

Abstract
Development of a general Ag(I)-promoted reaction for the direct conversion of thioamides to amidines is disclosed. This reaction was employed to prepare a key series of vancomycin aglycon residue 4 substituted amidines that were used to clarify their interaction with model ligands of peptidoglycan precursors and explore their resulting impact on antimicrobial properties.
The glycopeptide antibiotics are the most important class of drugs used in the treatment of resistant bacterial infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).1 After more than 50 years of clinical use, the emergence of resistant Gram-positive pathogens including vancomycin-resistant Enterococci (VRE) and vancomycin-resistant Staphylococcus aureus (VRSA) presents a serious public health problem at a time few new antibiotics are being developed.2 This has led to renewed interest in the search for additional effective treatments for resistant pathogens that display the durability of vancomycin, including the development of new derivatives of the glycopeptide antibiotics.3,4 Discovered at Eli Lilly, vancomycin (1, Figure 1) was disclosed in 19565 and introduced into the clinic in 1958 although its structure was not established until nearly 30 years later.6 With the emergence of MRSA, it has become the drug of last resort for the treatment of such resistant bacterial infections.1
Figure 1.

Structure of vancomycin.
The glycopeptide antibiotics inhibit bacterial cell wall synthesis by binding the precursor peptidoglycan peptide terminus D-Ala-D-Ala.7,8 In the two most prominent resistant phenotypes (VanA and VanB), this precursor is remodeled to D-Ala-D-Lac, incorporating an ester in place of the amide in the natural ligand.9 Synthesis of lipid intermediate I and II, containing the D-Ala-D-Ala termini, continues but vancomycin-resistant bacteria sense the antibiotic challenge10 and initiate a late stage remodeling from D-Ala-D-Ala to D-Ala-D-Lac to avoid the antibiotic action. The binding affinity of vancomycin for the altered ligand is reduced (1000-fold), resulting in a corresponding loss in antimicrobial activity (1000-fold). Thus, efforts to redesign the vancomycin binding pocket for its use against vancomycin-resistant bacteria must target compounds that not only bind D-Ala-D-Lac, but that also maintain binding to D-Ala-D-Ala.
Following an initial success with [Ψ[CH2NH]Tpg4]vancomycin aglycon (3)11 to achieve this dual binding by the removal of the lone pair repulsion between the vancomycin residue 4 carbonyl and D-Ala-D-Lac ester oxygens,12 we reported [Ψ[C(=NH)NH]-Tpg4]vancomycin aglycon (4)13 in a search for improved dual binding affinities and antimicrobial activities (Figure 2). Amidine 4 displayed effective, balanced binding affinity for both model ligands at a level that is within 2- to 3-fold that exhibited by vancomycin aglycon for D-Ala-D-Ala. Accurately reflecting these binding properties, 4 exhibited potent antimicrobial activity (MIC = 0.31 μg/mL, VanA E. faecalis) against VRE, being equipotent to the activity that vancomycin displays against sensitive bacterial strains. Although this represents a single atom exchange in the antibiotic (O→NH) to counter a corresponding single atom exchange in the cell wall precursors of resistant bacteria (NH→O), the modified antibiotic also maintains vancomycin’s ability to bind the unaltered peptidoglycan D-Ala-D-Ala by virtue of its apparent ability to serve as either a H-bond donor (for D-Ala-D-Lac) or H-bond acceptor (for D-Ala-D-Ala). Whereas the former entails binding of the expectedly protonated amidine (pKa = 12.5), the latter requires binding of the unprotonated amidine.
Figure 2.

Vancomycin aglycon residue 4 modifications and proposed dual binding behavior of the amidine 4.
Herein, we report the synthesis of a key series of substituted amidines designed to clarify their protonation state when bound to model ligands and explore additional questions on the potential behavior of such derivatives (Figure 3). Since selective modification of vancomycin at the residue 4 site is not yet possible, a divergent14 total synthesis based on our efforts targeting the naturally occurring aglycons15–19 was designed that proceeds through an intermediate capable of late-stage diversification. The approach incorporated a residue 4 thioamide, which could be selectively modified at the final stage of the divergent synthesis. In these studies, we found that the thioamide 5 could be selectively converted to the amidine 4 in a single step using a previously unexamined AgOAc-promoted reaction with NH3 in MeOH. Importantly, this reaction was successful (50–85%) on a fully functionalized and deprotected vancomycin aglycon.13
Figure 3.

Residue 4 substituted amidines.
Because of the magnitude of the effort involved, the survey herein was conducted on the advanced synthetic intermediate 9 bearing the residue 4 thioamide, but a C-terminus hydroxymethyl group in place of the carboxylic acid. This intermediate is available in 22 versus 26 steps and its derivatives, including the amidine 10, exhibit binding and in vitro antimicrobial properties indistinguishable from the corresponding vancomycin aglycon derivatives.13b
The first of the substituted amidines that we were especially interested in targeting was the N-methylamidine 11. Unexpectedly, efforts to convert thioamide 9 to 11 using AgOAc and MeNH2–MeOH under the reaction conditions used to prepare 4 and 10 were not successful. As a result, the various parameters of this reaction were examined first using the simpler substrate 15 (Figure 4).20,21
Figure 4.
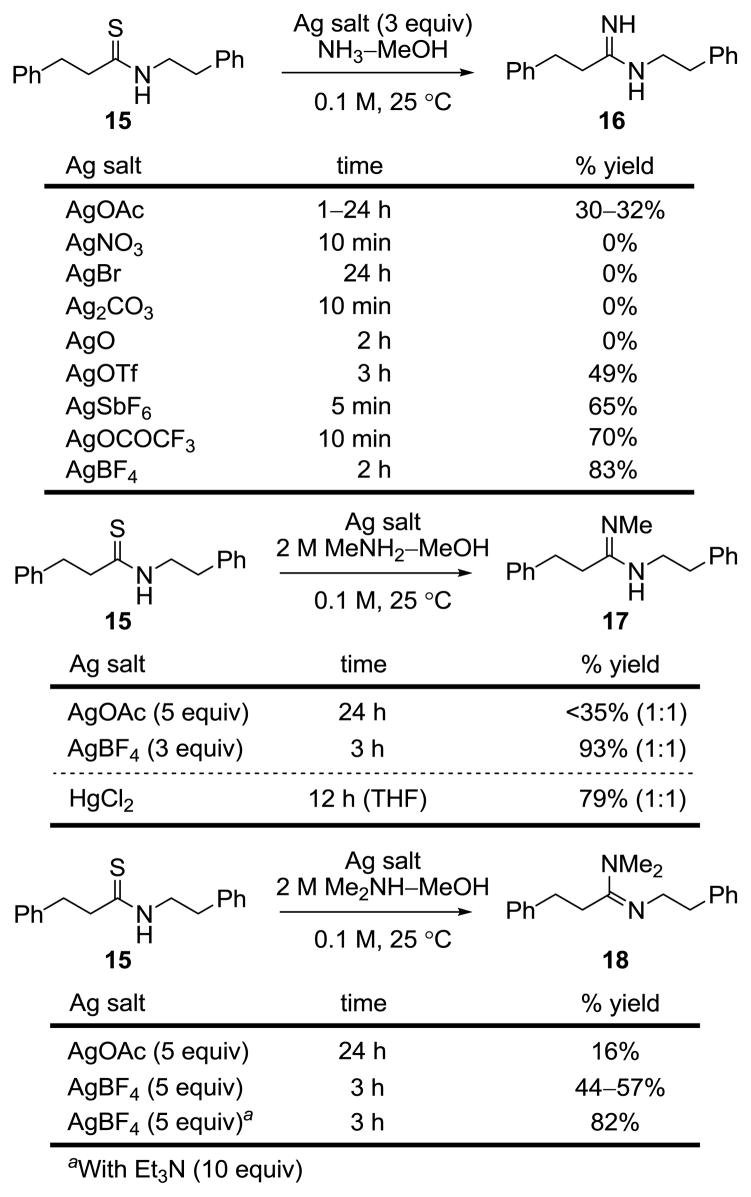
Amidine, N-methylamidine, and N,N-dimethylamidine formation.
Like the reaction with 9, attempts to convert 15 to 17 using MeNH2 (2 M in MeOH) and AgOAc (2–10 equiv) in MeOH were not especially successful. More surprisingly, we also found that AgOAc (3 equiv) in NH3–MeOH was not as effective in converting 15 to the parent amidine 16 although 15 is rapidly consumed.22 This led to an examination of a series of alternative Ag(I) salts. These studies revealed that the more reactive Ag(I) salts including AgBF4 and AgOCOCF3 were effective at promoting the conversion of 15 to the parent amidine 16 (83%), the N-methylamidine 17 (93%, 1:1 E:Z), or the N,N-dimethylamidine 18 (82%) in good yields in MeOH at room temperature (Figure 4). Moreover, these conditions were successful in converting the residue 4 thioamide in 9 to the N-methylamidine 11 as an inseparable or equilibrating 1:1 mixture of E:Z isomers (5 equiv AgBF4, 2 M MeNH2 in MeOH, 25 °C, 30 min), Figure 3.
Extension of the methodology to the preparation of the N-hydroxyamidine (amidoxime) 19 upon reaction of 15 with hydroxylamine is summarized in Figure 5. AgOAc proved modestly effective at promoting formation of 19 in MeOH, whereas the more reactive Ag(I) salts resulted in further reaction of the product amidoxime 19, leading to liberation of the N-hydroxyamidoxime and thioamide cleavage. This cleavage reaction of thioamide 15 was suppressed by running the reaction in less polar and aprotic solvents where 19 was isolated in excellent yields. Generation of 12, requiring the use of a protic solvent (MeOH), provided the easily handled residue 4 amidoxime as a single E-isomer.
Figure 5.

Amidoxime and amidrazone formation.
Similar observations were made in the preparation of the Boc protected N-aminoamidine (amidrazone) 20 upon reaction of 15 with BocNHNH2 (Figure 5). Due to the high nucleophilicity of BocNHNH2, most Ag(I)-promoted reactions led to double addition and cleavage of the thioamide. Short reaction times (5 min) with AgBF4 (5 equiv) and limiting the amount of BocNHNH2 (2 equiv, MeOH, 73%) or the use of aprotic, nonpolar solvents suppressed the overreaction and provided 20 in good yields. Such problems were less significant with 9, where the residue 4 thioamide is sterically hindered. The well behaved Boc protected precursor to the amidrazone 13 was isolated in good yield as a single isomer.
The amine anticipated to be most challenging was cyanamide, due to its lower nucleophilicity (Figure 6). Remarkably, use of AgOAc (5 equiv) in MeOH led to rapid conversion of 15 to N-cyanoamidine 21 (30 equiv H2NCN, 10 min, 85%). Extending this reaction to the preparation of the vancomycin aglycon N-cyanoamidine using AgOAc (5 equiv) provided 14 as a single isomer whose properties were consistent with the Z-configuration or equilibration to (Z)-14 under the assay conditions. The conversion of the thioamide 15 to the N-cycanoamidine 21 could also be conducted in aprotic solvents (THF > CH3CN > DMF). The further inclusion of Et3N (10 equiv) gave rise to a reaction that was complete in minutes and provided superb yields of 21 (91–94%).
Figure 6.

N-Cyanoamidine formation.
The results of the examination of the amidines 11–14 are summarized in Figure 3. N-Methylamidine 11 proved to be 30 to 50 times less effective than the parent amidine 10 at binding23 the model D-Ala-D-Ala and D-Ala-D-Lac ligands 6 and 7, respectively, but 11 bound both with near equal affinities. Accordingly, it was found to be active against VanA VRE (MIC = 20 μg/mL), albeit being 60-fold less potent than 10 precisely in line with its relative binding characteristics. Although the assessment was conducted with a sample composed of either an inseparable or equilibrating 1:1 mixture of E:Z isomers, the results still indicate that the substitution of the amidine with a small methyl group is sufficient to significantly diminish its binding and antimicrobial properties. Whereas it is difficult to infer details about the protonation state of an amidine when binding D-Ala-D-Ala, the comparison of 11 with 10 support expectations that it must be the protonated amidine that binds D-Ala-D-Lac. Unlike 10, the unprotonated state of 11 would be incapable of H-bonding to the ligand and suffers a further destabilizing lone pair/lone pair interaction, Figure 7.
Figure 7.

Dual binding of N-methylamidine 11. Effective binding to D-Ala-D-Lac must entail the protonated amidine.
The behavior of N-cyanoamidine 14, which cannot be protonated (pKa = 1), proved even more interesting. Although its affinities and activity were reduced relative to the amide 8, the relative behavior of 8 and 14 was identical and distinct from those of the amidines 10 and 11 (Figure 3). Like the amide 8, N-cyanoamidine 14 bound D-Ala-D-Ala much more effectively than D-Ala-D-Lac, which it failed to bind (≥120-fold). Accordingly, 14 lacked antimicrobial activity against VanA VRE (MIC > 40 μg/mL), but remained active against vancomycin-sensitive S. aureus (MIC = 10 μg/mL) at a level consistent with its affinity for D-Ala-D-Ala. Moreover, this affinity for D-Ala-D-Ala was found to be roughly equivalent to that of N-methylamidine 11, albeit 20-fold less than the parent amide 8 or amidine 10. The inability of the unprotonated amidine 14 to bind D-Ala-D-Lac confirms that the effective D-Ala-D-Lac binding of the parent amidine 10 and N-methylamidine 11 must entail binding of the protonated amidines, replacing the destabilizing lone pair repulsion with a stabilizing electrostatic interaction and weak reverse H-bond. Similarly, the comparable binding affinities of the unprotonated cyanoamidine 14 and the N-methylamidine 11 with D-Ala-D-Ala indicate both bind in their unprotonated state, accepting a H-bond from the linking amide in the bound ligand (Figure 8).
Figure 8.

N-Cyanoamidine 14 behavior paralleling that of amide 8. D-Ala-D-Ala (and lack of D-Ala-D-Lac) binding represents unprotonated amidine binding.
The amidoxime 12 and amidrazone 13 were important to examine for an additional reason. Both possess the potential of covalent attachment to bound D-Ala-D-Lac. Unlike the well-behaved physical properties of its N-Boc precursor, the amidrazone 13 obtained upon N-Boc deprotection (TFA, 25 °C, 12 h) proved unmanageable to work with. It was found to be insoluble in both protic (buffer, H2O and MeOH) and polar aprotic solvents (DMSO), preventing its true assessment in binding or antimicrobial assays where it proved ineffective (Figure 3). Even prolonged incubation of suspensions of 13 with D-Ala-D-Lac in the binding assay buffer (>4 months) failed to provide evidence of either reaction with the ligand (ester amidation) or ligand hydrolysis. In contrast, the amidoxime 12 was well behaved and easy to characterize. It was isolated as a single isomer, which we assigned as the E-isomer because of a potential stabilizing H-bond from the amide NH linking residues 3 and 4. Consistent with this assignment, both its binding and antimicrobial activity are reduced ≥200-fold relative to the parent amidine 10 (Figure 3). Prolonged incubation of 12 with D-Ala-D-Lac in the binding assay buffer (>6 months) also failed to provide evidence of either reaction with the ligand (transesterification)24 or ligand hydrolysis. Despite the lower activity of the amidoxime 12, it still represents a derivative class that merits future consideration as an effective in vivo antimicrobial agent. Its well behaved physical properties as an unprotonated amidine derivative (pKa = 6 vs 12.5), facilitating its absorption and permeability, as well as its likely rapid in vivo reduction to the active amidine suggest such amidoximes should continue to be examined in work going forward.25
Complementary to the studies detailed herein, the parent amidines 4 and 10 were shown to display identical dipeptide ligand binding selectivities and affinities as the corresponding amides 2 and 8, confirming that they (1) bind such ligands in the same manner, and (2) are subject to the same structural recognition features that dominate the vancomycin interaction with D-Ala-D-Ala and related ligands. This eliminated the possibility that the amidines may be interacting with the ligands in a unique manner.13 With the development of a general single step Ag(I)-promoted reaction applicable to amines with a wide range of nucleophilicities, the late-stage divergent synthesis of a key series of substituted amidines from a common residue 4 thioamide was conducted. The resulting amidine derivatives were used to define additional details of their interaction with model ligands, indicating that it requires the unprotonated amidine to bind D-Ala-D-Ala and the protonated amidine to bind D-Ala-D-Lac.
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of the National Institutes of Health (CA041101) and Skaggs Institute for Chemical Biology. A.O. is a JSPS Fellow (2010–2011), R.C.J. is a Skaggs Fellow (2006–2012), and J.G.P. was the recipient of a NIH postdoctoral fellowship (CA144333, 2009–2011).
Footnotes
Supporting Information Available: Experimental details are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Nagarajan R, editor. Glycopeptide Antibiotics. Marcel Dekker; New York: 1994. [Google Scholar]; (b) Kahne D, Leimkuhler C, Lu W, Walsh CT. Chem Rev. 2005;105:425. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 2.James RC, Pierce JG, Okano A, Xie J, Boger DL. ACS Chem Biol. 2012;7:000. doi: 10.1021/cb300007j. doi/pdf/10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Malabarba A, Nicas TI, Thompson RC. Med Res Rev. 1997;17:69. doi: 10.1002/(sici)1098-1128(199701)17:1<69::aid-med3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]; (b) Van Bambeke FV, Laethem YV, Courvalin P, Tulkens PM. Drugs. 2004;64:913. doi: 10.2165/00003495-200464090-00001. [DOI] [PubMed] [Google Scholar]
- 4.(a) Süssmuth RD. Chem Bio Chem. 2002;3:295. doi: 10.1002/1439-7633(20020402)3:4<295::AID-CBIC295>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]; (b) Walker S, Chen L, Helm J, Hu Y, Rew Y, Shin D, Boger DL. Chem Rev. 2005;105:449. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]; (c) von Nussbaum F, Brands M, Hinzen B, Weigand S, Häbich D. Angew Chem, Int Ed. 2006;45:5072. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]
- 5.McCormick MH, Stark WM, Pittenger GE, Pittenger RC, McGuire JM. Antibiot Annu. 1955–1956:606. [PubMed] [Google Scholar]
- 6.(a) Harris CM, Kopecka H, Harris TM. J Am Chem Soc. 1983;105:6915. [Google Scholar]; (b) Harris CM, Harris TM. J Am Chem Soc. 1982;104:4293. [Google Scholar]; (c) Williamson MP, Williams DH. J Am Chem Soc. 1981;103:6580. [Google Scholar]
- 7.Perkins HR. Pharmacol Ther. 1982;16:181. doi: 10.1016/0163-7258(82)90053-5. [DOI] [PubMed] [Google Scholar]
- 8.Williams DH, Williamson MP, Butcher DW, Hammond SJ. J Am Chem Soc. 1983;105:1332.X-ray: Schaefer M, Schneider TR, Sheldrick GM. Structure. 1996;4:1509. doi: 10.1016/s0969-2126(96)00156-6.
- 9.(a) Bugg TDH, Wright GD, Dutka–Malen S, Arthur M, Courvalin P, Walsh CT. Biochemistry. 1991;30:10408. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]; (b) Walsh CT. Science. 1993;261:308. doi: 10.1126/science.8392747. [DOI] [PubMed] [Google Scholar]
- 10.(a) Koteva K, Hong H-J, Wang XD, Nazi I, Hughes D, Naldrett MJ, Buttner MJ, Wright GD. Nat Chem Biol. 2010;6:327. doi: 10.1038/nchembio.350. [DOI] [PubMed] [Google Scholar]; (b) Hong H-J, Hutchings MI, Buttner MJ. Adv Exp Med Biol. 2008;631:200. doi: 10.1007/978-0-387-78885-2_14. [DOI] [PubMed] [Google Scholar]
- 11.Crowley BM, Boger DL. J Am Chem Soc. 2006;128:2885. doi: 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McComas CC, Crowley BM, Boger DL. J Am Chem Soc. 2003;125:9314. doi: 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- 13.(a) Xie J, Pierce JG, James RC, Okano A, Boger DL. J Am Chem Soc. 2011;133:13946. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL. J Am Chem Soc. 2012;134:1284. doi: 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boger DL, Brotherton CE. J Org Chem. 1984;49:4050. [Google Scholar]
- 15.Boger DL. Med Res Rev. 2001;21:356. doi: 10.1002/med.1014. [DOI] [PubMed] [Google Scholar]
- 16.(a) Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL. J Am Chem Soc. 1999;121:3226. [Google Scholar]; (b) Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. J Am Chem Soc. 1999;121:10004. [Google Scholar]
- 17.(a) Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng J-H, Mori Y, Rogel O, Castle SL, McAtee JJ. J Am Chem Soc. 2000;122:7416. [Google Scholar]; (b) Boger DL, Kim SH, Mori Y, Weng J-H, Rogel O, Castle SL, McAtee JJ. J Am Chem Soc. 2001;123:1862. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 18.Crowley BM, Mori Y, McComas CC, Tang D, Boger DL. J Am Chem Soc. 2004;126:4310. doi: 10.1021/ja039795a. [DOI] [PubMed] [Google Scholar]
- 19.(a) Garfunkle J, Kimball FS, Trzupek JD, Takazawa S, Shimamura H, Tomishima M, Boger DL. J Am Chem Soc. 2009;131:16036. doi: 10.1021/ja907193b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shimamura H, Breazzano SP, Garfunkle J, Kimball FS, Trzupek JD, Boger DL. J Am Chem Soc. 2010;132:7776. doi: 10.1021/ja102304p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Breazzano SP, Boger DL. J Am Chem Soc. 2011;133:18495. doi: 10.1021/ja208570q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Shibuya I, Taguchi Y, Tsuchiya T, Oishi A, Katoh E. Bull Chem Soc Jpn. 1994;67:3048. [Google Scholar]; (b) Avalos M, Babiano R, Duran CJ, Jimenez JL, Palacious JC. Tetrahedron Lett. 1994;35:477. [Google Scholar]; (c) Cacchi S, La Torre F, Misiti D. Chem Ind. 1978:669. [Google Scholar]; (d) Corey EJ, Boger DL. Tetrahedron Lett. 1978:5. [Google Scholar]
- 21.(a) Marchand–Brynaert J, Moya–Portuguez M, Huber I, Ghosez L. J Chem Soc, Chem Commun. 1983:818. [Google Scholar]; (b) Sauve G, Rao VS, Lajoie G, Belleau B. Can J Chem. 1985;63:3089. [Google Scholar]
- 22.In MeOH and in the absence of a reacting amine, treatment of 15 with AgOAc (3 equiv) rapidly provides the corresponding O-methylimidate (>90%). However, subsequent treatment of the in situ generated O-methylimidate with NH3 (7 M in MeOH) fails to provide 16, indicating that it is not an intermediate enroute to the amidine.
- 23.UV-difference titration assays were run as described: Nieto M, Perkins HR. Biochem J. 1971;124:845. doi: 10.1042/bj1240845.Nieto M, Perkins HR. Biochem J. 1971;123:773. doi: 10.1042/bj1230773.Nieto M, Perkins HR. Biochem J. 1971;123:789. doi: 10.1042/bj1230789.
- 24.Simanenko YS, Prokop’eva TM, Belousova IA, Popov AF, Karpichev EA. Theor Exp Chem. 2001;37:288. [Google Scholar]
- 25.Clement B. Drug Metab Rev. 2002;34:565. doi: 10.1081/dmr-120005643. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.