

Abstract
A simple protocol for the practical, scalable, catalytic asymmetric synthesis of γ-quaternary acylcyclopentenes in up to 91% overall yield and 92% ee has been developed. The reaction sequence employs a palladium-catalyzed enantioselective alkylation reaction and exploits the unusual stability of β-hydroxy cycloheptanones to achieve a general and robust method for performing two-carbon ring contractions. The resulting enantioenriched, highly-functionalized acylcyclopentenes provide a variable substituent and four additional functional group handles for chemoselective manipulation and potential application to the total synthesis of a wide array of natural products.
Keywords: ring contraction, rearrangement, allylic compounds, asymmetric catalysis, palladium, allylation, aldol reaction
Highly substituted cyclopentanes are a common structural motif integrated into thousands of natural products.[1] Selected examples of bioactive compounds containing this basic structural unit include the hamigerans (2),[2a] steroids (3),[2b] pleuromutilin antibiotics (4),[2c] cyathane diterpenoids (5),[2d] cyclic botryococcenes (6),[2e] and anti-HBV schisanwilsonenes (7a–c)[2f] (Figure 1). Synthetic methods for the asymmetric preparation of cyclopentanoid cores with multiple functional group handles are highly desirable because they allow for the strategic synthesis of these and other natural products.[3] Toward this goal, we envisioned that functionalized chiral units such as acylcyclopentene 1 could serve as valuable synthetic intermediates (Figure 1). In this report, we describe a general and enantioselective preparation of versatile chiral acylcyclopentenes[4, 5] that combines a catalytic asymmetric alkylation reaction[6] and a facile two-carbon ring contraction.
Figure 1.

Representative natural products possessing cyclopentanoid core structures with quaternary stereocenters.
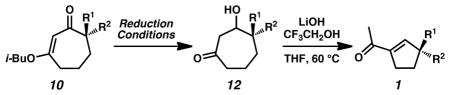
Our work in this area began with observation of the unusual reactivity of seven-membered ring vinylogous esters compared to their six-membered ring counterparts. Although LiAlH4 reduction of vinylogous ester 8 gave expected enone 9[7] as the major product after acidic workup (Scheme 1A), subjecting the analogous seven-membered ring vinylogous ester (10a) to identical reaction conditions led to cycloheptenone 11a as only a minor product (Scheme 1B). Interestingly, the major product was identified as stable β-hydroxyketone 12a.[ 8 ] The lack of appreciable β-elimination even under acidic conditions suggests that subtle, but fundamental differences in ring conformational preferences between six- and seven-membered rings may lead to the strikingly different product distributions.[9]
Scheme 1.

Anomalous reactivity of seven-membered ring vinylogous esters and discovery of a ring contraction reaction.
To further examine the inherent reactivity of β-hydroxyketone 12a, we exposed the compound to a variety of basic reaction conditions. Treatment of β-hydroxyketone 12a with LiOt-Bu in t-BuOH afforded acylcyclopentene 1a in 53% yield without any evidence of direct β-hydroxy elimination to enone 11a. Overall, the reaction constitutes a two-carbon ring contraction that likely proceeds via a retro-aldol fragmentation/aldol cyclization pathway. Although isolated examples of the preparation of acylcyclopentenes from seven-membered rings[10] are known, general ring contraction methods have not been demonstrated with γ-quaternary stereocenters and catalytic asymmetric routes are unprecedented.
Enticed by this initial finding, we investigated the effect of different bases on product formation (Table 1). Alcohol additives in combination with LiOH in THF improved the yield for the reaction (entries 2–4), with CF3CH2OH[11] enabling the production of 1a in 96% yield.[12] It is interesting to note that enone 11a was not observed under any of the surveyed conditions. Among the conditions that promote the desired ring contraction, the combination of LiOH and CF3CH2OH in THF offered a mild, efficient, and selective method for further studies (entry 4).
Table 1.
Ring contraction optimization.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | Base | Additive | Solvent | T [°C] | Yield [%][b] |
| 1 | LiOt-Bu | none | t-BuOH | 40 | 71 (53)[c] |
| 2 | LiOH | t-BuOH | THF | 60 | 78 |
| 3 | LiOH | HFIP[d] | THF | 60 | 87 |
| 4 | LiOH | CF3CH2OH | THF | 60 | 96 (84)[c] |
Conditions: β-hydroxyketone (1.0 equiv), base (1.5 equiv), additive (1.5 equiv) in solvent (0.1 M) at indicated temperature for 9–24 h.
GC yield using an internal standard.
Isolated yield in parentheses.
HFIP = 1,1,1,3,3,3-hexafluoro-2-propanol.
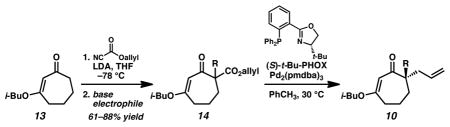
With an optimized procedure for the ring contraction, we turned our attention to the asymmetric synthesis of various quaternary substituted vinylogous esters (e.g., 10, Table 2).[13,14] A number of racemic β-ketoester substrates (e.g., 14) for catalytic enantioselective alkylation could be obtained by acylation of parent vinylogous ester 13 with allyl cyanoformate[15] and trapping with a range of electrophiles under basic conditions.[16] Application of our standard enantioselective decarboxylative alkylation reaction conditions[6,13] to substrate 14a produced chiral vinylogous ester 10a in 91% yield and 88% ee (entry 1).[17,18] Substituents such as ethyl, benzyl, propargyl, homoallyl, and 2,4-pentadienyl were well-tolerated in the reaction, giving similarly high yields and enantioselectivity (entries 2–6). A number of heteroatom containing substrates were explored to test if more diverse functionality could be incorporated into our target acylcyclopentenes (entries 7–11). β-Ketoesters bearing a 2-chloroallyl substitutent readily underwent the enantioselective alkylation reaction (entry 7). Gratifyingly, compounds that possess Lewis basic moieties readily furnished the desired products without complications (entries 8–9). Even indoles and free aldehydes could be incorporated into the cycloheptenone products (entries 10–11).
Table 2.
Scope of the Pd-catalyzed enantioselective alkylation of cyclic vinylogous esters.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate 14 |
R | Product 10 |
Yield [%][b] | ee [%][c] |
| 1 | 14a | –CH3 | 10a | 91 | 88 |
| 2 | 14b | –CH2CH3 | 10b | 89 | 92 |
| 3 | 14c | –CH2Ph | 10c | 98 | 86 |
| 4 | 14d | –CH2C≡CH | 10d | 88 | 89 |
| 5 | 14e | –CH2CH2CH=CH2 | 10e | 95 | 87 |
| 6 | 14f | 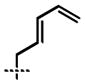 |
10f | 90 | 90 |
| 7 | 14g | 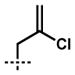 |
10g | 99 | 86 |
| 8 | 14h | –CH2CH2CN | 10h | 96 | 87 |
| 9 | 14i | 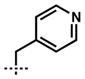 |
10i | 97 | 85 |
| 10 | 14j |  |
10j | 98 | 83 |
| 11 | 14k | 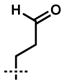 |
10k | 90 | 80 |
Conditions: β-ketoester (1.0 equiv), Pd2(pmdba)3 (2.5 mol %), (S)-t-Bu-PHOX (6.25 mol %) in PhCH3 (0.1 M) at 30 °C; pmdba = 4,4′-methoxydibenzylideneacetone.
Isolated yield.
Determined by chiral HPLC or SFC.
The chiral vinylogous esters (e.g., 10) prepared above allowed us to examine the scope of the ring contraction reaction (Table 3). Substrate reduction with LiAlH4 and base-promoted rearrangement of vinylogous esters bearing γ-alkyl substituents provided access to the corresponding acylcyclopentenes in excellent yields over the two-step protocol (entries 1–6). The chloroallyl, nitrile, and indole-containing substrates could be transformed with similarly high yields using the same conditions (entries 7–8, 10). Alternatively, DIBAL allowed smooth conversion of vinylogous ester 10i containing an N-basic pyridine (entry 9). Milder reduction under Luche conditions enabled facile conversion of silyl ether substrates (entries 11–12).[19] Furthermore, trans-propenyl substituted (entry 13) and spirocyclic (entry 14) substrates performed well in the ring contraction chemistry. With the combination of asymmetric alkylation and ring contraction, we achieved a route to substituted acylcyclopentenes with a wide range of functionality at the γ-quaternary stereocenter.
Table 3.
Ring contraction substrate scope.
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate 10 |
R1 | R2 | Product 1 |
Overall Yield [%][e] |
| 1[a,d] | 10a | –CH3 | –CH2CH=CH2 | 1a | 84 |
| 2[a,d] | 10b | –CH2CH3 | –CH2CH=CH2 | 1b | 90 |
| 3[a,d] | 10c | –CH2Ph | –CH2CH=CH2 | 1c | 86 |
| 4[a,d] | 10d | –CH2C≡CH | –CH2CH=CH2 | 1d | 95 |
| 5[a,d] | 10e | –CH2CH2CH=CH2 | –CH2CH=CH2 | 1e | 87 |
| 6[a,d] | 10f | 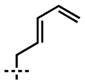 |
–CH2CH=CH2 | 1f | 91 |
| 7[a,d] | 10g | 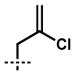 |
–CH2CH=CH2 | 1g | 92 |
| 8[a,d] | 10h | –CH2CH2CN | –CH2CH=CH2 | 1h | 85 |
| 9[b,d] | 10i | 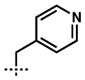 |
–CH2CH=CH2 | 1i | 80 |
| 10[a,d] | 10j | 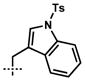 |
–CH2CH=CH2 | 1j | 87 |
| 11[c,d,f] | 10l | –CH2OTBDPS | –CH2CH=CH2 | 1l | 91 |
| 12[c,d,g] | 10m | –(CH2)3OTBDPS | –CH2CH=CH2 | 1m | 85 |
| 13[c,d,h] |
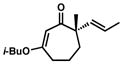 10n |
1n |
81 | ||
| 14[a,d,g] |
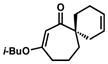 10o |
1o |
87 | ||
Reduction Conditions A: vinylogous ester (1.0 equiv), LiAlH4 (0.55 equiv) in Et2O (0.2 M) at 0 °C, then 10% aqueous HCl quench.
Reduction Conditions B: 1) vinylogous ester (1.0 equiv), DIBAL (1.2 equiv) in PHCH3 (0.03 M) at −78 °C; 2) oxalic acid•2H2O in MeOH (0.02 M).
Reduction Conditions C: vinylogous ester (1.0 equiv), CeCl3•7H2O (1.0 equiv), NaBH4 (3.0 equiv) in MeOH (0.02 M) at 0 °C, then 10% aqueous HCl in Et2O at 0 °C.
Ring Contraction Conditions: β-hydroxyketone (1.0 equiv), CF3CH2OH (1.5 equiv), LiOH (1.5 equiv) in THF (0.1 M) at 60 °C.
Isolated yield over 2–3 steps.
See Supporting Information for experimental procedures for substrate synthesis.
Prepared from 14k. See Supporting Information.
Prepared from 14a. See Supporting Information.
To demonstrate the practicality and scalability of the method, the α-methyl β-ketoester 14a was converted to the corresponding acylcyclopentene 1a in 69% yield over three steps on 15 g scale (Scheme 2A).[16] Notably, the multi-gram protocol proceeds with reduced catalyst loading and at higher reaction concentrations for the asymmetric alkylation step. Additionally, the enantiopurity of the acylcyclopentene 1a can be increased to 98% ee by recrystallization of semicarbazone 15 (Scheme 2B).[16] Hydrolysis of hydrazone 15 with aqueous HCl enabled facile recovery of 1a. Further derivatization afforded X-ray quality crystals of 16 for verification of absolute stereochemistry.[20] To enable access to β-substituted acylcyclopentenes, addition of n-BuMgBr to 10a resulted in formation of tertiary β-hydroxyketone 17 (Scheme 2C).[16] Application of modified ring contraction conditions allowed access to acylcyclopentene 18.
Scheme 2.

Multi-gram ring contraction, enrichment of ee by recrystallization, and organometallic modified ring contraction sequence.
With a versatile, enantioselective synthesis of γ-quaternary acylcyclopentenes 1 in hand, we sought to demonstrate the further synthetic utility of these compounds. By combining site selective manipulations in short reaction sequences (1–4 steps), any of five reactive handles present in acylcyclopentene 1 can be functionalized (Scheme 3, sites A–E). Through careful implementation of these transformations, diverse monocarbocyclic (1j, 18–23), spirocyclic (24–25), and fused polycyclic structures (26–27) can be obtained.[16]
Scheme 3.

Versatility and synthetic utility of acylcyclopentenes.
In summary, we have developed a catalytic enantioselective synthesis for the preparation of densely functionalized chiral acylcyclopentenes in excellent yields and enantioselectivities. The protocol exploits a highly efficient Pd-catalyzed asymmetric alkylation reaction and a newly-developed, mild two-carbon ring contraction. The important chiral building blocks formed using this method can undergo a variety of synthetic transformations and will serve as valuable intermediates for the total synthesis of natural products. Efforts directed toward these ends are currently underway and will be reported in due course.
Supplementary Material
Footnotes
This publication is based on work supported by Award No. KUS-11-006-02, made by King Abdullah University of Science and Technology (KAUST). The authors wish to thank NIH-NIGMS (R01GM080269-01), Amgen, Abbott, Boehringer Ingelheim, and Caltech for financial support. M. R. K. acknowledges Eli Lilly for a predoctoral fellowship. T. J. acknowledges the Danish Council for Independent Research/Natural Sciences for a postdoctoral fellowship. Materia, Inc. is gratefully acknowledged for the donation of catalysts. Lawrence Henling and Dr. Michael Day are gratefully acknowledged for X-ray crystallographic structure determination. The Bruker KAPPA APEXII X-ray diffractometer used in this study was purchased via an NSF CRIF:MU award to Caltech (CHE-0639094). Prof. Sarah Reisman, Dr. Scott Virgil, Dr. Christopher Henry, and Nathaniel Sherden are acknowledged for helpful discussions. Dr. David VanderVelde and Dr. Scott Ross are acknowledged for NMR assistance. The Varian 400 MR instrument used in this study was purchased via an NIH award to Caltech (NIH RR027690). Dr. Mona Shahgholi and Naseem Torian are acknowledged for high-resolution mass spectrometry assistance.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For a review of general methods for cyclopentane synthesis by ring contraction, see: Silva LF., Jr Tetrahedron. 2002;58:9137–9161.
- 2.a) Wellington KD, Cambie RC, Rutledge PS, Bergquist PR. J Nat Prod. 2000;63:79–85. doi: 10.1021/np9903494. [DOI] [PubMed] [Google Scholar]; b) Mills JS. J Chem Soc. 1956:2196–2202. [Google Scholar]; c) Kavanagh F, Hervey A, Robbins WJ. Proc Natl Acad Sci USA. 1951;37:570–574. doi: 10.1073/pnas.37.9.570. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Peng J, Walsh K, Weedman V, Bergthold JD, Lynch J, Lieu KL, Braude IA, Kelly M, Hamann MT. Tetrahedron. 2002;58:7809–7819. [Google Scholar]; e) Behrens A, Schaeffer P, Bernasconi S, Albrecht P. Org Lett. 2000;2:1271–1274. doi: 10.1021/ol0056980. [DOI] [PubMed] [Google Scholar]; f) Ma WH, Huang H, Zhou P, Chen DF. J Nat Prod. 2009;72:676–678. doi: 10.1021/np8007864. [DOI] [PubMed] [Google Scholar]
- 3.For a review discussing our strategy of using natural product structures to drive the development of enantioselective catalysis, see: Mohr JT, Krout MR, Stoltz BM. Nature. 2008;455:323–332. doi: 10.1038/nature07370.
- 4.For an example of the preparation of substituted acylcyclopentenes from five-membered rings, see: Trost BM, Schroeder GM. Chem Eur J. 2005;11:174–184. doi: 10.1002/chem.200400666.
- 5.For selected examples of the preparation of substituted acylcyclopentenes from six-membered rings, see: Wilson SR, Turner RB. J Org Chem. 1973;38:2870–2873. doi: 10.1021/jo00956a030.Gibbons EG. J Am Chem Soc. 1982;104:1767–1769.Paquette LA, Dahnke K, Doyon J, He W, Wyant K, Friedrich D. J Org Chem. 1991;56:6199–6205.
- 6.a) Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem. 2005;117:7084–7087. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; Angew. Chem., Int. Ed. 2005, 44, 6924–6927; c) Mohr JT, Stoltz BM. Chem Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Seto M, Roizen JL, Stoltz BM. Angew Chem. 2008;120:6979–6982. doi: 10.1002/anie.200801424. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2008;47:6873–6876. doi: 10.1002/anie.200801424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stork G, Danheiser RL. J Org Chem. 1973;38:1775–1776. [Google Scholar]
- 8.While a recent report shows a single example of a similar β-hydroxyketone, we believe the unusual reactivity and synthetic potential of these compounds has not been fully explored: Rinderhagen H, Mattay J. Chem Eur J. 2004;10:851–874. doi: 10.1002/chem.200304827.
- 9.Differences in ring conformational preferences can also be observed in the 1H NMR spectra of 1,3-cyclohexadione (exclusively ketoenol form) and 1,3-cycloheptadione (exclusively diketo form), see: Do N, McDermott RE, Ragan JA. Org Synth. 2008;85:138–146.
- 10.For selected examples of the preparation of substituted acylcyclopentenes from seven-membered rings, see: Frankel JJ, Julia S, Richard-Neuville C. Bull Soc Chim Fr. 1968:4870–4875.Jun C-H, Moon CW, Lim S-G, Lee H. Org Lett. 2002;4:1595–1597. doi: 10.1021/ol025816e.
- 11.For a discussion of the properties of fluorinated alcohols and their use, see: Begue J-P, Bonnet-Delpon D, Crousse B. Synlett. 2004:18–29.
- 12.A lithium alkoxide species is presumably generated in situ based on pKa values, see: Bordwell FG. Acc Chem Res. 1988;21:456–463.b) The use of LiOCH2CF3 as base provided comparable results (90% isolated yield), supporting the likelihood of its in situ formation under the reaction conditions.[16]
- 13.For examples of the asymmetric alkylation of vinylogous esters or thioesters from our laboratory, see: White DE, Stewart IC, Grubbs RH, Stoltz BM. J Am Chem Soc. 2008;130:810–811. doi: 10.1021/ja710294k.Levine SR, Krout MR, Stoltz BM. Org Lett. 2009;11:289–292. doi: 10.1021/ol802409h.Petrova KV, Mohr JT, Stoltz BM. Org Lett. 2009;11:293–295. doi: 10.1021/ol802410t.
- 14.For a related example of the asymmetric alkylation of vinylogous thioesters, see: Trost BM, Bream RN, Xu J. Angew Chem. 2006;118:3181–3184. doi: 10.1002/anie.200504421.Angew Chem, Int Ed. 2006;45:3109–3112. doi: 10.1002/anie.200504421.
- 15.a) Mander LN, Sethi SP. Tetrahedron Lett. 1983;24:5425–5428. [Google Scholar]; b) Donnelly DMX, Finet JP, Rattigan BA. J Chem Soc Perkin Trans 1. 1993:1729–1735. [Google Scholar]
- 16.See Supporting Information for experimental procedures.
- 17.a) Tani K, Behenna DC, McFadden RM, Stoltz BM. Org Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]; b) Krout MR, Mohr JT, Stoltz BM. Org Synth. 2009;82:181–193. doi: 10.15227/orgsyn.086.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pd2(pmdba)3 is preferable to Pd2(dba)3 in this reaction for ease of separation of pmdba from the reaction products during purification. dba = dibenzylideneacetone.
-
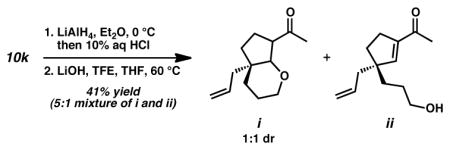
19.Exposure of aldehyde 10k (Table 2, entry 11) to our standard reduction/ring contraction conditions produced a mixture of products as shown below:
- 20.Crystallographic data have been deposited at the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK and copies can be obtained on request, free of charge, by quoting the publication citation and the deposition number 686849.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.