

Abstract
Although Grb2-Gab1 macromolecular complex mediates a multitude of cellular signaling cascades, the molecular basis of its assembly remains hitherto largely elusive. Herein, using an array of biophysical techniques, we show that while Grb2 exists in a monomer-dimer equilibrium, the proline-rich (PR) domain of Gab1 is a monomer in solution. Of particular interest is the observation that although the PR domain appears to be structurally disordered, it nonetheless adopts a more or less compact conformation reminiscent of natively-folded globular proteins. Importantly, the structurally flexible conformation of the PR domain appears to facilitate the binding of Gab1 to Grb2 with a 1:2 stoichiometry. More specifically, the formation of Grb2-Gab1 signaling complex is driven via a bivalent interaction through the binding of cSH3 domain within each monomer of Grb2 homodimer to two distinct RXXK motifs, herein designated G1 and G2, located within the PR domain of Gab1. Strikingly, in spite of the key role of bivalency in driving this macromolecular assembly, the cSH3 domains bind to G1 and G2 motifs in an independent manner with zero cooperativity. Taken together, our study sheds new light on the physicochemical forces driving the assembly of a key macromolecular signaling complex pertinent to cellular health and disease.
Keywords: Macromolecular assembly, SH3-ligand interactions, Intrinsic disorder, Multivalent binding, Zero cooperativity
INTRODUCTION
The Grb2-Gab1 interaction mediates signaling between upstream cell surface receptor tyrosine kinases (RTKs) and downstream effectors such as Ras and Akt involved in a diverse array of cellular activities including growth, survival, proliferation and oncogenic transformation [1–6]. Indeed, the disruption of grb2 and gab1 genes results in numerous developmental defects in mice and their over-expression is implicated in the genesis of human breast cancer [7–12]. Importantly, proteins involved in mediating cellular signaling usually contain a modular architecture and this evolutionary design also holds true of Grb2 and Gab1. Thus, while Grb2 is constructed on the nSH3-SH2-cSH3 signaling module where SH2 and SH3 are Src homology domains, Gab1 is comprised of the PH-PR signaling cassette where PH and PR respectively denote the pleckstrin homology and proline-rich domains.
The modular design of Grb2 and Gab1 exquisitely befits their participation in a diverse array of signaling cascades central to health and disease. In particular, Grb2 adaptor recognizes activated cell surface RTKs such as HGFR, EGFR, FGFR and PDGFR by virtue of its SH2 domain’s ability to bind to tyrosine-phosphorylated (pY) sequences in the context of pYXN motifs located within the receptor tails on the cytoplasmic face of the membrane [13, 14]. Upon binding to RTKs, the SH3 domains of Grb2 recruit a wide variety of proteins containing proline-rich sequences to the inner membrane surface — the site of initiation of a multitude of signaling cascades [15–23]. Among them, the Sos1 guanine nucleotide exchange factor and the Gab1 docking protein are by far the best characterized downstream partners of Grb2 [15–18]. Upon recruitment to the inner membrane surface, Sos1 facilitates the GDP-GTP exchange within the membrane-bound Ras GTPase and thereby switches on a key signaling circuit that involves the activation of MAPK cascade central to cellular growth and proliferation [24]. In contrast, the recruitment of Gab1 to the inner membrane surface provides docking platforms for the Shp2 tyrosine phosphatase and the PI3 kinase, which respectively account for further amplification of Ras activity, as sustained activation of Ras requires both the Sos1-dependent and Gab1-dependent pathways [1, 25, 26], and the activation of Akt serine-threonine kinase, which plays an important role in cell growth and survival [27]. Although the Grb2-Gab1 interaction is believed to occur through the binding of cSH3 domain of Grb2 to an atypical RXXK motif within the PR domain of Gab1 [18, 28], the precise mechanism underlying the assembly of this key signaling complex remains hitherto poorly understood. Of particular note is the demonstration that the cSH3 domain of Grb2 recognizes two distinct RXXK motifs, herein designated G1 and G2 (Figure 1), within the PR domain of Gab1 in a physiologically-relevant manner [29]. This salient observation, coupled with the knowledge that Grb2 exists in a dimer-monomer equilibrium in solution [30], raises the possibility that the assembly of Grb2-Gab1 signaling complex may be driven through multivalent binding leading to the formation of higher-order Grb2-Gab1 multimers rather than a simple binary complex.
Figure 1.

An overview of the various constructs of full-length Grb2 and the proline-rich (PR) domain of Gab1 used in this study. (a) Gab1 contains an N-terminal PH (Pleckstrin homology) domain and a C-terminal proline-rich (PR) domain flanked by long stretches of uncharacterized regions. The PR domain contains two distinct RXXK motifs, herein designated G1 and G2. The amino acid sequence of these motifs and flanking residues within Gab1 is provided. The numbering of various residues within and flanking the RXXK motifs is based on the nomenclature suggested by Feller and coworkers [47]. (b) Maps of the various constructs of PR domain of Gab1. In addition to the wildtype PR domain construct (PR_WT), which contains both native G1 and G2 motifs, constructs were also designed in which the G2 (PR_mG2) and G1 (PR_mG1) motifs were individually mutated, through alanine substitution of arginine and lysine residues within the corresponding RXXK consensus sequence, so as to abrogate their binding to the cSH3 domain of Grb2. Note that the numerals at the ends of each construct indicate amino acid sequence number within human Gab1. (c) 3D structure of Grb2 homodimer with a two-fold axis of symmetry as solved by Ducruix and coworkers [31]. One monomer of Grb2 is shown in green (MonA) and the other in blue (MonB). The various domains within the nSH3-SH2-cSH3 modular cassette of each Grb2 monomer are labeled for clarity. The sidechain moieties of F61/F182/R207, the three most likely candidates involved in buttressing intermolecular contacts at the monomer-monomer interface and subjected to alanine substitution, are colored red (MonA) and yellow (MonB).
In an attempt to further our understanding of the assembly of Grb2-Gab1 signaling complex, the present study was undertaken. Herein, using an array of biophysical techniques, we show that while Grb2 exists in a monomer-dimer equilibrium, the proline-rich (PR) domain of Gab1 is a monomer in solution. Of particular interest is the observation that although the PR domain appears to be structurally disordered, it nonetheless adopts a more or less compact conformation reminiscent of natively-folded globular proteins. Importantly, the structurally flexible conformation of the PR domain appears to facilitate the binding of Gab1 to Grb2 with a 1:2 stoichiometry. More specifically, the formation of Grb2-Gab1 signaling complex is driven via a bivalent interaction through the binding of cSH3 domain within each monomer of Grb2 homodimer to two distinct RXXK motifs, herein designated G1 and G2, located within the PR domain of Gab1. Strikingly, in spite of the key role of bivalency in driving this macromolecular assembly, the cSH3 domains bind to G1 and G2 motifs in an independent manner with zero cooperativity. Taken together, our study sheds new light on the physicochemical forces driving the assembly of a key macromolecular signaling complex pertinent to cellular health and disease.
RESULTS and DISCUSSION
The PR domain of Gab1 is a monomer in solution
In order to understand the assembly of Grb2-Gab1 complex, we first conducted ALS analysis on the wildtype full-length Grb2 (Grb2_WT) and the wildtype PR domain (PR_WT) of Gab1 so as to assess their propensities to associate into higher-order oligomers in solution (Figure 2). Our data suggest that while Grb2 exists in a monomer-dimer equilibrium in agreement with our previous study [30], the PR domain of Gab1 is a monomer in solution (Tables 1 and 2). In an attempt to gain insights into the macromolecular polydispersity and shape of the various species, we also determined the corresponding Mw/Mn ratios and hydrodynamic radii from our data. Our analysis reveals that both the monomeric and dimeric forms of Grb2 as well as the PR domain display Mw/Mn ratio close to unity, implying that they are all highly monodisperse in solution. Furthermore, the hydrodynamic radii observed for the Grb2 monomer and dimer are consistent with their tightly packed αβ-folds [31].
Figure 2.

ALS analysis of full-length Grb2 and the PR domain of Gab1 (Gab1_PR). (a) Elution profiles as monitored by the differential refractive index (Δn) plotted as a function of elution volume (V) for Grb2 (top panel) and Gab1_PR (bottom panel). Note that Grb2 elutes as two distinct species corresponding to a dimer and a monomer, while Gab1_PR domain elutes as a single monomeric species. (b) Partial Zimm plots obtained from analytical SLS measurements at a specific protein concentration for Grb2 dimer and monomer (top panel) and Gab1_PR monomer (bottom panel). The solid lines through the data points represent linear fits. (c) Autocorrelation function plots obtained from analytical DLS measurements at a specific protein concentration for Grb2 dimer and monomer (top panel) and Gab1_PR monomer (bottom panel). The solid lines through the data points represent non-linear least squares fits to Eq [6].
Table 1.
Hydrodynamic parameters obtained from ALS measurements for wildtype (WT) and various mutant constructs of full-length Grb2
| Associativity | Mw/kD | Mn/kD | Mw/Mn | Dt/μm2.s−1 | Rh/Å | |
|---|---|---|---|---|---|---|
| Grb2_WT | Monomer | 31 ± 1 | 30 ± 1 | 1.02 ± 0.03 | 64 ± 1 | 38 ± 1 |
| Dimer | 63 ± 2 | 61 ± 2 | 1.02 ± 0.01 | 46 ± 1 | 53 ± 1 | |
| Grb2_F61A | Monomer | 34 ± 2 | 33 ± 1 | 1.01 ± 0.01 | 58 ± 1 | 41 ± 1 |
| Dimer | 58 ± 1 | 57 ± 1 | 1.02 ± 0.01 | 45 ± 1 | 53 ± 1 | |
| Grb2_F182A | Monomer | 31 ± 1 | 31 ± 1 | 1.00 ± 0.01 | 62 ± 1 | 39 ± 1 |
| Dimer | 54 ± 1 | 54 ± 1 | 1.00 ± 0.01 | 43 ± 1 | 56 ± 1 | |
| Grb2_R207A | Monomer | 30 ± 1 | 30 ± 1 | 1.00 ± 0.02 | 62 ± 1 | 39 ± 1 |
| Dimer | 56 ± 1 | 55 ± 1 | 1.02 ± 0.01 | 42 ± 1 | 57 ± 1 |
Note that the molar mass of recombinant full-length Grb2 calculated from amino acid sequence alone is 30 kD. Errors were calculated from at least three independent measurements. All errors are given to one standard deviation.
Table 2.
Hydrodynamic parameters obtained from ALS measurements for wildtype (WT) and various mutant constructs of the PR domain of Gab1
| Associativity | Mw/kD | Mn/kD | Mw/Mn | Dt/μm2.s−1 | Rh/Å | |
|---|---|---|---|---|---|---|
| PR_WT | Monomer | 28 ± 1 | 27 ± 2 | 1.04 ± 0.02 | 53 ± 1 | 47 ± 1 |
| PR_mG2 | Monomer | 26 ± 2 | 26 ± 3 | 1.01 ± 0.01 | 55 ± 1 | 44 ± 1 |
| PR_mG1 | Monomer | 29 ± 1 | 28 ± 1 | 1.03 ± 0.01 | 53 ± 1 | 47 ± 1 |
Note that the molar mass of recombinant PR domain of Gab1 calculated from amino acid sequence alone is 27 kD. Errors were calculated from at least three independent measurements. All errors are given to one standard deviation.
Remarkably, the hydrodynamic radius observed for the PR domain of Gab1 lies somewhere between that of monomeric and dimeric Grb2 (Table 2), implying that the PR domain adopts a compact shape in a manner akin to that adopted by globular proteins rather than an extended random coil-like conformation devoid of any structural features. This observation is particularly surprising in that the proline-rich proteins such as the PR domain of Gab1 are believed to be devoid of any intrinsic structure in solution. This is due to the rigidity of proline sidechain that is structurally-destabilizing but nevertheless allows proline-rich proteins to adopt a rigid conformation ideally suited for binding to cognate ligands at the expense of little entropic penalty, which is often the bottle-neck in protein-protein interactions pertinent to cellular signaling cascades. The fact that the PR domain of Gab1 appears to be compact and globular in solution suggests strongly that the lack of secondary structural elements such as α-helices and β-strands alone may not necessarily equate to lack of intrinsic structure. On the contrary, our data support the notion that proline-rich proteins may be able to adopt a compact shape and that such adoption may be a necessity to avoid chaos within the cellular environment, where the high concentration of proline-rich sequences and proteins in general may otherwise increase cellular entropy through intrinsic disorder and formation of structural knots.
In order to evaluate the extent to which the integrity of G1 and G2 motifs is required for the PR domain of Gab1 to adopt a compact globular-like conformation, we also conducted ALS analysis on PR_mG2 and PR_mG1 mutant constructs of the PR domain and compared various hydrodynamic parameters with those obtained for the wildtype PR_WT construct (Figure 1b). Our data show that the mutation of G1 and G2 motifs has no apparent effect on the hydrodynamic radius of the PR domain (Table 2), implying that these motifs do not play a direct role in the ability of PR domain to attain a compact fold. Next, we also made an attempt to disrupt the monomer-monomer interface within Grb2 homodimer so as to generate a mutant protein that is largely monomeric in solution for subsequent analysis. The 3D structure of Grb2 homodimer, as solved by Ducruix and co-workers [31] (Figure 1c), suggests that the F61/F182/R207 trio within each monomer may play a key role in reinforcing and buttressing the monomer-monomer contacts. Thus, not only the phenyl moiety of F61 within one monomer stacks against the phenyl moiety of F182 within the other monomer via van der Waals contacts at the monomer-monomer interface, the aliphatic sidechains of R207 within each monomer also experience a similar fate. In light of this tuition, we generated Grb2_F61A, Grb2_F182A and Grb2_R207A single-alanine mutant constructs and analyzed and compared their hydrodynamic properties to the wildtype Grb2_WT construct (Table 1). To our surprise, all three mutant constructs of Grb2 failed to shift the dimer-monomer equilibrium in favor of the latter. Further work is clearly warranted to unearth the molecular basis of Grb2 dimerization as this not only bears direct relevance to understanding how Grb2 binds its ligands but could also potentially lead to the development of a protein with therapeutic potential.
Intrinsic disorder reigns within the PR domain of Gab1
The PR domain of Gab1 is abundant in residues, such as proline as well as polar and charged residues, which are usually found in structurally-disordered proteins. Given its key role in the Ras/MAPK pathway, it is fitting that the PR domain of Gab1 shares such a virtue with other structurally-disordered proteins believed to play a central role in mediating cellular signaling cascades [32–36]. In an attempt to further analyze the extent of structural disorder within the PR domain of Gab1, we performed secondary structure prediction analysis using POODLE [37]. As shown in Figure 3a, our analysis reveals that the PR domain is indeed predominantly disordered and lacks any identifiable α-helical and/or β-strand features characteristic of well-folded αβ proteins. This salient observation is further corroborated by our far-UV CD analysis (Figure 3b), wherein the spectrum of the PR domain shows a minimum centered around 208nm, a signature usually characteristic of proteins containing random coil and polyproline type II (PPII) helical conformations [38, 39]. Additionally, the rather broad far-UV CD spectrum of the PR domain suggests that it is conformationally heterogeneous as would be expected of structurally-disorderd proteins devoid of a well-defined compact fold.
Figure 3.
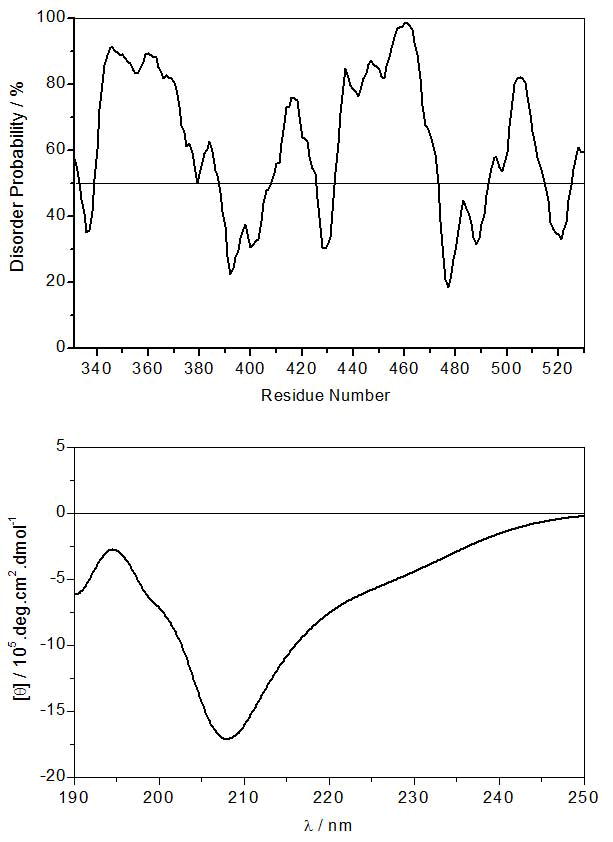
Secondary structure analysis of the PR domain of Gab1. (a) In silico prediction of intrinsic disorder within the PR domain. (b) Experimentally-determined far-UV CD spectrum of the PR domain.
Grb2 binds to the PR domain of Gab1 with a 2:1 stoichiometry
Grb2-Gab1 interaction is believed to occur through the binding of cSH3 domain of Grb2 to an atypical RXXK motif within the PR domain of Gab1 [18, 28]. Importantly, we previously demonstarted that the cSH3 domain of Grb2 recognizes two distinct RXXK motifs, herein designated G1 and G2 (Figure 1a), within the PR domain of Gab1 in a physiologically-relevant manner [29]. To understand stoichiometry and the underlying thermodynamic forces driving the binding of Grb2 to Gab1, we next measured the binding of full-length Grb2 to the PR domain of Gab1 using ITC (Figure 4). Our analysis reveals that Grb2 binds to the wildtype PR domain (PR_WT), containing both G1 and G2 sites, with a 2:1 stoichiometry (Table 3). Coupled with previous data [18, 28, 29], the most straightforward interpretation of this finding is that Grb2 binds in a bivalent manner to Gab1 to form the Grb2-Gab1 complex with a 2:1 stoichiometry and that this interaction is mediated through the binding of cSH3 domain within each Grb2 molecule to each of the two distinct RXXK motifs (G1 and G2) within Gab1. Given that the PR domain of Gab1 adopts a compact shape and that Grb2 displays the propensity to homodimerize in solution (Figure 2), the 2:1 stoichiometry observed here however may not necessarily be due to the binding of cSH3 domain within each molecule of Grb2 to both G1 and G2 sites. On the contrary, it is highly plausible that the binding of PR domain through one of its two RXXK motifs to the cSH3 domain of one monomer within Grb2 homodimer sterically blocks the binding of second monomer to another PR domain so as to prevent the formation of Grb2-Gab1 complex with a 2:2 stoichiometry.
Figure 4.

ITC analysis of the binding of full-length Grb2 to PR_WT (a), PR_mG2 (b) and PR_mG1 (c) constructs of Gab1. The upper panels show raw ITC data expressed as change in thermal power with respect to time over the period of titration. In the lower panels, change in molar heat is expressed as a function of molar ratio of the corresponding PR construct to Grb2. The solid lines in the lower panels show the fit of data to a one-site model, as embodied in Eq [11], using the ORIGIN software.
Table 3.
Thermodynamic parameters obtained from ITC measurements for the binding of wildtype and mutant constructs of the PR domain of Gab1 to full-length Grb2
| n | Kd/μM | ΔH/kcal.mol−1 | TΔS/kcal.mol−1 | ΔG/kcal.mol−1 | ΔΔGc/kcal.mol−1 | |
|---|---|---|---|---|---|---|
| PR_WT | 1.94 ± 0.05 | 7.21 ± 0.44 | −34.47 ± 0.09 | −20.42 ± 0.16 | −14.04 ± 0.07 | +0.09 ± 0.08 |
| PR_mG2 | 1.02 ± 0.04 | 20.26 ± 1.70 | −13.21 ± 0.12 | −6.80 ± 0.07 | −6.41 ± 0.05 | NA |
| PR_mG1 | 1.07 ± 0.03 | 2.24 ± 0.23 | −23.37 ± 0.24 | −15.65 ± 0.18 | −7.72 ± 0.06 | NA |
Note that n is the stoichiometry of full-length Grb2 bound to each PR construct. Errors were calculated from at least three independent measurements. All errors are given to one standard deviation.
To test the extent to which the formation of Grb2-Gab1 complex with a 2:1 stoichiometry may be influenced by steric hindrance, we also measured the binding of full-length Grb2 to mutant PR constructs, in which either G1 site (PR_mG1) or G2 site (PR_mG2) has been mutated (Table 3). Our data show that Grb2 binds to both PR_mG1 and PR_mG2 constructs with a 1:1 stoichiometry, implying that the formation of Grb2-Gab1 complex with a 2:1 stoichiometry is indeed driven through the binding of both G1 and G2 motifs within the PR domain to cSH3 domain within each molecule of Grb2. Furthermore, our data also suggest that the formation of Grb2-Gab1 complex is driven by favorable enthalpic changes accompanied by entropic penalty. This implies that specific intermolecular hydrogen bonding and ion pairing interactions predominate over hydrophobic forces in mediating the association of Grb2 with Gab1 in agreement with our previous report [29].
Bivalent binding of Grb2 to the PR domain of Gab1 is not governed by cooperativity
Our ITC data suggest that Grb2 binds to G2 motif with an affinity that is nearly an order of magnitude greater than that observed for binding to G1 motif (Table 3), implying that the PR domain of Gab1 contains a high-affinity G2 site and a low-affinity G1 site for latching onto Grb2. Given that the formation of Grb2-Gab1 complex occurs through bivalent binding, it is tempting to invoke the role of cooperativity in driving the assembly of this key signaling complex. Thus, for cooperative binding, the affinity of binding of Grb2 to PR_WT domain, containing both G1 and G2 sites, should be expected to be much higher than that observed for binding to each individual site due to entropic advantage. To our surprise, our data indicate that binding of Grb2 to the PR_WT domain occurs with an affinity that is greater than that observed for its binding to G1 site but lower than G2 site, or essentially an average of the two constituent affinity terms (Table 3). In order to understand the role of cooperativity in driving the formation of Grb2-Gab1 complex in more quantitative terms, we also calculated the contribution of cooperative interactions to the overall free energy of binding. Consistent with the foregoing argument, the free energy of cooperativity (ΔΔGc) associated with the formation of Grb2-Gab1 complex is close to zero (Table 3), implying that the cSH3 domains within two individual Grb2 molecules bind to two RXXK motifs within Gab1 in an independent manner. That this is so is further corroborated by the fact that the underlying enthalpic (ΔH) and entropic (TΔS) contributions to the overall free energy (ΔG) of binding of Grb2 to the PR_WT domain more or less equal the sum of corresponding thermodynamic terms associated with the binding of Grb2 to G1 and G2 sites in the context of mutant PR domains.
We also note that although binding of RXXK motifs within the PR domain of Gab1 to Grb2 appears to occur in an independent manner with zero cooperativity, it is nonetheless context-dependent. Thus, in conjunction with our previous study [29], it seems that RXXK motifs in the context of PR domain bind to cSH3 domains of Grb2 with affinities that are an order of magnitude greater than those observed for their binding in the context of isolated peptides. That this is so strongly argues that regions outside the canonical RXXK motifs play an integral role in mediating Grb2-Gab1 interaction in addition to site-specific interactions. It may well also be the case that the RXXK motifs depart from their physiological behavior when treated as short peptides due to the loss of local conformational constraints that they may be subject to in the context of PR domain or full-length Gab1. This notion is further supported by the observation that the PR domain adopts a compact shape and is therefore likely to impart entropic advantage by virtue of its ability to restrict the degrees of freedom available to RXXK motifs. Importantly, such a synergistic effect has also been observed upon the binding of a proline-rich sequence within the PI3 kinase to the SH3 domain of Fyn kinase [40]. In short, our data suggest that the bivalent binding of cSH3 domains of two individual Grb2 molecules to two distinct RXXK motifs within Gab1 occurs in an independent manner without any cooperative interactions.
Structural basis of the binding of Grb2 to the PR domain of Gab1 as two independent monomers versus a homodimer
Our thermodynamic data suggest that the bivalent binding of Grb2 to Gab1 with a 2:1 stoichiometry is not under cooperative control but rather driven through the binding of cSH3 domains within two individual molecules of Grb2 to two distinct RXXK motifs within the PR domain of Gab1 in an independent manner. On the other hand, our hydrodynamic data indicate that Grb2 exists in a monomer-dimer equilibrium in solution. In light of these observations, it is thus conceivable that Grb2 could bind to the PR domain of Gab1 either as two independent monomers (Grb2-PR-Grb2), or alternatively as a homodimer ([Grb2]2-PR). To gain insights into the structural basis of the binding of Grb2 to the PR domain as two independent monomers and to compare this mode of binding to that of a homodimer, we built appropriate structural models of Grb2 in complex with the PR domain of Gab1 (Figure 5). Our structural models suggest that the canonical hydrophobic grooves within the cSH3 domains of Grb2 that accommodate the RXXK ligands are fully exposed to solution in the context of both the monomers and homodimer, implying that it is indeed physically feasible for Grb2 to bind to the PR domain both as two independent monomers or as a homodimer. Importantly, the monomers within Grb2 homodimer adopt a two-fold axis of symmetry such that the SH2 domain of one monomer docks against the cSH3 domain of the other and vice versa in a head-to-tail fashion as observed in the crystal structure determined by Ducruix and co-workers [31].
Figure 5.

Structural models of Grb2 bound to G1 and G2 sites within the PR domain of Gab1 either as two isolated monomers, Grb2-PR-Grb2 (a), or in the context of a homodimer, [Grb2]2-PR (b). One monomer of Grb2 is shown in green (MonA) and the other in blue (MonB). The PR domain of Gab1 is colored brown. The interfaces between the hydrophobic grooves within the cSH3 domains of Grb2 monomers accommodating the RXXK motifs within G1 and G2 sites are marked by dashed circles. Notably, the sidechain moieties of arginine and lysine residues within the RXXK motifs at G1 and G2 sites are colored yellow. The sidechian moieties of residues within the cSH3 domains of Grb2 that interact with arginine and lysine residues within the RXXK motifs are colored red.
It should be noted that the G1 and G2 motifs within the PR domain of Gab1 bind to the cSH3 domain of Grb2 with distinct mechanisms as reported previously [29]. Thus, while the G1 motif strictly requires the PPRPPKP consensus sequence for high-affinity binding to the cSH3 domain, the G2 motif displays preference for the PXVXRXLKPXR consensus. Although both G1 and G2 motifs are accommodated within the same hydrophobic groove located within the β-barrel fold of each cSH3 domain, there are some notable differences in their modes of binding. Thus, while the G1 motif adopts the relatively open PPII-helical conformation upon binding to the cSH3 domain, the G2 motif assumes a much tighter 310-helical conformation (Figures 5a and 5b). Remarkably, despite such distinguishing conformations, the nature of residues within each cSH3 domain involved in interacting with G1 and G2 motifs bear substantial similarities. Importantly, the R344/R521 and K347/K524 basic residues within the RXXK consensus sequence at G1 and G2 sites are respectively engaged in close intermolecular hydrogen bonding and/or ion pairing contacts with E174 and E171 acidic residues located within the hydrophobic groove of each cSH3 domain. Additionally, aliphatic sidechains of R344/R521 and K347/K524 within RXXK motifs also respectively associate with benzyl ring of F167 and indole moiety of W193 within each cSH3 domain through intermolecular van der Waals contacts. Finally, numerous residues within and flanking the RXXK motifs at both G1 and G2 sites further engage in an intricate network of intermolecular contacts with their counterparts lining the hydrophobic groove within each SH3 domain in stabilizing these key SH3-ligand interactions as fully discussed earlier [29].
We note that although the spatial orientations of both RXXK motifs relative to the cSH3 domains within Grb2 monomers or homodimer can be relied upon with a high degree of confidence at atomic level, due to the fact that they were modeled on the basis of high sequence identity with corresponding templates, there is a high probability of uncertainty in the relative orientation and conformation of the intervening polypeptide chain spanning G1 and G2 sites within the PR domain of Gab1. Notably, this intervening polypeptide chain was folded into a compact globular-like topology, in agreement with our hydrodynamic data, on the basis of ab initio modeling without any template. Despite such shortcomings, our structural models lend physical insights into the bivalent binding of Grb2 adaptor to Gab1 docking protein and provide a much anticipated structural framework for further understanding the assembly of this key signaling complex.
MD simulations support the binding of Grb2 to the PR domain as a homodimer in lieu of two independent monomers
In an attempt test the validity of our structural models and to shed light on macromolecular dynamics underlying the assembly of Grb2-Gab1 signaling complex, we performed MD simulations on structural models of Grb2 bound to the PR domain of Gab1 either as two independent monomers (Grb2-PR-Grb2), or alternatively as a homodimer ([Grb2]2-PR) (Figure 6). Importantly, we assessed the stability and dynamics of various complexes and their constituent components in terms of both the root mean square deviation (RMSD) of backbone atoms as a function of simulation time and root mean square fluctuation (RMSF) of backbone atoms as a function of residue number along each protein chain. As shown in Figure 6a, the MD trajectories reveal that while the [Grb2]2-PR complex asymptotically equilibrates with an RMSD of ~7Å after about 20ns, the Grb2-PR-Grb2 complex displays structural instability with an RMSD of greater than 15Å even after 100ns of simulation time. Simply put, these observations strongly argue that the Grb2 homodimer bound to PR domain of Gab1 is structurally more stable than Grb2 monomers.
Figure 6.

MD analysis of Grb2 bound to G1 and G2 sites within the PR domain of Gab1 either as two independent monomers (Grb2-PR-Grb2) or in the context of a homodimer ([Grb2]2-PR). (a) Root mean square deviation (RMSD) of backbone atoms (N, Cα and C) within each simulated structure relative to the initial modeled structures of Grb2-PR-Grb2 (top panel) and [Grb2]2-PR (bottom panel) complexes as a function of simulation time. Note that the overall RMSD for each complex (black) is deconvoluted into PR domain (brown) and each of the two Grb2 monomers, designated MonA (green) and MonB (blue). (b) Root mean square fluctuation (RMSF) of backbone atoms (N, Cα and C) averaged over the entire course of corresponding MD trajectories of Grb2-PR-Grb2 (top panel) and [Grb2]2-PR (bottom panel) complexes as a function of residue number within each of the two Grb2 monomers, designated MonA (green) and MonB (blue). (c) Root mean square fluctuation (RMSF) of backbone atoms (N, Cα and C) averaged over the entire course of corresponding MD trajectories of Grb2-PR-Grb2 (top panel) and [Grb2]2-PR (bottom panel) complexes as a function of residue number within PR domain (brown). Note that the vertical arrows indicate the location of G1 and G2 motifs within the PR domain.
To understand the origin of such differential stabilities, we next deconvoluted the overall RMSD of each complex into its three constituent components: the PR domain of Gab1 and Grb2 monomers, designated MonA and MonB. Remarkably, our analysis reveals that the enhanced stability of [Grb2]2-PR complex arises from both the enhanced stability of the PR domain as well as monomers within the Grb2 dimer relative to their stabilities within the Grb2-PR-Grb2 complex. Thus, while Grb2 monomers in the context of a Grb2 homodimer bound to the PR domain display relatively high stability with an RMSD of ~1.5Å over the entire course of MD simulation, they appear to be highly unstable with an RMSD of ~5Å when bound to the PR domain as two independent monomers. Likewise, the PR domain of Gab1 structurally stabilizes with an RMSD of ~8Å, it remains relatively unstable with an RMSD of ~15Å over the course of MD simulation. These observations are further corroborated through our RMSF analysis (Figure 6b), wherein the distribution of atomic fluctuations within each residue of Grb2 monomers is monitored over the entire course of MD simulation. Thus, while a majority of residues within Grb2 monomers in the context of [Grb2]2-PR complex fluctuate with an RMSF of less than 2Å, this value rises to greater than 4Å in the context of Grb2-PR-Grb2 complex. Strikingly, our RMSF analysis also reveals that the residues within the PR domain of Gab1 in the context of [Grb2]2-PR complex display lower fluctuations than those in the context of Grb2-PR-Grb2 complex (Figure 6c). These differences are particularly pronounced for residues within and flanking the G1 and G2 motifs located within the PR domain. Thus, while residues within and flanking the G1 and G2 motifs in the context of [Grb2]2-PR complex fluctuate with RMSF of less than 3Å, the corresponding residues in the context of Grb2-PR-Grb2 complex do so with an RMSF of greater than 5Å. Taken together, our MD analysis supports the notion that Grb2 most likely binds to the PR domain of Gab1 as a homodimer so as to form the [Grb2]2-PR complex in lieu of two independent monomers.
CONCLUSIONS
Multivalent binding is a hallmark of biological systems and, in particular, a common feature of proteins involved in mediating cellular signaling cascades. It is generally believed that multivalency provides a physical mechanism to drive the binding of cellular partners in a synergistic and cooperative manner. Such allosteric behavior allows proteins to fine tune their activity in response to rapidly fluctuating cellular conditions and external stimuli. Thus, for example, binding at one site not only increases the local concentration of two interacting partners but may also lower entropic penalty for binding at a second site in a cooperative manner. The resulting enhancement in binding affinity provides the basis for positive cooperativity. On the other hand, binding at one site may either induce conformational changes within a protein such that binding at a second site is compromised, or alternatively sterically hinder binding at a second site, through negative cooperativity. Finally, multivalent binding may also be driven in a manner such that binding at multiple sites occurs independent of each other in the absence of any cooperativity. Our present study shows that the bivalent binding of Grb2 adaptor to two distinct RXXK sites within Gab1 docking protein is governed by this so-called zero cooperativity.
We note that such lack of cooperativity observed in driving the assembly of Grb2-Gab1 macromolecular complex does not necessarily imply lack of allosteric communication. On the contrary, our previous work has shown that the formation of Sos1-Grb2-Gab1 ternary complex is under tight allosteric control [41]. Specifically, our previous findings revealed that the binding of one molecule of Sos1 to the nSH3 domain allosterically induces a conformational change within Grb2 such that the loading of a second molecule of Sos1 onto the cSH3 domain is blocked and, in so doing, allows Gab1 access to the cSH3 domain in an exclusively non-competitive manner to generate the Sos1-Grb2-Gab1 ternary signaling complex. Indeed, it is unlikely that Grb2-Gab1 complex exists in isolation in the context of cellular milieu and, under any given conditions, Gab1 most likely binds to Grb2 in association with other cellular components such as Sos1.
Tellingly, our reductionist approach to understanding the assembly Grb2-Gab1 complex in isolation from other interacting partners has provided novel insights that would have been difficult to infer from studies on live cells and the complexity of cellular milieu would have blurred the clear picture presented here. Indeed, despite its key role in driving a multitude of cellular signaling cascades [1–3], the mechanism underpinning the assembly of Grb2-Gab1 complex remains hitherto poorly understood. In an effort to lay the groundwork toward this goal, we have demonstrated here that the Grb2-Gab1 interaction is mediated by the binding of cSH3 domain within two individual Grb2 molecules to two distinct RXXK motifs, designated G1 and G2, within the PR domain of Gab1. Although it is physically feasible for the binding of Grb2 to Gab1 as two independent monomers (Grb2-PR-Grb2), our MD simulations support a model where the formation of this complex would be preferred via the binding of Grb2 homodimer to Gab1 to generate the [Grb2]2-PR ternary complex. The fact that Grb2 exists in a monomer-dimer equilibrium in solution, it is equally plausible that binding of Gab1 to Grb2 homodimer simply shifts this equilibrium in its favor. Nonetheless, the possibility that Grb2 monomers associate into a homodimer upon binding to Gab1 cannot be excluded. Importantly, the monomers within Grb2 homodimer adopt a two-fold axis of symmetry such that the SH2 domain of one monomer docks against the cSH3 domain of the other and vice versa in a head-to-tail fashion as observed in the crystal structure determined by Ducruix and co-workers [31]. Our previous studies have shown that Grb2 monomers most likely undergo structural rearrangement, with respect to the spatial orientation of various domains within the nSH3-SH2-cSH3 modular cassette, so as to adopt a conformation that is somewhat topologically distinct and thermodynamically more stable from that observed in the context of Grb2 homodimer [30]. Although no experimental data are available, we speculate that the integrity of both the SH2 and cSH3 domains is critical for the dimerization of Grb2.
We add that our present study fails to adequately address whether binding of Grb2 to two RXXK motifs within Gab1 occurs in an ordered or random manner. Although our data presented herein unequivocally demonstrate that Gab1 contains a high-affinity G2 site and a low-affinity G1 site for latching onto Grb2, it is unlikely that binding of these sites to Grb2 occurs in an ordered manner in light of the fact that both of these sites appear to bind to Grb2 in an independent manner with zero cooperativity. It is noteworthy that G1 and G2 motifs flank the binding site for MET and RON receptor tyrosine kinases within the PR domain of Gab1 [17, 42, 43]. Given that Gab1 can be recruited to the inner membrane surface via binding to Grb2 or through its direct interaction with the cytoplasmic tails of MET and RON receptors, we believe that Gab1-MET/RON and Gab1-Grb2 interactions are likely to be mutually exclusive. Thus, binding of Grb2 to Gab1 would most likely prevent subsequent binding of MET/RON via steric hindrance and vice versa. Accordingly, the cellular ratio of Grb2 likely determines whether coupling of Gab1 to MET/RON occurs directly or via the Grb2-Gab1 interaction with important consequences on downstream signaling pathways involved in a diverse array of cellular activities [42, 44]. In addition to competition between Grb2 adaptor and MET/RON receptors for binding to Gab1, post-translational modifications may also provide additional regulatory layers for mediating this competition and/or providing access to a plethora of other cellular partners of Gab1. Thus, for example, recent work by Eulenfeld and Schaper showed that MAPK-dependent phosphorylation of S551 located within the C-terminal of Gab1 is required for its recruitment to the inner membrane surface via its PH domain [45]. It is thus conceivable that the C-terminal, including the PR domain, somehow associates with the N-terminal PH domain in an intramolecular auto-inhibitory fashion under quiescent cellular state and such inhibition is only relieved upon the activation of MAPK cascade. Likewise, access of other cellular partners to the PR domain of Gab1 may also be blocked until after receptor stimulation. Collectively, these observations epitomize the role of interacting partners and regulatory switches in dictating the accessibility of Grb2 to G1 and G2 motifs within the PR domain of Gab1.
Together with Gab1, Gab2 and Gab3 constitute the Gab family of proteins implicated in coupling activated RTKs to downstream signaling cascades involved in a myriad of cellular activities [1–3]. Importantly, the role of Gab proteins in RTK signaling does not appear to be redundant nor are they dispensable. Could functional differences between Gab proteins be accounted for in terms of their structural organization? Interestingly, while all three members of Gab family contain highly conserved G1 and G2 motifs for the recruitment of Grb2, the binding site for MET/RON receptors is unique to Gab1 and absent from Gab2 and Gab3. Consistent with this observation, disruption of Gab1 gene in mice results in developmental defects in a manner akin to MET gene-knockout [7, 8, 46], implying that Gab1 has a non-redundant role in coupling signaling from the MET receptor. Feller and co-workers recently characterized the structural basis of the binding of cSH3 domain of Grb2 to G1 and G2 motifs derived from Gab2 [47]. Their work indicated that while G1 motif adopts PPII helical conformation upon binding to the cSH3 domain, the G2 motif attains a 310-helical conformation. Given that G1 and G2 motifs are virtually conserved in both Gab1 and Gab2, we have proposed herein a similar mechanism for the interaction of Grb2 to Gab1 in line with this previous study. Thus, although Gab1 may be non-redundant for signaling through MET receptor, we believe that it may be dispensable for signaling through Grb2. However, we note that other functional differences between Gab proteins may also arise from their ability to be targeted for post-translational modifications by upstream effectors such as the MAPK cascade.
Finally, we note that, due to difficulties associated with purification of full-length Gab1, the insights into the assembly of Grb2-Gab1 signaling complex presented here are largely drawn from the isolated PR domain. Although the possibility that the PR domain might behave in a differential manner in the context of intact Gab1, both in terms of its conformation and ligand binding, we believe that our new findings lay the groundwork for future studies despite such shortcomings. In sum, our study sheds new light on the physicochemical forces driving the assembly of a key macromolecular signaling complex pertinent to cellular health and disease.
MATERIALS and METHODS
Sample preparation
Full-length human Grb2 (residues 1–217; UniProt# P62993) and the PR domain of human Gab1 (residues 331–530; UniProt# Q13480) were cloned into pET30 bacterial expression vectors with an N-terminal His-tag using Novagen LIC technology (Novagen, Madison, WI, USA). Additionally, various mutant constructs of full-length Grb2 and the PR domain of Gab1 were generated through de novo cDNA synthesis, courtesy of GenScript Corporation (GenScript, Piscataway, NJ, USA), or alternatively using the PCR primer extension method [48] (Figure 1). All recombinant constructs were expressed in Escherichia coli BL21*(DE3) bacterial strain (Invitrogen, Carlsbad, CA, USA) and purified on a Ni-NTA affinity column using standard procedures. Briefly, bacterial cells were grown at 20°C in Terrific Broth to an optical density of greater than unity at 600nm prior to induction with 0.5mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The bacterial culture was further grown overnight at 20°C and the cells were subsequently harvested and disrupted using a BeadBeater (Biospec). After separation of cell debris at high-speed centrifugation, the cell lysate was loaded onto a Ni-NTA column and washed extensively with 20mM imidazole to remove non-specific binding of bacterial proteins to the column. The recombinant proteins were subsequently eluted with 200mM imidazole and dialyzed against an appropriate buffer to remove excess imidazole. Further treatment on a Hiload Superdex 200 size-exclusion chromatography (SEC) column coupled in-line with GE Akta FPLC system (GE Healthcare, Milwaukee, WI, USA) led to purification of various constructs of Grb2 and the PR domain of Gab1 to an apparent homogeneity as judged by SDS-PAGE analysis. Final yields were typically between 10–20mg protein of apparent homogeneity per liter of bacterial culture. Treatment with thrombin protease to remove the N-terminal His-tag destabilized the recombinant proteins and they appeared to be proteolytically unstable. For this reason, except for control experiments to ensure that the His-tag had no effect on protein conformation and ligand binding, all experiments reported herein were carried out on fusion constructs with an N-terminal His-tag. Protein concentration was determined by the fluorescence-based Quant-It assay (Invitrogen) and spectrophotometrically on the basis of extinction coefficients calculated for each recombinant construct using the online software ProtParam at ExPasy Server [49]. Results from both methods were in an excellent agreement.
Analytical light scattering
Analytical light scattering (ALS) experiments were conducted on a Wyatt miniDAWN TREOS triple-angle static light scattering detector (Wyatt Technology Corp., Santa Barbara, CA, USA) and Wyatt QELS dynamic light scattering detector coupled in-line with a Wyatt Optilab rEX differential refractive index detector and interfaced to a Hiload Superdex 200 size-exclusion chromatography column under the control of a GE Akta FPLC system within a chromatography refrigerator at 10°C. Protein samples of various constructs of full-length Grb2 and the PR domain of Gab1 were prepared in 50mM Tris, 200mM NaCl, 1mM EDTA and 5mM β-mercaptoethanol at pH 8.0 and loaded onto the column at a flow rate of 1ml/min and the data were automatically acquired using the ASTRA software. The starting concentrations injected onto the column were typically between 40–50μM of each protein construct. The angular- and concentration-dependence of static light scattering (SLS) intensity of each protein species resolved in the flow mode was measured by the Wyatt miniDAWN TREOS detector. The SLS data were analyzed according to the following built-in Zimm equation in ASTRA software [50, 51]:
| [1] |
where Rθ is the excess Raleigh ratio due to protein in the solution as a function of protein concentration c (mg/ml) and the scattering angle θ (42°, 90° and 138°), M is the observed molar mass of each protein species, A2 is the second virial coefficient, λ is the wavelength of laser light in solution (658nm), Rg is the radius of gyration of protein, and K is given by the following relationship:
| [2] |
where n is the refractive index of the solvent, dn/dc is the refractive index increment of the protein in solution and NA is the Avogadro’s number (6.02×1023mol−1). Under dilute protein concentrations (c → 0), Eq [1] reduces to:
| [3] |
Thus, a plot of [Kc/Rθ] versus sin2(θ/2) yields a straight line with slope 16π2Rg2/3Mλ2 and y-intercept 1/M. Accordingly, molar mass can be obtained in a global analysis from the y-intercept of linear fits of a range of [Kc/Rθ]−sin2(θ/2) plots as a function of protein concentration along the elution profile of each protein species using SLS measurements at three scattering angles. Weighted-average molar mass (Mw) and number-average molar mass (Mn) were calculated from the following relationships:
| [4] |
| [5] |
where ci is the protein concentration and Mi is the observed molar mass at the ith slice within an elution profile. It should however be noted that Rg could not be determined for any protein species due to the lack of angular-dependence of scattered light. The time- and concentration-dependence of dynamic light scattering (DLS) intensity fluctuation of each protein species resolved in the flow mode was measured by the Wyatt QELS detector positioned at 90° with respect to the incident laser beam. The DLS data were iteratively fit using non-linear least squares regression analysis to the following built-in equation in ASTRA software [52–54]:
| [6] |
where G(τ) is the autocorrelation function of dynamic light scattering intensity fluctuation I, τ is the delay time of autocorrelation function, Γ is the decay rate constant of autocorrelation function, α is the initial amplitude of autocorrelation function at zero delay time, and β is the baseline offset (the value of autocorrelation function at infinite delay time). Thus, fitting the above equation to a range of G(τ)-τ plots as a function of protein concentration along the elution profile of each protein species computes the weighted-average value of Γ using DLS measurements at a scattering angle of 90°. Accordingly, the translational diffusion coefficient (Dt) of each protein species was calculated from the following relationship:
| [7] |
where λ is the wavelength of laser light in solution (658nm), n is the refractive index of the solvent and θ is the scattering angle (90°). Additionally, the hydrodynamic radius (Rh) of each protein construct can be determined from the Stokes-Einstein relationship:
| [8] |
where kB is Boltzman’s constant (1.38×10−23JK−1), T is the absolute temperature and η is the solvent viscosity. We note that the Rh reported here represents the weighted-average value as defined by the following expression:
| [9] |
where ci is the protein concentration and Rh,i is the observed hydrodynamic radius at the ith slice within an elution profile. It should also be noted that, in both the SLS and DLS measurements, protein concentration (c) along the elution profile of each protein species was automatically quantified in the ASTRA software from the change in refractive index (Δn) with respect to the solvent as measured by the Wyatt Optilab rEX detector using the following relationship:
| [10] |
where dn/dc is the refractive index increment of the protein in solution.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) measurements were performed on a Microcal VP-ITC instrument (MicroCal, Inc., Northampton, MA, USA) and data were acquired and processed using fully automized features in Microcal ORIGIN software. Briefly, protein samples were prepared in 50mM Tris, 200mM NaCl, 1mM EDTA and 5mM β-mercaptoethanol at pH 8.0. The experiments were initiated by injecting 25 × 10μl aliquots of 0.5–1mM of each construct of the PR domain of Gab1 from the syringe into the calorimetric cell containing 1.8ml of 50–100μM of full-length Grb2 solution at 25°C. The change in thermal power as a function of each injection was automatically recorded using the ORIGIN software and the raw data were further processed to yield binding isotherms of heat release per injection as a function of molar ratio of each PR construct to full-length Grb2. The heats of mixing and dilution were subtracted from the heat of binding per injection by carrying out a control experiment in which the same buffer in the calorimetric cell was titrated against each PR construct in an identical manner. To extract binding affinity (Kd) and binding enthalpy (ΔH), the ITC isotherms were iteratively fit using non-linear least squares regression analysis to the following built-in equation in the ORIGIN software:
| [11] |
where q(i) is the heat release (kcal/mol) for the ith injection, V is the effective volume of protein solution in the calorimetric cell (1.46ml), P is the total concentration of full-length Grb2 in the calorimetric cell, L is the total concentration of each PR construct added from the syringe for the ith injection, and n is the stoichiometry of full-length Grb2 bound to each PR construct at equilibrium. The above equation is derived from the binding of a ligand to a macromolecule using the law of mass action assuming a one-site model [55]. The free energy change (ΔG) upon the binding of Grb2 to each PR construct was calculated from the relationship:
| [12] |
where n is the integral number of binding sites within the PR domain or the observed stoichiometry of binding of Grb2 to the PR domain, R is the universal molar gas constant (1.99 cal/K/mol), and T is the absolute temperature. The entropic contribution (TΔS) to the free energy of binding was calculated from the relationship:
| [13] |
where ΔH and ΔG are as defined above. The free energy of cooperativity (ΔΔGc), resulting from the bivalent binding of Grb2 to G1 and G2 sites within the PR domain of Gab1, was calculated from the following relationship:
| [14] |
where ΔGm2 is the free energy change associated with the binding of Grb2 to G1 site within the PR_mG2 construct (G2 site mutated), ΔGm1 is the free energy change associated with the binding of Grb2 to G2 site within the PR_mG1 construct (G1 site mutated), and ΔGwt is the free energy change associated with the binding of Grb2 to both G1/G2 sites within the PR_WT construct.
Circular dichroism
Far-UV circular dichroism (CD) measurements were conducted on a Jasco J-815 spectrometer (Jasco, Inc., Easton, MD, USA) thermostatically controlled at 25°C. Experiments were conducted on 5μM of wildtype PR domain of Gab1 in 10mM Sodium phosphate containing 5mM β-mercaptoethanol at pH 8.0. Notably, despite its undesirable absorption in the far-UV region, the inclusion of β-mercaptoethanol in the buffer was necessary to prevent inter-chain cross-linking of the PR domain of Gab1 due to rapid oxidation of thiol moieties of several cysteine residues. Data were collected using a quartz cuvette with a 2-mm pathlength in the 190–250nm wavelength range. Data were normalized against reference spectra to remove the contribution of buffer. Data were recorded with a slit bandwidth of 2nm at a scan rate of 10nm/min. Each data set represents an average of four scans acquired at 0.1nm intervals. Data were converted to molar ellipticity, [θ], as a function of wavelength (λ) of electromagnetic radiation using the equation:
| [15] |
where Δε is the observed ellipticity in mdeg, c is the protein concentration in μM and l is the cuvette pathlength in cm.
Molecular modeling
Molecular modeling (MM) was employed to construct structural models of Grb2 in complex with the PR domain of Gab1 either as two independent monomers (Grb2-PR-Grb2), or in the context of a homodimer ([Grb2]2-PR), using the MODELLER software based on homology modeling [56]. Briefly, the atomic models were built in various stages. First, the intervening polypeptide chain spanning G1 and G2 sites within the PR domain of Gab1 was folded into a compact globular-like topology using the QUARK server based on ab initio modeling. The QUARK server can be accessed online at http://zhanglab.ccmb.med.umich.edu/quark. Notably, ab initio modeling was motivated by the fact that the PR domain of Gab1 appears to be structurally compact on the basis of our hydrodynamic data presented here. Next, the [Grb2]2-PR structural model was built using the ab initio structural model of the PR domain in combination with the crystal structure of Grb2 homodimer alone (PDB# 1GRI), the crystal structure of isolated cSH3 domain of Grb2 bound to an RXXK motif homologous to G1 site (PDB# 2W0Z), and the crystal structure of isolated cSH3 domain of Grb2 bound to an RXXK motif homologous to G2 site (PDB# 2VWF) using multiple-template alignment strategy in MODELLER [56]. Additionally, hydrogen bonding restraints between consensus arginine and lysine (R344/K347) within the G1 site and E171/E174 within the cSH3 domain of one monomer of Grb2 as well as between consensus arginine and lysine (R521/K524) within the G2 site and E171/E174 within the nSH3 domain of second monomer of Grb2 were introduced as described previously [29]. The Grb2-PR-Grb2 structural model was derived from the pre-built structural model of [Grb2]2-PR. Here, the intervening polypeptide chain spanning G1 and G2 sites within the PR domain of Gab1 was excised out and the two monomers within Grb2 homodimer bound to isolated G1 and G2 sites were spatially displaced and moved apart laterally in a rigid-body fashion so as to devoid them of any inter-monomer contacts in MOLMOL [57]. The resulting conformation of two non-interacting Grb2 monomers in complex with isolated G1 and G2 sites in combination with the ab initio structural model of the PR domain were subsequently used as templates to construct the Grb2-PR-Grb2 structural model in MODELLER [56]. In each case, a total of 100 structural models were calculated and the structure with the lowest energy, as judged by the MODELLER Objective Function, was selected for further analysis. The modeled structures were rendered using RIBBONS [58]. All calculations and data processing were performed on a Linux workstation equipped with a dual-core processor.
Molecular dynamics
Molecular dynamics (MD) simulations were performed with GROMACS [59, 60] using the integrated OPLS-AA force field [61, 62]. Briefly, the modeled structures of Grb2 in complex with the PR domain of Gab1 as two independent monomers (Grb2-PR-Grb2) and in the context of a homodimer ([Grb2]2-PR) were centered within a cubic box and hydrated using the extended simple point charge (SPC/E) water model [63, 64]. The hydrated structures were energy-minimized with the steepest descent algorithm prior to equilibration under the NPT ensemble conditions, wherein the number of atoms (N), pressure (P) and temperature (T) within the system were respectively kept constant at ~105, 1 bar and 300 K. The Particle-Mesh Ewald (PME) method was employed to compute long-range electrostatic interactions with a 10Å cut-off [65] and the Linear Constraint Solver (LINCS) algorithm to restrain bond lengths [66]. All MD simulations were performed under periodic boundary conditions (PBC) using the leap-frog integrator with a time step of 2fs. For the final MD production runs, data were collected every 10ps over a time scale of 50ns. All simulations were run on a Linux workstation using parallel processors at the High Performance Computing facility within the Center for Computational Science of the University of Miami.
Acknowledgments
This work was supported by funds from the National Institutes of Health (Grant# R01-GM083897) and the USylvester Braman Family Breast Cancer Institute to AF. CBM is a recipient of a postdoctoral fellowship from the National Institutes of Health (Award# T32-CA119929).
ABBREVIATIONS
- ALS
Analytical light scattering
- CD
Circular dichroism
- DLS
Dynamic light scattering
- EGFR
Epidermal growth factor receptor
- FGFR
Fibroblast growth factor receptor
- Gab1
Grb2-associated binder 1
- Grb2
Growth factor receptor binder 2
- HGFR
Hepatocyte growth factor
- ITC
Isothermal titration calorimetry
- LIC
Ligation-independent cloning
- MAPK
Mitogen-activated protein kinase
- MD
Molecular dynamics
- MM
Molecular modeling
- PDGF
Platelet-derived growth factor
- PPII
Polyproline type II
- PR
Proline-rich
- RTK
Receptor tyrosine kinase
- SEC
Size-exclusion chromatography
- SH2
Src homology 2
- SH3
Src homology 3
- SLS
Static light scattering
- Sos1
Son of sevenless 1
References
- 1.Gu H, Neel BG. The “Gab” in signal transduction. Trends Cell Biol. 2003;13:122–130. doi: 10.1016/s0962-8924(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Rohrschneider LR. The gift of Gab. FEBS Lett. 2002;515:1–7. doi: 10.1016/s0014-5793(02)02425-0. [DOI] [PubMed] [Google Scholar]
- 3.Nishida K, Hirano T. The role of Gab family scaffolding adapter proteins in the signal transduction of cytokine and growth factor receptors. Cancer Sci. 2003;94:1029–1033. doi: 10.1111/j.1349-7006.2003.tb01396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wohrle FU, Daly RJ, Brummer T. How to Grb2 a Gab. Structure. 2009;17:779–781. doi: 10.1016/j.str.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Vaughan TY, Verma S, Bunting KD. Grb2-associated binding (Gab) proteins in hematopoietic and immune cell biology. Am J Blood Res. 2011;1:130–134. [PMC free article] [PubMed] [Google Scholar]
- 6.Simister PC, Feller SM. Order and disorder in large multi-site docking proteins of the Gab family--implications for signalling complex formation and inhibitor design strategies. Mol Biosyst. 2012;8:33–46. doi: 10.1039/c1mb05272a. [DOI] [PubMed] [Google Scholar]
- 7.Itoh M, Yoshida Y, Nishida K, Narimatsu M, Hibi M, Hirano T. Role of Gab1 in heart, placenta, and skin development and growth factor- and cytokine-induced extracellular signal-regulated kinase mitogen-activated protein kinase activation. Mol Cell Biol. 2000;20:3695–3704. doi: 10.1128/mcb.20.10.3695-3704.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sachs M, Brohmann H, Zechner D, Muller T, Hulsken J, Walther I, Schaeper U, Birchmeier C, Birchmeier W. Essential role of Gab1 for signaling by the c-Met receptor in vivo. J Cell Biol. 2000;150:1375–1384. doi: 10.1083/jcb.150.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng AM, Saxton TM, Sakai R, Kulkarni S, Mbamalu G, Vogel W, Tortorice CG, Cardiff RD, Cross JC, Muller WJ, et al. Mammalian Grb2 regulates multiple steps in embryonic development and malignant transformation. Cell. 1998;95:793–803. doi: 10.1016/s0092-8674(00)81702-x. [DOI] [PubMed] [Google Scholar]
- 10.Meng S, Chen Z, Munoz-Antonia T, Wu J. Participation of both Gab1 and Gab2 in the activation of the ERK/MAPK pathway by epidermal growth factor. Biochem J. 2005;391:143–151. doi: 10.1042/BJ20050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daly RJ, Binder MD, Sutherland RL. Overexpression of the Grb2 gene in human breast cancer cell lines. Oncogene. 1994;9:2723–2727. [PubMed] [Google Scholar]
- 12.Tari AM, Hung MC, Li K, Lopez-Berestein G. Growth inhibition of breast cancer cells by Grb2 downregulation is correlated with inactivation of mitogen-activated protein kinase in EGFR, but not in ErbB2, cells. Oncogene. 1999;18:1325–1332. doi: 10.1038/sj.onc.1202422. [DOI] [PubMed] [Google Scholar]
- 13.Rozakis-Adcock M, McGlade J, Mbamalu G, Pelicci G, Daly R, Li W, Batzer A, Thomas S, Brugge J, Pelicci PG, et al. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature. 1992;360:689–692. doi: 10.1038/360689a0. [DOI] [PubMed] [Google Scholar]
- 14.Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D, Schlessinger J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70:431–442. doi: 10.1016/0092-8674(92)90167-b. [DOI] [PubMed] [Google Scholar]
- 15.Chardin P, Camonis JH, Gale NW, van Aelst L, Schlessinger J, Wigler MH, Bar-Sagi D. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science. 1993;260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- 16.Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P, Bar-Sagi D, Margolis B, Schlessinger J. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature. 1993;363:85–88. doi: 10.1038/363085a0. [DOI] [PubMed] [Google Scholar]
- 17.Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W. Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J Cell Biol. 2000;149:1419–1432. doi: 10.1083/jcb.149.7.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewitzky M, Kardinal C, Gehring NH, Schmidt EK, Konkol B, Eulitz M, Birchmeier W, Schaeper U, Feller SM. The C-terminal SH3 domain of the adapter protein Grb2 binds with high affinity to sequences in Gab1 and SLP-76 which lack the SH3-typical P-x-x-P core motif. Oncogene. 2001;20:1052–1062. doi: 10.1038/sj.onc.1204202. [DOI] [PubMed] [Google Scholar]
- 19.Seedorf K, Kostka G, Lammers R, Bashkin P, Daly R, Burgess WH, van der Bliek AM, Schlessinger J, Ullrich A. Dynamin binds to SH3 domains of phospholipase C gamma and GRB-2. J Biol Chem. 1994;269:16009–16014. [PubMed] [Google Scholar]
- 20.Vidal M, Montiel JL, Cussac D, Cornille F, Duchesne M, Parker F, Tocque B, Roques BP, Garbay C. Differential interactions of the growth factor receptor-bound protein 2 N-SH3 domain with son of sevenless and dynamin. Potential role in the Ras-dependent signaling pathway. J Biol Chem. 1998;273:5343–5348. doi: 10.1074/jbc.273.9.5343. [DOI] [PubMed] [Google Scholar]
- 21.Odai H, Sasaki K, Iwamatsu A, Hanazono Y, Tanaka T, Mitani K, Yazaki Y, Hirai H. The proto-oncogene product c-Cbl becomes tyrosine phosphorylated by stimulation with GM-CSF or Epo and constitutively binds to the SH3 domain of Grb2/Ash in human hematopoietic cells. J Biol Chem. 1995;270:10800–10805. doi: 10.1074/jbc.270.18.10800. [DOI] [PubMed] [Google Scholar]
- 22.Park RK, Kyono WT, Liu Y, Durden DL. CBL-GRB2 interaction in myeloid immunoreceptor tyrosine activation motif signaling. J Immunol. 1998;160:5018–5027. [PubMed] [Google Scholar]
- 23.Moeller SJ, Head ED, Sheaff RJ. p27Kip1 inhibition of GRB2-SOS formation can regulate Ras activation. Mol Cell Biol. 2003;23:3735–3752. doi: 10.1128/MCB.23.11.3735-3752.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 25.Cunnick JM, Meng S, Ren Y, Desponts C, Wang HG, Djeu JY, Wu J. Regulation of the mitogen-activated protein kinase signaling pathway by SHP2. J Biol Chem. 2002;277:9498–9504. doi: 10.1074/jbc.M110547200. [DOI] [PubMed] [Google Scholar]
- 26.Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J Biol Chem. 2003;278:41677–41684. doi: 10.1074/jbc.M306461200. [DOI] [PubMed] [Google Scholar]
- 27.Kim D, Chung J. Akt: versatile mediator of cell survival and beyond. J Biochem Mol Biol. 2002;35:106–115. doi: 10.5483/bmbrep.2002.35.1.106. [DOI] [PubMed] [Google Scholar]
- 28.Lock LS, Royal I, Naujokas MA, Park M. Identification of an atypical Grb2 carboxyl-terminal SH3 domain binding site in Gab docking proteins reveals Grb2-dependent and -independent recruitment of Gab1 to receptor tyrosine kinases. J Biol Chem. 2000;275:31536–31545. doi: 10.1074/jbc.M003597200. [DOI] [PubMed] [Google Scholar]
- 29.McDonald CB, Seldeen KL, Deegan BJ, Bhat V, Farooq A. Binding of the cSH3 domain of Grb2 adaptor to two distinct RXXK motifs within Gab1 docker employs differential mechanisms. J Mol Recognit. 2011;24:585–596. doi: 10.1002/jmr.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald CB, Seldeen KL, Deegan BJ, Lewis MS, Farooq A. Grb2 adaptor undergoes conformational change upon dimerization. Arch Biochem Biophys. 2008;475:25–35. doi: 10.1016/j.abb.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Maignan S, Guilloteau JP, Fromage N, Arnoux B, Becquart J, Ducruix A. Crystal structure of the mammalian Grb2 adaptor. Science. 1995;268:291–293. doi: 10.1126/science.7716522. [DOI] [PubMed] [Google Scholar]
- 32.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW. Intrinsically Disordered Protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 33.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uversky VN. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol. 2011;43:1090–1103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu K, Hirose S, Noguchi T. POODLE-S: web application for predicting protein disorder by using physicochemical features and reduced amino acid set of a position-specific scoring matrix. Bioinformatics. 2007;23:2337–2338. doi: 10.1093/bioinformatics/btm330. [DOI] [PubMed] [Google Scholar]
- 38.Rabanal F, Ludevid MD, Pons M, Giralt E. CD of proline-rich polypeptides: application to the study of the repetitive domain of maize glutelin-2. Biopolymers. 1993;33:1019–1028. doi: 10.1002/bip.360330704. [DOI] [PubMed] [Google Scholar]
- 39.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 40.Renzoni DA, Pugh DJ, Siligardi G, Das P, Morton CJ, Rossi C, Waterfield MD, Campbell ID, Ladbury JE. Structural and thermodynamic characterization of the interaction of the SH3 domain from Fyn with the proline-rich binding site on the p85 subunit of PI3-kinase. Biochemistry. 1996;35:15646–15653. doi: 10.1021/bi9620969. [DOI] [PubMed] [Google Scholar]
- 41.McDonald CB, Seldeen KL, Deegan BJ, Bhat V, Farooq A. Assembly of the Sos1-Grb2-Gab1 ternary signaling complex is under allosteric control. Arch Biochem Biophys. 2010;494:216–225. doi: 10.1016/j.abb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaeper U, Vogel R, Chmielowiec J, Huelsken J, Rosario M, Birchmeier W. Distinct requirements for Gab1 in Met and EGF receptor signaling in vivo. Proc Natl Acad Sci U S A. 2007;104:15376–15381. doi: 10.1073/pnas.0702555104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaudhuri A, Xie MH, Yang B, Mahapatra K, Liu J, Marsters S, Bodepudi S, Ashkenazi A. Distinct involvement of the Gab1 and Grb2 adaptor proteins in signal transduction by the related receptor tyrosine kinases RON and MET. J Biol Chem. 2011;286:32762–32774. doi: 10.1074/jbc.M111.239384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagh PK, Peace BE, Waltz SE. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv Cancer Res. 2008;100:1–33. doi: 10.1016/S0065-230X(08)00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eulenfeld R, Schaper F. A new mechanism for the regulation of Gab1 recruitment to the plasma membrane. J Cell Sci. 2009;122:55–64. doi: 10.1242/jcs.037226. [DOI] [PubMed] [Google Scholar]
- 46.Rosario M, Birchmeier W. How to make tubes: signaling by the Met receptor tyrosine kinase. Trends Cell Biol. 2003;13:328–335. doi: 10.1016/s0962-8924(03)00104-1. [DOI] [PubMed] [Google Scholar]
- 47.Harkiolaki M, Tsirka T, Lewitzky M, Simister PC, Joshi D, Bird LE, Jones EY, O’Reilly N, Feller SM. Distinct binding modes of two epitopes in Gab2 that interact with the SH3C domain of Grb2. Structure. 2009;17:809–822. doi: 10.1016/j.str.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 48.Gao X, Yo P, Keith A, Ragan TJ, Harris TK. Thermodynamically balanced inside-out (TBIO) PCR-based gene synthesis: a novel method of primer design for high-fidelity assembly of longer gene sequences. Nucleic Acids Res. 2003;31:e143. doi: 10.1093/nar/gng143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; Totowa, New Jersey, USA: 2005. pp. 571–607. [Google Scholar]
- 50.Zimm BH. The Scattering of Light and the Radial Distribution Function of High Polymer Solutions. J Chem Phys. 1948;16:1093–1099. [Google Scholar]
- 51.Wyatt PJ. Light Scattering and the Absolute Characterization of Macromolecules. Anal Chim Acta. 1993;272:1–40. [Google Scholar]
- 52.Berne BJ, Pecora R. Dynamic Light Scattering. Wiley; New York: 1976. [Google Scholar]
- 53.Chu B. Laser Light Scattering: Basic Principles and Practice. Academic; Boston: 1991. [Google Scholar]
- 54.Koppel DE. Analysis of Macromolecular Polydispersity in Intensity Correlation Spectroscopy. J Chem Phys. 1972;57:4814–4820. [Google Scholar]
- 55.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 56.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative Protein Structure Modeling of Genes and Genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 57.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 58.Carson M. Ribbons 2.0. J Appl Crystallogr. 1991;24:958–961. [Google Scholar]
- 59.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 60.Hess B. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 61.Jorgensen WL, Tirado-Rives J. The OPLS Force Field for Proteins: Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 62.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 63.Toukan K, Rahman A. Molecular-dynamics study of atomic motions in water. Physical Review B. 1985;31:2643–2648. doi: 10.1103/physrevb.31.2643. [DOI] [PubMed] [Google Scholar]
- 64.Berendsen HJC, Grigera JR, Straatsma TP. The Missing Term in Effective Pair Potentials. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- 65.Darden TA, York D, Pedersen L. Particle mesh Ewald: An N. log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 66.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]