama S S, Dohi T. Cellular and molecular aspects of monoamine neurotransmitter transporters. J Pharmacol. 1996;72:195. 195–208. doi: 10.1254/jjp.72.195. [DOI] [PubMed] [Google Scholar]
- 7.Barker EL, Moore KR, Rakhshan F, Blakely RD. Transmembrane domain I contributes to the permeation pathway for serotonin and ions in the serotonin transporter. J. Neurosci. 1999;19:4705–4717. doi: 10.1523/JNEUROSCI.19-12-04705.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barker EL, Perlman MA, Adkins EM, Houlihan WJ, Pristupa ZB, Niznik HB, Blakely RD. High affinity recognition of serotonin transporter antagonists defined, by species-scanning mutagenesis. An aromatic residue in transmembrane domain I dictates, species-selective recognition of citalopram and mazindol. J. Biol. Chem. 1998;273:19459–19468. doi: 10.1074/jbc.273.31.19459. [DOI] [PubMed] [Google Scholar]
- 9.Iversen L. Neurotransmitter transporters: fruitful targets for CNS drug discovery. Mol. Psychiatry. 2000;5:357–362. doi: 10.1038/sj.mp.4000728. [DOI] [PubMed] [Google Scholar]
- Stahl SM. Mechanism of action of serotonin selective reuptake inhibitors: Serotonin receptors and pathways mediate therapeutic effects and side effects. J. Aff. Dis. 1998;5:215–235. doi: 10.1016/s0165-0327(98)00221-3. [DOI] [PubMed] [Google Scholar]
- 11.Homberg JR, Schubert D, Gaspar P. New perspectives on the neurodevelopmental effects of SSRIs. Trends in Pharmacol. Sci. 2010;31:60–65. doi: 10.1016/j.tips.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Blier P, de Montigny C. Current advances and trends in treatment of depression. Trends Pharmacol. Sci. 1994;15:220–226. doi: 10.1016/0165-6147(94)90315-8. [DOI] [PubMed] [Google Scholar]
- 13.Jones BJ, Blackburn TP. The medical benefit of 5-HT research. Pharmacol. Biochem. Behav. 2002;71:555–568. doi: 10.1016/s0091-3057(01)00745-6. [DOI] [PubMed] [Google Scholar]
- 14.Thomas DR, Nelson DR, Johnson AM. Biochemical effects of the antidepressant paroxetine, a specific 5-hydroxytryptamine uptake inhibitor. Psychopharmacology. 1987;93:193–200. doi: 10.1007/BF00179933. [DOI] [PubMed] [Google Scholar]
- 15.Wong DT, Perry KW, Bymaster FP. The discovery of fluoxetine hydrochloride (Prozac) Nature Reviews Drug Discovery. 2005;4:764–774. doi: 10.1038/nrd1821. [DOI] [PubMed] [Google Scholar]
- 16.Chilmonczyk Z, Leś A, Woźniakowska A, Cybulski J, Kozioł AE, Gdaniec M. Buspirone Analogues as Ligands of the 5-HT1A Receptor. 1. The Molecular Structure of Buspirone and its Two Analogues. J. Med. Chem. 1995;38:1701–1710. doi: 10.1021/jm00010a015. [DOI] [PubMed] [Google Scholar]
- 17.Krajewski KJ, Leś A, Cybulski J, Chilmonczyk Z, Bronowska A, Szelejewska-Woźniakowska A. Novel Buspirone-Like 5-HT1A Receptor Ligands. Pol. J. Chem. 2001;75:71–78. [Google Scholar]
- 18.Chilmonczyk Z, Cybulski M, Iskra-Jopa J, Chojnacka-Wójcik E, Tatarczyńska E, Kłodzińska A, Leś A, Bronowska A, Sylte I. Interaction of 1,2,4-substituted piperazines, new serotonin receptor ligands, with 5-HT1A and 5-HT2A receptors. Il Farmaco. 2002;57:285–301. doi: 10.1016/s0014-827x(02)01205-3. [DOI] [PubMed] [Google Scholar]
- 19.Krajewski JK. PhD thesis (polish) Warsaw; 2001. Structure-activity relationship in a series of new 1,4-disubstituted piperazines as potential anxiolytic drugs. [Google Scholar]
- 20.Jarończyk M, Chilmonczyk Z, Mazurek AP, Nowak G, Ravna AW, Kristiansen K, Sylte I. The molecular interactions of buspirone analogues with the serotonin transporter. Bioorg. Med. Chem. 2008;16:9283–9294. doi: 10.1016/j.bmc.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto K, Goromaru T. High affinity binding of [3H]6-nitroquipazine to cortical membranes in the rat: Inhibition by 5-hydroxytryptamine and 5-hydroxytryptamine uptake inhibitors. Neuropharmacology. 1991;30:113–117. doi: 10.1016/0028-3908(91)90193-f. [DOI] [PubMed] [Google Scholar]
- 22.Hashimoto K, Goromaru T. High-affinity binding of [‘H]6- nitroquipazine to 5-hydroxytryptamine transporter in human platelets. Eur. J. Pharmacol. 1990;187:295–302. doi: 10.1016/0014-2999(90)90356-b. [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto K, Goromaru T. Reduction of [3H]6-nitroquipazine-labelled 5-hydroxytryptamine uptake sites in rat brain by 3,4-methylenedioxymethamphetamine. Fundam. Clin. Pharmacol. 1990;4(6):635–641. doi: 10.1111/j.1472-8206.1990.tb00043.x. [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto K, Goromaru T. In vivo labeling of 5-hydroxytryptamine uptake sites in mouse brain with [3H]6-nitroquipazine. Pharmacol. Exper. Ther. 1990;255:146–153. [PubMed] [Google Scholar]
- 25.Cappelli A, Giuliani G, Gallelli A, Valenti S, Anzini M, Mennuni L, Makovec F, Cupello A, Vomero S. Structure-affinity relationship studies on arylpiperazine derivatives related to quipazine as serotonin transporter ligands. Molecular basis of the selectivity SERT/5HT3 receptor. Bioorg. Med. Chem. Lett. 2005;13:3455–3460. doi: 10.1016/j.bmc.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 26.Lee BS, Chu S, Lee BC, Chi DY, Choe YS, Jeong KJ, Jin C. Syntheses and binding affinities of 6-nitroquipazine analogues for serotonin transporter. Part 1. Bioorg. Med. Chem. Lett. 2000;10:1559–1562. doi: 10.1016/s0960-894x(00)00290-0. [DOI] [PubMed] [Google Scholar]
- 27.Gerdes JM, DeFina SC, Wilson PA, Taylor SE. Serotonin transporter inhibitors: Synthesis and Binding Potency of 2’-methyl- and 3’-methyl-6nitroquipazine. Bioorg. Med. Chem. Lett. 2000;10:2643–2646. doi: 10.1016/s0960-894x(00)00546-1. [DOI] [PubMed] [Google Scholar]
- 28.Lee BS, Chu S, Lee B-S, Chi DY, Song YS, Jin C. Syntheses and binding affinities of 6-nitroquipazine analogues for serotonin transporter. Part 2: 4-substituted 6-nitroquipazines. Bioorg. Med. Chem. Lett. 2002;12:811–815. doi: 10.1016/s0960-894x(02)00028-8. [DOI] [PubMed] [Google Scholar]
- 29.Moon BS, Lee BS, Chi DY. Syntheses and binding affinities of 6-nitroquipazine analogues for serotonin transporter. Part 2: 3-alkyl-4-halo-6-nitroquipazines. Bioorg. Med. Chem. 2005;13:4952–4959. doi: 10.1016/j.bmc.2005.05.031. [DOI] [PubMed] [Google Scholar]
- 30.Zhou D, Stack GP, Lo J, Failli AA, Evrard DA, Harrison BL, Hatzenbuhler NT, Tran M, Croce S, Yi S, Golembieski J, Hornby GA, Lai M, Lin Q, Schechter LE, Smith DL, Shilling AD, Huselton C, Mitchell P, Beyer CE, Andree TH. Synthesis, Potency, and in Vivo Evaluation of 2-Piperazin-1-ylquinoline Analogues as Dual Serotonin Reuptake Inhibitors and Serotonin 5-HT1A Receptor Antagonists. J. Med. Chem. 2009;52:4955–4959. doi: 10.1021/jm900374r. [DOI] [PubMed] [Google Scholar]
- 31.Perrone R, Berardi F, Colabufo NA, Lacivita E, Larizza C, Leopoldo M, Tortorella V. Design and synthesis of long-chain arylpiperazines with mixed affinity for serotonin transporter (SERT) and 5-HT1A receptor. JPP. 2005;57:1319–1327. doi: 10.1211/jpp.57.10.0011. [DOI] [PubMed] [Google Scholar]
- 32.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl- dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 33.Gabrielsen M, Ravna AW, Kristiansen K, Sylte I. Substrate binding and translocation of the serotonin transporter studied by docking and molecular dynamics simulations. J. Mol. Model. doi: 10.1007/s00894-011-1133-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satinder SK, Pisticelli CL, Yamashita A, Gouaux E. A comprehensive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1660. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J. Pharmacol. Exp. Ther. 1997;283:1305–1322. [PubMed] [Google Scholar]
- 36.Middlemiss DN, Fozard JR. 8-Hydroxy-2-(di-n-propylamino)-tetralin discriminates between subtypes of the 5-HT1 recognition site. Eur. J. Pharmacol. 1983;90:151–153. doi: 10.1016/0014-2999(83)90230-3. [DOI] [PubMed] [Google Scholar]
- 37.Gabrielsen M, Kurczab R, Ravna AW, Kufareva I, Abagyan R, Chilmonczyk Z, Bojarski AJ, Sylte I. Molecular mechanism of serotonin transporter inhibition elucidated by a new flexible docking protocol. Eur. J. Med. Chem. 2011 doi: 10.1016/j.ejmech.2011.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:115–119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abagyan R, Totrov M, Kuznetsow DN. ICM: a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comp. Chem. 1994;15:488–506. [Google Scholar]
- 40.Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/ Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 2006;70:1630–1642. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- 41.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 42.Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J. Med. Chem. 2009;52:397–406. doi: 10.1021/jm8009958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins Suppl. 1997;1:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 44.Schapira M, Totrov M, Abagyan R. Prediction of the binding energy for small molecules, peptides and proteins. J. Mol. Recog. 1999;12:177–190. doi: 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 45.Forrest LR, Rudnick G. The Rocking bundle: A mechanism for ion-coupled solute flux by symmetrical transporters. Physiology. 2009;24:377–386. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch J, Mchaourab HS. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter: sodium symporters. Nature Struct. Mol. Biol. 2010;7:822–830. doi: 10.1038/nsmb.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chothia C, Lesk AM AM. The relation between the divergence of sequence and structure in proteins. Embo J. 1986;5(4):823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forrest LR, Tang CL, Honig B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys. J. 2006;91(2):508–517. doi: 10.1529/biophysj.106.082313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henry LK, Field JR, Adkins EM, Parnas ML, Vaughan RA, Zou M, Newman AH, Blakely RD. Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J. Biol. Chem. 2006;281:2012–2023. doi: 10.1074/jbc.M505055200. [DOI] [PubMed] [Google Scholar]
- 50.Barker EL, Perlman MA, Adkins EM, Houlihan WJ, Pristupa ZB, Niznik HB, Blakely RD. High affinity recognition of serotonin transporter antagonists defined by species-scanning mutagenesis. An aromatic residue in transmembrane domain I dictates species-selective recognition of citalopram and mazindol. J. Biol. Chem. 1998;273:19459–19468. doi: 10.1074/jbc.273.31.19459. [DOI] [PubMed] [Google Scholar]
- 51.Barker EL, Moore KR, Rakhshan F, Blakely RD. Transmembrane Domain I Contributes to the Permeation Pathway for Serotonin and Ions in the Serotonin Transporter. J. Neurosci. 1999;19:4705–4717. doi: 10.1523/JNEUROSCI.19-12-04705.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henry LK, Adkins EM, Han O, Blakely RD. Serotonin and Cocaine-sensitive Inactivation of Human Serotonin Transporters by Methanethiosulfonates Targeted to Transmembrane Domain I. J. Biol. Chem. 2003;278:37052–37063. doi: 10.1074/jbc.M305514200. [DOI] [PubMed] [Google Scholar]
- 53.Lin Z, Wang W, Kopajtic T, Revay RS, Uhl GR. Dopamine transporter: transmembrane phenylalanine mutations can selectively influence dopamine uptake and cocaine analog recognition. Mol. Pharmacol. 1999;56:434–447. doi: 10.1124/mol.56.2.434. [DOI] [PubMed] [Google Scholar]
- 54.Mortensen OV, Kristensen AS, Wiborg O. Species-scanning mutagenesis of the serotonin transporter reveals residues essential in selective, high-affinity recognition of antidepressants. J. Neurochem. 2001;79:237–247. doi: 10.1046/j.1471-4159.2001.00587.x. [DOI] [PubMed] [Google Scholar]
- 55.Chen J-G, Sachpatzidis A, Rudnick G. The third transmembrane domain of the serotonin transporter contains residues associated with substrate and cocaine binding. J. Biol. Chem. 1997;272:28321–28327. doi: 10.1074/jbc.272.45.28321. [DOI] [PubMed] [Google Scholar]
- 56.Roubert C, Cox PJ, Bruss M, Hamon M, Bonisch H, GiroS B. Determination of residues in the norepinephrine transporter that are critical for tricyclic antidepressant affinity. J. Biol. Chem. 2001;276:8254–8260. doi: 10.1074/jbc.M009798200. [DOI] [PubMed] [Google Scholar]
- 57.Lin Z, Wang W, Uhl GR. Dopamine transporter tryptophan mutants highlight candidate dopamine- and cocaine-selective domains. Mol. Pharmacol. 2000;58:1581–1592. doi: 10.1124/mol.58.6.1581. [DOI] [PubMed] [Google Scholar]
- 58.Andersen J, Taboureau O, Hansen KB, Olsen L, Egebjerg J, Strømgaard K, Kristensen AS. Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J. Biol. Chem. 2009;284:10276–10284. doi: 10.1074/jbc.M806907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang YW, Rudnick G. The cytoplasmic substrate permeation pathway of serotonin transporter. J. Biol. Chem. 2006;281:36213–36220. doi: 10.1074/jbc.M605468200. [DOI] [PubMed] [Google Scholar]
- 60.Mitchell SM, Lee E, Garcia ML, Stephan MM. Structure and function of extracellular loop 4 of the serotonin transporter as revealed by cysteine-scanning mutagenesis. J. Biol. Chem. 2004;279:24089–24099. doi: 10.1074/jbc.M311173200. [DOI] [PubMed] [Google Scholar]
- 61.Plevin MJ, Bryce DL, Boisbouvier J. Direct detection of CH/π interactions in proteins. Nature Chemistry. 2010;2:466–471. doi: 10.1038/nchem.650. [DOI] [PubMed] [Google Scholar]
- 62.Nishio M, Hirota M, Umezawa Y. Evidence, Nature and Consequences. New York: Wiley-VCH; 1998. The CH/π Interaction. [Google Scholar]
- 63.New JS, Christopher WL, Yevich JP, Butler R, Schlemmer RF, Jr, VanderMaelen CP, Cipollina JA. The thieno[3,2-c]pyridine and furo[3,2-c]pyridine rings: new pharmacophores with potential antipsychotic activity. J. Med. Chem. 1989;32:1147–1156. doi: 10.1021/jm00126a002. [DOI] [PubMed] [Google Scholar]
- 64.Podona T, Guardiola-Lemaitre B, Caignard DH, Adam G, Pfeiffer B, Renard P, Guillaumet G. 3,4-Dihydro-3-amino-2H-1-benzopyran derivatives as 5-HT1A receptor ligands and potential anxiolytic agents. 1. Synthesis and structure-activity relationship studies. J. Med. Chem. 1994;37:1779–1793. doi: 10.1021/jm00038a007. [DOI] [PubMed] [Google Scholar]
- 65.Hou DR, Cheng HY, Wang EC. Efficient synthesis of oncinotine and neooncinotine. J. Org. Chem. 2004;69:6094–6099. doi: 10.1021/jo049526i. [DOI] [PubMed] [Google Scholar]
- 66.Thao Z, Zhang Y-W, Agyiri A, Rudnick G. Ligand effects on cross-linking support a conformational mechanism for serotonin transport. J. Biol. Chem. 2009;284:33807–33814. doi: 10.1074/jbc.M109.071977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.An J, Totrov M, Abagyan R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol. Cell Proteomics. 2005;4:752–761. doi: 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.