

Background: Interactions of nitric oxide with hemoglobin are critical to NO function.
Results: Reductive nitrosylation of hemoglobin is faster at low NO concentrations and sensitive to allosteric modifications.
Conclusion: Hemoglobin can catalyze faster reductive nitrosylation reactions at physiological, low NO concentrations.
Significance: Reductive nitrosylation may be more relevant in vivo than previously recognized.
Keywords: Allosteric Regulation, Enzyme Kinetics, Hemoglobin, Nitric Oxide, Nitrosylation, Nitrite, Nitrosothiols, Reductive Nitrosylation
Abstract
The reductive nitrosylation of ferric (met)hemoglobin is of considerable interest and remains incompletely explained. We have previously observed that at low NO concentrations the reaction with tetrameric hemoglobin occurs with an observed rate constant that is at least 5 times faster than that observed at higher concentrations. This was ascribed to a faster reaction of NO with a methemoglobin-nitrite complex. We now report detailed studies of this reaction of low NO with methemoglobin. Nitric oxide paradoxically reacts with ferric hemoglobin with faster observed rate constants at the lower NO concentration in a manner that is not affected by changes in nitrite concentration, suggesting that it is not a competition between NO and nitrite, as we previously hypothesized. By evaluation of the fast reaction in the presence of allosteric effectors and isolated β- and α-chains of hemoglobin, it appears that NO reacts with a subpopulation of β-subunit ferric hemes whose population is influenced by quaternary state, redox potential, and hemoglobin dimerization. To further characterize the role of nitrite, we developed a system that oxidizes nitrite to nitrate to eliminate nitrite contamination. Removal of nitrite does not alter reaction kinetics, but modulates reaction products, with a decrease in the formation of S-nitrosothiols. These results are consistent with the formation of NO2/N2O3 in the presence of nitrite. The observed fast reductive nitrosylation observed at low NO concentrations may function to preserve NO bioactivity via primary oxidation of NO to form nitrite or in the presence of nitrite to form N2O3 and S-nitrosothiols.
Introduction
The reactions between hemoglobin (Hb)3 and nitric oxide (NO) are fundamental to the vascular biology of NO. According to calculations based on experimental data, most of the NO produced by the endothelial nitric-oxide synthases is consumed by reaction with hemoglobin (1–4). The main NO-consuming reaction in oxygenated blood is the reaction of oxyhemoglobin with NO to produce methemoglobin and nitrate; a fast reaction with rates that are diffusion controlled (5). Nitric oxide can form tight complexes with ferrous deoxyhemoglobin, but can also form complexes with the oxidized ferric form of hemoglobin (methemoglobin). In this case a methemoglobin-NO complex is formed transiently and decays to ferrous nitrosylhemoglobin. This reaction is called reductive nitrosylation and was first described for hemoglobin in 1937 by Keilin and Hartree (6). A number of studies suggest the following mechanism (7–9).
It has been noted that for hemoglobin, the Fe3+-NO complex can also react with water at a noticeable rate (8).
The reductive nitrosylation reaction is classically studied in the presence of saturating concentrations of nitric oxide (7–9). In biological conditions hemoglobin is present in a large excess and the steady state concentration of NO available for these reactions is very low. The reaction has been studied under biologically relevant conditions only in a few cases (11–14). Unfortunately, although important phenomenological observations have been made, these studies have not provided information about the reaction mechanisms that can be used to make reliable predictions about the reaction kinetics or product yields. A noteworthy observation in several works is the presence of different responses to similar amounts of NO administered at different rates. For example, Herold and Rock (13) observed that higher yields of S-nitroso-Hb (βCys93-SNO, SNO-Hb) were formed after addition of 0.1 eq of NO to MetHb than after the addition of 1 eq of NO; also faster addition of NO resulted in lower RS-NO yields than slower addition experiments. Luchsinger et al. (11) also reported differences in the yields of SNO-Hb and proposed that this nitrosation reaction was more important at lower NO/Hb ratios, and attributed this fact to different reactivities of tetranitrosyl MetHb (MetNO4) versus MetHb that is nitrosylated only on the β-hemoglobin subunits (αMet2βMetNO2). Although these observations are important, no kinetic data for the reactivities of these species and no chemical basis for the formation of different products was provided.
Previous studies indicate that nitrite has the ability to speed up the reaction (9) and mixtures of methemoglobin/nitrite/nitric oxide can catalyze the formation of nitrosating species (14–17). Although the amounts of methemoglobin in the blood are usually low, normally less than 1% of total hemoglobin, with one study reporting 0.24 ± 0.02% (18), it is possible that in some in vivo circumstances the reaction of NO with MetHb can be of biological relevance. This may be the case for red blood cell membrane-associated methemoglobin, which can bind to the red cell membrane anion exchange protein.
In some aspects the reductive nitrosylation reaction can be considered the reverse of the nitrite reductase reaction (19–25), where nitrite reacts with deoxyhemoglobin to form methemoglobin and NO, shown in Equations 5–8, going from the left-hand to right-hand sides.
For a better comprehension of the kinetic processes involved, Equations 1–8 (and other additional processes, such as possible N2O3 formation) are summarized in Scheme 1. For hemoglobin, the nitrite reduction reaction shows a marked allosteric effect (19, 22) and therefore we speculated that similar effects may be relevant to the reductive nitrosylation reaction. For the sake of comparison, there is also a clear dependence of the nitrite reduction rates on [H+] (20), whereas the rate of reductive nitrosylation increases with increasing [OH−] (8, 26–28).
FIGURE 5.

Effect of IHP on the reductive nitrosylation observed rate contants. We observe that the effect of IHP is small at 37 °C but becomes more noticeable at lower temperatures. A, observed rates of HbFe2+-NO formation when HbFe3+ (≈100 μm) was mixed with 100 μm NO from ProliNONOate at 15, 25, or 37 °C in the presence of 5 mm IHP. B, observed rate constants of HbFe2+-NO formation when HbFe3+ (≈100 μm) was mixed with 50 μm NO from ProliNONOate at 15, 25, or 37 °C in the presence of 5 mm IHP.
FIGURE 6.

pH effects on reductive nitrosylation observed rate constants. Ferric Hb chains (HbFe3+) were mixed with NO from ProliNONOate. Reactions were performed at 37 °C in 100 mm sodium phosphate (pH 6.5 or 7.4) or in 100 mm Tris-HCl buffer (pH 9.0). The observed rate constants increase at higher pH but the dependence is smaller than what would be expected if the reaction were completely base-catalyzed (expectation is a 10-fold increase in rate per pH unit increase). The inset shows the observed rate constants in the high NO range.
FIGURE 7.

Effect of dimer-promoting agents on reductive nitrosylation. Panels A–C show selected spectra during the reductive nitrosylation of 100 μm methemoglobin in 200 μm PMB at various ProliNONOate concentrations. Arrows indicate the direction of change in the spectra. The insets show the change in absorption versus time at the given wavelength along with the first order kinetics fit. A, 500 μm ProliNONOate spectra taken every 30 s for 45 min. B, 50 μm ProliNONOate spectra taken every minute for 1 h. C, 20 μm ProliNONOate spectra taken every 30 s for 45 min. D, the observed rate constants for the reductive nitrosylation of hemoglobin with different effectors.
FIGURE 8.

The presence of small amounts of methemoglobin greatly reduces the stability of NO in solution. A, time course of injections of 125 nm ProliNO (in PBS) (pH 7.4), in the absence or presence of 25 μm methemoglobin into PBS in a purge vessel of NOA. The data are representative of three separate experiments. B, half-life of increasing concentrations of ProliNO (in PBS) (pH 7.4) injected into PBS in a purge vessel of NOA in the absence or presence of 25 μm methemoglobin. Data shown are from the average of three independent experiments.
FIGURE 1.

Reductive nitrosylation of methemoglobin (HbFe3+). HbFe3+ (≈25 μm) was mixed with variable amounts of ProliNONOate to achieve the indicated NO concentrations. Reactions were performed at 37 °C in 100 mm sodium phosphate (pH 7.4), except reactions in panels D and F, which were performed at 25 °C. A, 24 μm HbFe3+ and reacted with the NO donor ProliNONOate (final [NO] = 21 μm). B, concentration of different species following spectral deconvolution for the reaction shown in panel A (HbFe3+, red; HbFe2+-NO, black; HbFe3+-NO, green). C, observed rate constants of HbFe2+-NO formation from experiments where HbFe3+ (≈25 μm) was mixed with increasing amounts of ProliNONOate. The red line shows the calculated rates from a simulation with the model in Equations 1–8 (see text for details). D, observed rate constants of HbFe2+-NO formation from experiments where ≈20 μm ProliNONOate was mixed with increasing concentrations of HbFe3+. E, observed initial rates of HbFe2+-NO formation for the experiments in panel C. F, observed initial rates of HbFe2+-NO formation for the experiments in panel D.
FIGURE 2.

Observed rate constants at low NO are not due to NO bolus effect or nitrite contamination. A, comparison of observed rate constants from experiments with NO from either ProliNONOate or NO-saturated buffer. HbFe3+ (≈25 μm) was mixed with variable amounts of ProliNONOate or NO-saturated buffer to achieve the indicated NO concentrations. Reactions were performed at 37 °C in 100 mm sodium phosphate (pH 7.4). B, fast observed rate constants at low [NO] are not due to nitrite contamination of ProliNONOate. Methemoglobin (HbFe3+) solutions (23 μm) containing variable amounts of sodium nitrite were mixed with ProliNONOate to yield a final NO concentration of 5 μm. Reactions were carried out at 37 °C in 100 mm sodium phosphate (pH 7.4). The line is a linear fit of the data. C, high concentrations of nitrite do have an appreciable effect on observed rate constants at high NO. Methemoglobin (HbFe3+) solutions (30 μm) were mixed with 1 mm NO from ProliNONOate in the presence of a range of nitrite concentrations at 37 °C in 100 mm sodium phosphate (pH 7.4).
FIGURE 3.

Reductive nitrosylation observed rate constants for isolated Hb chains. Ferric Hb (HbFe3+) chains were mixed with NO (final concentrations 2–1000 μm) from ProliNONOate. Protein concentrations used (in heme) were 10–45 μm (α-chains) and 10–90 μm (β-chains). Reactions were performed at 37 °C in 100 mm sodium phosphate (pH 7.4). A, Hb α-chains. B, Hb β-chains. The insets show the observed rate constants in the low NO range.
FIGURE 4.

Reductive nitrosylation observed rate constants for NEM-modified β-chains and HbA. Ferric Hb (HbFe3+) chains were mixed with NO (final concentrations 2–1000 μm) from ProliNONOate. Protein concentrations used (in heme) were 7–20 μm (NEM-modified β-chains) and 25–50 μm (NEM-modified HbA). Reactions were performed at 37 °C in 100 mm sodium phosphate (pH 7.4). A, NEM-modified HbA; B, NEM-modified β-chains. The insets show the observed rates in the low NO range.
SCHEME 1.

To advance our understanding of the reductive nitrosylation of Hb, we examined a number of issues. 1) We studied the reductive nitrosylation reaction in conditions of excess hemoglobin. 2) We investigated the possible relevance of allosteric and chain effects on the kinetics of the reaction of NO with hemoglobin. 3) We examined the effects of nitrite on the reductive nitrosylation reaction, both on the reaction rate and the production of S-nitrosated species (S-nitrosothiols). Our data suggest that reductive nitrosylation is fastest in a subpopulation of β hemes the proportion of which are increased by T-state formation, increased redox potential and possibly hemoglobin dimerization, whereas formation of S-nitrosothiols are largely dependent on the presence of nitrite.
EXPERIMENTAL PROCEDURES
Reagents and Protein Preparation
Reagents were obtained from Sigma unless stated otherwise. ProliNONOate was purchased from Alexis Biochemicals (San Diego, CA) or Cayman Chemical (Ann Arbor, MI). SpermineNONOate was purchased from Cayman Chemical. Sodium nitrite and sodium iodide were purchased from Fisher Scientific. Reduced glutathione was purchased from Alexis Biochemicals (San Diego, CA). Copper (I) chloride and mercury chloride were purchased from Acros Organics (Morris Plains, NJ). NO gas was from Valley National Gases (West Mifflin, PA). Human hemoglobin was purified from erythrocytes as described (22, 29). α and β-hemoglobin chains were purified from human hemoglobin according to the method of Geraci et al. (29) with the modifications of Parkhurst and Parkhurst (30). The purity of α- and β-chain preparations was assessed by analytical isoelectrofocusing using ReadyGel IEF Gels from Bio-Rad. NEM (N-ethylmaleimide)-modified Hb was prepared as described by Huang et al. (22). Complexes of inositol hexaphosphate (IHP) and hemoglobin were studied in the presence of 5 mm IHP.
Reductive Nitrosylation Experiments
Protein samples were prepared in a glove box under nitrogen atmosphere to avoid oxygen contamination. MetHb samples were prepared by incubation of Hb with excess potassium ferricyanide. Oxidized heme proteins were run through PD-10 columns (Bio-Rad) to remove ferricyanide. Samples were diluted to the assay concentration (25 to 500 μm Hb) and transferred to 3.5-ml optical glass cuvettes closed by a silicon septum (Starna Cells, Atascadero, CA). Unless otherwise stated, reactions were followed at 37 °C. The reactions were initiated by addition of either NO from an NO-saturated solution or argon-saturated solutions of the NO donor ProliNONOate. The NO or NO donor (10–100 μl) was added to a final reaction volume of 2000 μl. Spectra were taken every second during 1000–2000 s in a diode array Agilent HP8453 spectrophotometer or at a similar rate using Cary 50 or 100 spectrophotometers. All solutions were prepared in 100 mm sodium phosphate of the noted pH (6.5–7.4), phosphate-buffered saline (pH 7.4), or 100 mm Tris-HCl buffer (pH 9.0), except ProliNONOate solutions that were made in 0.1 m NaOH to prevent hydrolysis.
Nitrite Reduction Experiments
The reactions were carried out in 3.5-ml optical glass cuvettes (Starna Cells, Atascadero, CA) closed by a silicon septum. Reactions were followed at 37 °C unless stated otherwise. The experiments were carried out in the presence or absence of dithionite. When dithionite was used, samples (25 or 100 μm Hb) were made anaerobic by addition of excess sodium dithionite (2.5 mm) and the reaction was initiated by addition of aliquots of an anaerobic solution of sodium nitrite (100 mm stock). When the reaction was studied in the absence of dithionite, samples were made anaerobic inside a glove box (Coy Laboratory Products, Grass Lake, MI) by mixing the protein with sodium dithionite to produce the deoxy protein and then run through a PD-10 column (Bio-Rad), to remove sodium dithionite.
Spectral Deconvolution
The kinetic data were analyzed by spectral deconvolution. The spectra of the reaction mixture at any time point was calculated as a linear combination of the spectra of the following standard spectra: oxy-Hb, deoxy-Hb, aquo-MetHb, hydroxy-MetHb, MetNOHb, nitrosyl Hb, and MetHb-nitrite. Best fit was calculated by the least-squares method as implemented in the Microsoft Excel Solver routine. The standard spectra are shown in supplemental Fig. S1. In the case of Hb α- and Hb β-chains we did not use spectral deconvolution due to the instability of the Fe3+ species; the rates for the HbFe3+ to HbFe2+-NO transitions were calculated from the traces of the absorbance at 575 nm minus the absorbance at 700 nm.
Measurements of NO Disappearance
ProliNO solutions were injected into PBS buffer under aerobic conditions in the presence or absence of 25 μm MetHb. The amount of NO remaining was then determined by injection into a Sievers Nitric Oxide Analyzer (NOA 280i, Sievers, GE Analytical Instruments, Boulder, CO) at various time points.
Nitrite-mediated RS-NO Formation during Reductive Nitrosylation
For all reactions, 100 μm of metal chelator diethylene triamine pentaacetic acid was added to PBS before addition of methemoglobin from a stock solution. At the end of a reaction, high molecular weight S-nitrosothiol (SNO-Hb) was determined by treatment with SNO-stop solution (4 mm potassium ferricyanide, 10 mm NEM, 100 μm diethylene triamine pentaacetic acid) for 60 min, passed through 2 columns (Sephadex G-25, GE Healthcare) and low molecular weight S-nitrosothiol glutathione (GSNO) by treatment with 5 mm NEM for 40 min followed by filtering in Microcon-10 or Amicon Ultra 10 (Millipore, Billerica, MA) for 20 min at 10,000 × g. This was followed by analysis of GSNO by the modified 2C method using ozone chemiluminescence by the NOA as described previously (31, 32). All reactions under anaerobic conditions were carried out in septum-sealed cuvettes under either nitrogen gas or argon gas pressure at 37 °C. Nitrite concentrations were determined by the sodium iodide (NaI) assay by the NOA 280i as prescribed by the manufacturer. In the presence of an NO donor, the assay can detect any free NO released along with nitrite. Simultaneous concentrations of nitrite and nitrate were determined by ENO-20 (EICOM-USA, San Diego, CA). Total S-nitrosothiol (total RS-NO) is the sum of high molecular weight (S-nitrosated hemoglobin, SNO-Hb) and low molecular weight S-nitrosothiols (S-nitrosoglutathione, GSNO) made during a reaction.
Experiments designed to explore the role of nitrite in RS-NO formation employed a nitrite oxidase system that includes catalase, glucose oxidase, and glucose (33, 34). All reactions of catalase with glucose and glucose oxidase were initiated under aerobic conditions immediately followed by monitoring of oxygen saturation by an OXY-Meter with a needle electrode (OX-N-7969, Unisense, Aahrus, Denmark) to ensure that the reaction was depleted of oxygen before addition of methemoglobin to continue the reactions under anaerobic conditions. In every case the addition of glucose oxidase to catalase and glucose at 37 °C led to rapid depletion of oxygen in the reaction mixtures.
RESULTS
Kinetic Considerations
Following Equations 1–4, the expected kinetics of Fe3+ reduction can be derived (8, 35). We have that,
where [Fe3+]total is the total concentration of Fe3+, which includes that which is unbound and bound to NO, and we employ [Fe3+] to refer to free/unbound ferric heme.
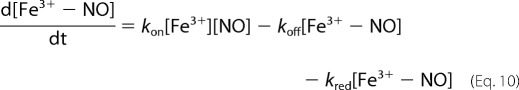 |
where kred is the rate constant for the reaction of Fe3+-NO with OH− or water given by kOH[OH−] + kH2O, and kon and koff refer to the association and dissociation rate constants for NO and ferric heme.
Substituting Equations 10 and 11 into 9, we have Equation 12.
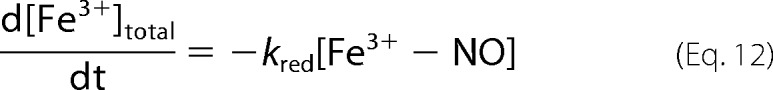 |
If we assume that the amount of free NO and that bound to MetHb is always in equilibrium during the reaction, we can substitute the equilibrium constant Kassoc,
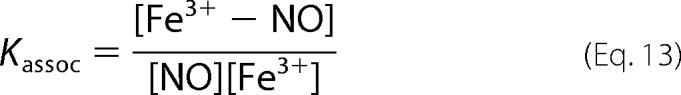 |
in Equation 12 and we get Equation 14.
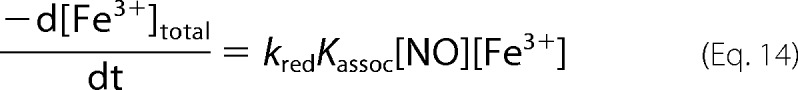 |
We can write,
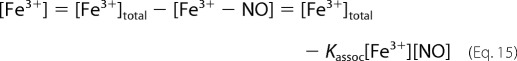 |
so that,
and therefore,
Then, substituting [Fe3+] into Equation 14 we get Equation 18.
This gives the observed rate constant, when NO is in excess to MetHb as Equation 19.
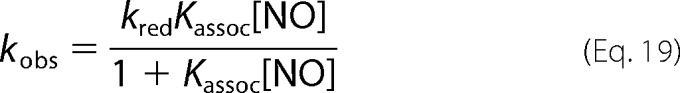 |
Based on the stoichiometry of the reaction, Equation 4, we also have the following.
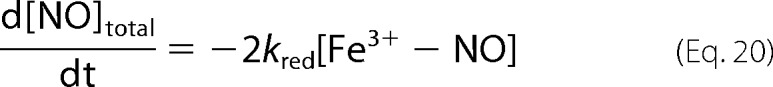 |
Assuming Equation 13 also holds when MetHb is in excess to NO we get,
and
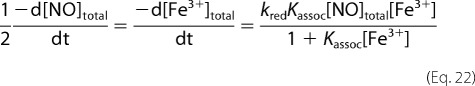 |
So that when MetHb is in excess to NO we expect pseudo-first order kinetics with rate constants of,
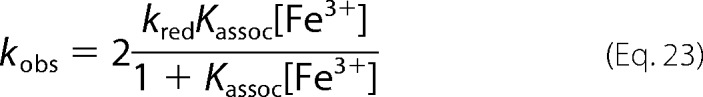 |
in analogy to Equation 19.
Equations 19 and 23 assume that Equation 13 holds throughout the reactions and that the concentration of MetHb is in dynamic equilibrium with NO through the reductive nitrosylation reactions. Given that the rate that equilibrium is established will be governed by the sum of the association and dissociation rates, and the dissociation rate constant is on the order of 1 s−1 (36), we suggest that our dynamic equilibrium assumption holds for all the reactions we study.
Reductive Nitrosylation of Hemoglobin
We first examined experimentally the reductive nitrosylation of hemoglobin (25 μm) at different concentrations of NO. The fast NO donor proliNONOate (t½ = 1.8 s at 37 °C (37) was used as NO source to limit possible bolus artifacts of rapid NO solution mixing with hemoglobin (38, 39). A typical reaction is shown in Fig. 1, A and B. The spectral data were deconvoluted using a set of standard Hb spectra, which included oxy-Hb, deoxy-Hb, metOH Hb, metH2O Hb, metNO, HbNO, and met-nitrite. After the deconvolution, the formation of HbNO versus time was fitted to a single-exponential equation to determine the kobs. When the observed rate constants were plotted versus the concentration of NO, we did not observe a constant value at low [NO] as expected by Equation 23, but rather a large observed rate constant at low NO that decays sharply and then increases slowly at higher NO concentrations (Fig. 1C, see also supplemental Fig. S2). The formation of HbNO is better described by a single-exponential process at low NO but as NO increases the time course more closely resembles a double-exponential process. This kinetic behavior and the observed rate constants from a single exponential fit are concordant with our previous report of unexpectedly rapid reaction of MetHb with low NO concentrations in the presence of NO2− (15).
We further studied the reaction at a fixed ProliNONOate concentration (20 μm) while increasing the Met Hb concentration (Fig. 1D). In this case the observed rate constant of HbNO formation follows what is expected from Equation 23, where the observed rate constant increases as MetHb increases and reaches a maximal value. Fig. 1, E and F, shows the initial rates for the reaction (measured as HbNO formation per second) obtained from the experiments shown in Fig. 1, C and D. In both cases, the initial rates increase as NO or Hb increases. The different trends observed in comparing Fig. 1, C and E, can be explained by the fact that Fig. 1C is a plot of observed rate constants and Fig. 1E plots the initial rates, nevertheless, the values of Fig. 1, D and F, are more consistent while also comparing observed rate constants and initial rates. One would not expect to have these different trends, but the fact that they are observed indicates complexity in the reaction consistent with the proposal that Equation 23 does not apply at low NO. It is well documented that when non-pseudo-first order conditions are used, the apparent kobs does increase at lower reactant concentrations (40, 41). To compare the experimental results with the expected rates from the reaction in Scheme 1, we carried out different computer simulations using different rates and kinetic models (supplemental Figs. S3–S9). In all cases we observe a pattern that does not match our experimental results. As illustrated by the red trace in Fig. 1C, our simulations do show an increase in kobs at low NO, but it is also apparent these simulated rates at low NO are much lower than the observed values in our experiments. The known effects due to hyperbolic kinetics do not predict that kobs when [NO] ≪ [MetHb] would actually become greater than that observed when [NO] ≫ [MetHb]. We modified different rates in our simulations (supplemental Figs. S3–S9) to explore if some kinetic parameters could increase the rates at low [NO]. The only noticeable changes were actually in the opposite direction, with a decreased effect (see supplemental Figs. S5C or S6A). It must be noted that in both cases (especially in supplemental Fig. S5C) the values used are probably closer to the actual rates of the reaction, at least for the Fe3+ + NO ⇌ Fe3+-NO equilibrium and the reaction rate at high NO. We conclude that additional mechanisms must operate so our kinetic model is incomplete and therefore the derived equations fail to fit the observed rates. These results are further described under “Discussion.”
Effects of Nitrite on Reductive Nitrosylation Reaction of Hemoglobin
As nitrite is known to catalyze the reductive nitrosylation of hemoglobin (9) and can also promote the formation of N2O3 in the presence of MetHb and NO (15), nitrite contamination of the samples could be a possible explanation of the increase in the observed rate constant at low NO. Solutions of NO donors such as ProliNONOate usually contain significant amounts of nitrite, so we conducted additional experiments to examine the effect of nitrite. We conducted similar experiments adding NO from an NO-saturated solution. The observed rate constants obtained (Fig. 2A) match those from the experiments with ProliNONOate. Although NO-saturated buffers can also contain low amounts of other nitrogen oxides, the observed rate constants obtained with the two methods are remarkably similar and appear to support the idea that the low-NO observed rate constants are independent of nitrite. In Fig. 2B, the observed rate constants for a mixture of 23 μm MetHb and 2.5 μm ProliNO spiked with different amounts of nitrite are shown. Even with a 100-fold NO/nitrite ratio, no significant increase in the observed rate constants is apparent. This confirms that the increase in observed rate constants seen at low NO concentrations is not due to nitrite, contrary to our earlier hypothesis (15). However, as mentioned above there is evidence that nitrite does influence the reductive nitrosylation rates under some conditions (9, 15). To verify these observations we conducted the reactions with a fixed concentration of ProliNONOate and MetHb in the presence of increasing concentrations of nitrite. In Fig. 2C the effect of nitrite at high NO concentrations is shown. When 30 μm MetHb and 1 mm NO (from ProliNONOate) are reacted in the presence of millimolar concentrations of nitrite, a linear increase of the observed rate constants is apparent in good agreement with previous studies (9, 15). Thus, our results suggest that the increase in the observed rate constant of reductive nitrosylation at low NO concentrations is not due to nitrite but arises intrinsically from the reductive nitrosylation reaction.
Reductive Nitrosylation of Hemoglobin α- and β-Chains
To investigate the mechanism for the unexpected dependence of the observed rate constants with the concentration of NO we focused on molecular aspects of hemoglobin such as differences in NO reactivity with the α- and β-globin chains. When MetHb reacts with NO, the individual rates of each α- or β-subunit to form MetNO and then to react with OH− or water may be different enough to yield predominantly αHbNO or βHbNO. The reductive nitrosylation in isolated β-chains has been described as “considerably faster” than in α-chains, but not quantitatively measured (36). This inequality has been proposed as a basis for increased formation of βHbNO versus αHbNO (11). Binding studies have shown that NO binds around 4 times faster to β-chains than α-chains of MetHb (36) but to date no values for the reductive nitrosylation rates have been provided. We studied the reductive nitrosylation of isolated α- and β-chains in search of noticeable differences. The study of these reactions is hampered by the instability of the isolated ferric α- and β-chains. As previously reported (36) we observed that α- and β-chains were stable in their oxy and deoxy forms, but denatured at different rates when oxidized to the ferric state. Denaturation of the ferric α-chains was slow but the ferric β-chains denature at a faster rate and the protein solution becomes noticeably cloudy during the experiments. Binding of NEM to the β-chains did not improve their stability. This denaturation of the protein through the reaction, more perceptible in the β-chains, leads to some imprecision in the rate determination.
Despite these challenges our measurements were reproducible and results are shown in Fig. 3, A and B. The reaction of ferric Hb α-chains with NO shows little differences with increasing NO concentrations and minimal differences in the rates when the Hb is in excess (Fig. 3A). The increase of the observed rates at low NO is not marked (as compared with HbA or β-chains) and may be due to the expected kobs increase described earlier (see also supplemental Figs. S3–S9). The patterns for the ferric β-chains are very different, with an overall behavior that resembles the HbA tetramer (Fig. 3B). The observed rate constants also seem faster in the ferric β-chains than for α-chain (usually above 0.025 s−1 for β-chains and below 0.02 s−1 for α-chains), in agreement with other reports (36).
Effects of R-state Stabilization on Reductive Nitrosylation Rates
We next tested the effects of NEM on the reductive nitrosylation observed rate constants; NEM reacts with βCys93 causing a conformational change in Hb that stabilizes the R-state and reduces the heme redox potential. NEM has been shown to accelerate the nitrite reductase reaction, which we consider may represent the reverse reaction of reductive nitrosylation. NEM may also block any possible reactions of the βCys93 thiol with reactive oxygen or nitrogen species. The results for NEM-modified HbA and NEM-modified β-chains are shown in Fig. 4, A and B, respectively. In both cases, NEM treatment inhibited the fast reactivity at low NO concentrations. It is also notable that the NEM-treated β-chain rates are similar to the values for native β-chains if we exclude from the analysis the low NO/Hb ratios. Treatment of the HbA tetramer with NEM also abolishes the low NO effects (Fig. 4A). The NEM-treated HbA rates in the low NO/Hb range are roughly equal to the average of α-chains and NEM-treated β-chains. These results suggest that the lower and variable redox potential of subpopulations of β-chains may regulate the fast reductive nitrosylation at low NO, and also supports the thesis that this is the reverse reaction of nitrite reduction, the rate of which is increased by NEM binding.
Effects of T-state Stabilization and pH on Reductive Nitrosylation rates
We used IHP to promote the T-state in the hemoglobin tetramer. Our first experiments did detect a very small increase in the observed rate constants (Fig. 5, 37 °C), whereas experiments at 25 °C showed a more noticeable effect (Fig. 5, 25 °C). Experiments run at 15 °C confirmed this trend, with IHP being more effective at increasing the rate constants at lower temperature. The reasons for these temperature effects of IHP on rates are not clear, but we speculate that IHP has two effects: it speeds up the reaction through the stabilization of the T-state but also may promote the formation of HbA dimers when used at high concentrations (42–47). These hemoglobin dimers can also cause the reaction to speed up as discussed below.
Effects of pH on Reductive Nitrosylation Rates
The observed rate constants for reductive nitrosylation experiments at pH 6.5, 7.4, and 9.0 are shown in Fig. 6. As the reductive nitrosylation is dominated by a reaction involving OH−, the rates are expected to increase as pH increases. We did observe a noticeable increase in the observed rate constants at increasing pH at low NO/hemoglobin ratios (Fig. 6). This pH dependence is opposite to the one observed in the nitrite reductase reaction (19, 22), but the magnitude of the increase is less than what is observed for the nitrite reductase reaction. The nitrite reduction requires a proton and the rates increase linearly with [H+] so a decrease of 1 pH unit causes a 10-fold increase in the rates. Here we observe an increase of only 2–3-fold per pH unit. This weaker dependence has been attributed to the reaction of the ferric nitrosyl-Hb with water in addition to OH−. Thus the pH dependence observed is consistent with that reported previously by Hoshino et al. (8) who fit the dependence of the Hb observed rate constants versus [OH−] at high NO/Hb ratios including a term for the reaction with water (8).
Effect of Dimer/Tetramer Equilibrium-modifying Agents
To test the hypothesis that the increase in the observed rates at low NO is, at least partially, due to the formation of methemoglobin dimers, the observed rate constant for the reductive nitrosylation of methemoglobin dimers was determined at various concentrations of NO. Hemoglobin was almost completely dissociated into dimers using either 2 m NaCl or a 2:1 paramercuribenzoate (PMB) to hemoglobin ratio (48). Upon addition of excess (500 μm) ProliNONOate to 100 μm MetHb in the presence of 200 μm PMB, ferric nitrosyl hemoglobin is transiently formed, this species decays to ferrous nitrosyl hemoglobin due to reductive nitrosylation as shown in Fig. 7A. Kinetic spectra at ProliNONOate concentrations of 50 and 20 μm show the same reductive nitrosylation, however, the initial spectra do not show complete nitrosylation (Fig. 7, B and C). Kinetic spectra for reductive nitrosylation of methemoglobin using NaCl to induce dimerization showed a similar trend of ferric nitrosyl to ferrous nitrosyl and thus resembled those for shown in Fig. 7, A–C, using PMB (data not shown). Methemoglobin dissociation into dimers increased the observed rate constant of reductive nitrosylation at all concentrations in comparison to methemoglobin in PBS (Fig. 7D). Interestingly IHP, an allosteric effector that also pushes the tetramer-dimer equilibrium toward tetramer at low concentrations, but toward dimers when used at high concentrations as was the case in our studies (42–47, 49), showed an increased observed rate constant similar to those seen by dimers at both 50 and 25 μm ProliNO (see Fig. 5). The effect of IHP can then potentially be seen as a combination of an allosteric and dimer/tetramer equilibrium phenomenon.
Fast NO Disappearance at Low NO Concentration in the Presence of MetHb
To confirm our observation of fast reductive nitrosylation at low concentrations, we explored the lifetime of NO itself in the presence or absence of 25 μm MetHb. Based on our measurements of HbNO formation during reductive nitrosylation, we expect that, in the presence of the MetHb, the observed rate constant will increase (i.e. the half-life of the reaction will decrease) as the concentration of NO is lowered. To compare these kinetics to more traditional ones, we conducted these experiments under aerobic conditions so we could also observe the disappearance of NO (and formation of nitrite) when oxygen reacts with NO, as described previously (50). Fig. 8A shows the concentration of NO as measured by injections into a Sievers nitric oxide analyzer as a function of time when 125 nm of the NO donor ProliNO was added to buffer in the presence or absence of 25 μm MetHb. It is seen that, in the presence of MetHb, NO disappears much faster than in its absence, where much of the NO has disappeared during the time taken to make the first injection. Fig. 8B plots the lifetimes of NO in the presence or absence of MetHb for different concentrations of NO added. As expected, when MetHb is absent the reaction of NO with oxygen becomes much slower because the reaction rate is determined by the concentration of oxygen multiplied by the NO concentration squared (50). On the other hand, contrary to what is expected from Equation 23, but consistent with our reductive nitrosylation data of Fig. 1C, we observe that the reaction with MetHb accelerates at low concentrations of NO with a half-life of 0.7 min using 125 nm ProliNO.
Formation of RS-NO during Reductive Nitrosylation and Role of Nitrite
To measure S-nitrosothiol (RS-NO) formation during reductive nitrosylation, total RS-NO (the sum of GSNO and SNO-Hb) was measured in the reaction of 100 μm methemoglobin with 100 μm of the NO donor, spermineNONOate (t½ = 39 min at 37 °C) in the presence of 1 mm GSH with either no nitrite or 1 mm nitrite, for 60 min under anaerobic conditions at 37 °C at pH 7.4. The amount of total RS-NO made in the absence of nitrite was 7.4 ± 1.8 μm, whereas that made in the presence of 1 mm nitrite was 10.4 ± 1.5 μm (p = 0.01). The significant, but moderate, increase in RS-NO yield in the presence of nitrite is consistent with previous proposed mechanisms of nitrite-mediated RS-NO formation, requiring formation of N2O3 (9) (see discussion below). Although these results seem to indicate that more RS-NO is formed through a nitrite-independent pathway, the NO donors usually contain contaminating nitrite, and nitrite is also formed during the reductive nitrosylation reaction (Equation 4). Therefore to more critically evaluate the role of nitrite we developed a nitrite oxidase system, comprised of enzyme catalase in the presence of glucose and glucose oxidase, which oxidizes nitrite to nitrate during the reaction.
To investigate whether the nitrite from NO donor stock solutions can be oxidized by the system comprised of enzyme catalase in the presence of glucose and glucose oxidase, 8 μm catalase was reacted with 100 μm nitrite, in the presence of 10 mm glucose and 100 μg of glucose oxidase at 37 °C, in PBS (pH 7.4), and aliquots were injected into the NOA. Supplemental Fig. S10A shows the time course of nitrite concentration determined for 120 min. Nitrite was eliminated in a time-dependent manner by catalase demonstrating that nitrite can be scavenged by catalase in the presence of H2O2 generated by glucose and glucose oxidase. Therefore if the NO donor is incubated with this nitrite scavenging system it should eliminate nitrite contamination in the NO donor stock solutions.
To determine the fate of nitrite in the presence of the nitrite scavenging system, 100 μm nitrite was treated with 8 μm catalase in the presence of 20 mm glucose and 200 μg of glucose oxidase at 37 °C, in PBS (pH 7.4), and aliquots were injected into the ENO-20 for the measurement of nitrite and nitrate concentrations with time (from the same injection) for a total of 80 min (supplemental Fig. S10B). With time, along with the decrease of nitrite concentration, there was a simultaneous increase in nitrate concentrations, whereas the total concentration of nitrite and nitrate remained constant. This indicates that nitrite is oxidized to nitrate by the nitrite oxidase system.
To show that nitrite contaminants in the NO donor solution or formed during reductive nitrosylation itself, can be eliminated by catalase in the presence of a H2O2 generating system, glucose, and glucose oxidase, nitrite concentrations in the NO donor stock solution were measured. In supplemental Fig. S10C, about 60 μm nitrite and free NO released from 100 μm of the NO donor ProliNONOate was measured at 0 min when 100 μm methemoglobin was reacted with 1 mm GSH and 100 μm ProliNONOate in PBS (pH 7.4) at 37 °C under anaerobic conditions. When 8 μm catalase was added along with 20 mm glucose and 200 μg of glucose oxidase to the reaction, there was a time-dependent decrease in nitrite concentration to less than 1 μm in 60 min, whereas in the reaction without catalase and glucose/glucose oxidase, the nitrite + NO level at 0 min (61 μm) remained close to that at 60 min (59 μm). (Note that NO released by the NO donor is converted over time to nitrite under the reaction conditions employed here.) These data suggest that nitrite in the ProliNONOate solution, and perhaps any made by reductive nitrosylation, was scavenged by the catalase and glucose/glucose oxidase system.
Having established the efficacy of the nitrite oxidase system, we investigated whether it would lead to a decrease in SNO yield from reductive nitrosylation, supporting a role for nitrite-dependent pathways. To test the role of nitrite in RS-NO formation we examined the effect of the nitrite oxidase system during reductive nitrosylation to eliminate nitrite. We compared the amount of GSNO and SNO-Hb in the presence or absence of the nitrite oxidase system (Fig. 9). We found 4.6 ± 0.9 μm RS-NO made in the absence of the nitrite oxidase system and only 0.4 ± 0.2 μm in the presence of the oxidase. As shown in supplemental Fig. S11, the increase in SNO is not due to any effect of the oxidase system on NO itself, in fact more NO was observed in the presence of the oxidase system. In control experiments examining RS-NO formation from all pairs of reactants, such as MetHb + NO donor, GSH + NO donor, MetHb + GSH, in the presence or absence of the nitrite oxidase system, no SNO was measured (determined as a mercury susceptible transient peak in the NOA) except for the mixture of NO donor and GSH in the presence of the nitrite oxidase system, where 0.9 ± 0.5 μm RS-NO was made (data not shown). This RS-NO made in the presence, but not absence of the nitrite oxidase system may have been due to small amounts of oxygen in the system. In any case the trend in that control experiment (where more SNO is made in the presence of the nitrite oxidase system than in its absence), is the opposite of that observed in Fig. 9. These data support the hypothesis that RS-NO formation during reductive nitrosylation is substantially dependent on nitrite, likely via N2O3 formation pathways (9, 15).
FIGURE 9.

Nitrite enhances RS-NO formation during reductive nitrosylation. Nitrite-enhanced S-nitrosothiol formation during reductive nitrosylation following elimination of nitrite contamination in NO donor solution by treatment with catalase in the presence of glucose and glucose oxidase (nitrite scavenging system). SpermineNONOate (100 μm) preincubated for 30 min at 37 °C with 16 μm catalase in PBS (pH 7.4), with or without 20 mm glucose, 1 mg of glucose oxidase was then reacted with 100 μm methemoglobin and 1 mm GSH under anaerobic conditions for 60 min. Total RS-NO made in the presence of nitrite in the NO donor solution and without glucose/glucose oxidase was 4.6 ± 0.9 μm (3.54 ± 0.8 μm GSNO and 1.1 ± 0.1 μm, SNO-Hb) and total RS-NO made in the presence of the nitrite scavenging system, with glucose/glucose oxidase, was 0.4 ± 0.2 μm (0.4 ± 0.2 μm GSNO, and 0 μm SNO-Hb). The figure shows mean ± S.E. of three independent experiments.
DISCUSSION
Observation of Fast Reductive Nitrosylation at Low NO
We studied the reaction of NO with methemoglobin and observed an unexpected increase in the apparent rate constants of ferrous heme-NO formation at low NO/Hb ratios. A summary of kinetic effects of different effectors and Hb forms is given in Supplementary Table 1 and Supplementary Fig. 12. Similar observations have been documented (11, 13, 14), but never pursued in detail or over a range of NO concentrations. We have previously observed a similar behavior for MetHb and NO in the presence of nitrite and attributed this to a higher reactivity of the methemoglobin-nitrite complexes (15). Here we observe the same reaction in the absence of nitrite (or at least independent of low nitrite concentrations). We must conclude that this behavior is not caused by nitrite but is inherent to the reductive nitrosylation reaction. Although the presence of nitrite in low concentrations has little effect on the reductive nitrosylation rates, nitrite does increase the rate at higher concentrations, as reported by Fernandez and Ford (9). Consistent with their proposed mechanism for an inner sphere electron transfer from Fe3+-NO to nitrite we observe that nitrite increases RS-NO yields (9) (Fig. 10). Although our observations of fast kinetics at low NO concentrations do not appear to be dependent on a methemoglobin-nitrite complex, the increase in RS-NO yield observed in the presence of nitrite could still represent a reaction of methemoglobin-bound nitrite and NO as we previously proposed (15).
FIGURE 10.

Possible pathways for nitrite catalysis of the reductive nitrosylation and routes for formation of nitrosating species through ferric-nitrite complexes. The two proposed pathways for the nitrite catalysis of reductive nitrosylation lead to the transient formation of nitrosating species such as NO2· and N2O3 (nitrite outer/inner sphere electron transfer routes). Similar routes with the formation of a Fe2+-NO2· intermediate that can react with NO have been proposed (nitric oxide-inner electron transfer route). Note that the routes where the inner sphere electron transfer occurs yield similar products and can hypothetically start from either Fe3+-NO or Fe3+-NO2− complexes.
Mechanism of Fast Reductive Nitrosylation at Low NO
One may think that our observation of increasing observed rate constants at low NO concentrations is due to the simple expected phenomenon that arises when the concentrations of two reactants in a second-order reaction are of nearly equal concentration. When the two reactants are at nearly equal concentrations, one expects the reaction to take a long time for completion as the concentration of both reactants slowly approaches zero. This phenomenon has also been discussed in the context of enzyme kinetics (40, 51). However, our measurements include conditions where MetHb is in substantial excess to NO and hence one would expect pseudo-first order kinetics (as depicted in Equations 22 and 23) rather than hyperbolic time dependence that results when two reactants are nearly equal in concentration. Moreover, the increase is not observed when Hb α-subunits were used, or after NEM treatment. We carried out a number of simulations to test if the kinetic model in Equations 1–8 can predict this behavior (supplemental Figs. S3–S9). It was evident from these calculations that the increase in kobs is too large to be predicted from the kinetic model. Moreover, when conditions apparently closer to the actual reaction rates are used (supplemental Fig. S5C), the rate increases at low [NO] are negligible. The calculated trend is in fact similar to that observed in NEM-treated HbA (Fig. 4A). We conclude that our observed rates are not explained by the kinetic model, and additional reactions are needed. Such reactions are apparently mediated by β-chains of Hb, and inhibited by increasing concentrations of [NO].
A general explanation for the observed kinetics is that it is due to a subpopulation of hemes. In this scenario, the ferric hemes in the subpopulation undergo reductive nitrosylation rapidly compared with others. As the concentration of NO increases, reactivity with the fast subpopulation saturates (that is the fast population is used up), making it take longer for the reaction to come to completion. This scheme is consistent with our observation that at low NO concentrations the kinetics are well described by a single exponential process, whereas at higher NO concentrations two exponential processes are needed to fit the data well (15).
This scheme is also consistent with our observation that as the concentration of MetHb increases while the concentration of NO is kept constant (Fig. 1D), the observed rate constant increases to the point of saturation as predicted by Equation 23. As the concentration of MetHb increases, the concentration of all the subpopulations also increases so that the reaction is expected to come to completion faster. Finally, this scheme is also consistent with the fact that the rate of reductive nitrosylation (as opposed to the observed rate constant) increases as the concentration of NO increases, even at low NO concentrations (Fig. 1, E and F). The rate of the reaction will be equal to the sum of the rates of the reactions with each subpopulation. Thus, increasing the concentration of NO will always increase the rate of the reaction (up to the point where all MetHb is saturated with NO as predicted by Equation 14). At low concentrations of NO, most of the NO will react with the fast subpopulation and the reaction is completed more rapidly then the observed rate constant will be greater. Increasing the concentration of NO will increase the proportion of the slower subpopulation participating in the reaction, making the reaction take a longer to come to completion, but the rate that HbNO formed per second will increase as both subpopulations participate.
Our data provide several clues into the nature of the fast subpopulation. As shown in Fig. 3, the fast subpopulation of hemes is likely in the β rather than α-subunits only. In addition, the proportion of β-subunit hemes in this fast reacting state seems to be greater in dimeric as compared with tetrameric hemoglobin. Similar observations have been made in relationship of ligand binding to hemoglobin where a fast binding population within β-subunits was proposed, perhaps due to a rotationally disordered heme, and the abundance of these reactive hemes was influenced by dimer/tetramer equilibrium (52). An allosteric effect in the reaction is expected by comparison to the nitrite reductase reaction, which can be considered the backwards reaction (19, 22). We used IHP and NEM as T-state and R-state promoting compounds and studied the reactions for isolated α- and β-chains. We note that NEM decreases the observed rate constant, whereas IHP increases it, so that the fast reacting subpopulation is favored in T-state tetramers compared with R-state tetramers, the opposite of nitrite reduction. These data are consistent with recent observations from the Rifkind lab showing greater reactivity of T-state ferric heme with NO than R-state hemes (53).
We must note that the inequality of α- and β-subunits has been observed in different contexts. Both pMB (54) and IHP (55) cause changes from low to high spin of the aquo- and hydroxymethemoglobin. These effects are not the same in both subunits. It is feasible that the change in spin state also causes a change in the reactivity of the hemoglobin; for instance, azide and cyanide bind faster to β-subunits in the presence of IHP (55). Other studies have shown increased dissociation rates for O2 (56, 57) and CO (57) in the presence of IHP. It is questionable if these observations have any relevance for the observed effects on reductive nitrosylation, nevertheless, these effects may provide new hypothesis to be pursued in subsequent studies.
Nitrite Dependence of RS-NO Formation in Reductive Nitrosylation
Formation of nitrosothiols upon nitrite administration has been observed both in vitro and in vivo (11, 14, 15, 58, 59). The formation of nitrosothiols could arise from transfer of NO+ from the transient MetHb-NO complex to the β93 thiol (11, 14, 59). Alternatively (or in addition), nitrosothiols could be formed via N2O3 formation that results from either reaction of nitrite with Hb Fe3+-NO (9) or reaction of NO with nitrite-bound hemoglobin (15). Whether or not additional nitrite is required to produce nitrosothiols from a mixture of MetHb and NO addresses the question of whether the nitrosothiols are formed by direct NO+ transfer via a nitrite-independent path or via a nitrite-dependent path such as that involving N2O3. Our data in Fig. 9 suggests that most nitrosothiol formation is via a nitrite-dependent pathway. Thus, it appears that nitrite does not speed up reductive nitrosylation at low [NO], but it does enhance RS-NO formation (changes product yields). In the absence of nitrite the Fe3+-NO complex reacts with water to form nitrite, whereas in the presence of nitrite more RS-NO is made suggesting the Fe3+-NO complex reacts with nitrite to form N2O3 (Fig. 10, Equation 24).
Biological Implications
The observation of an apparent increase in the reaction rates of NO and MetHb at low NO/Hb ratios has potential signaling implications, as the interactions of Hb and NO in vivo are almost always related to very low NO/Hb ratios. The reaction of NO with oxyhemoglobin leads to the formation of nitrate and methemoglobin (5, 60); as we observe here, further addition of NO to an environment rich in methemoglobin can lead to the formation of nitrite, nitrosating species, and RS-NO formation. This reaction has been proposed to produce stable signaling intermediates (53) and a fast reaction at the β-hemes to form SNO-Hb has been suggested as a mechanism for preserving NO bioactivity (11). Nitrite is an important signaling molecule (61), and 70% of plasma nitrite is thought to derive from endogenously produced NO (62, 63), yet (as data in Fig. 8 confirm) the reaction of NO with oxygen is too slow to account for nitrite production under physiological (low NO) conditions. An intriguing alternative pathway for nitrite formation involves NO oxidation by ceruloplasmin (10), and this may indeed account for nitrite homeostasis. We suggest that another contribution to nitrite production in vivo could be reductive nitrosylation as this reaction produces nitrite from NO and is faster at low NO (Fig. 1). As shown in Fig. 9, nitrite also appears to facilitate RS-NO formation upon reductive nitrosylation. Compartmentalization of the ferric hemoglobin species can thus provide new routes to overcome NO scavenging by the erythrocyte. This could be accomplished either by nitrite-dependent nitrosothiol formation or nitrite formation, which in turn might partially account for export of nitrite-dependent NO bioactivity from red blood cells and maintenance of nitrite homeostasis.
Supplementary Material
Acknowledgment
We acknowledge an anonymous reviewer for helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grants HL058091, HL098032, HL096973, and DK085852, and the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.

This article contains supplemental Figs. S1–S12 and Table S1.
- Hb
- hemoglobin
- IHP
- inositol hexaphosphate
- NEM
- N-ethylmaleimide
- PMB
- p-mercuribenzoate
- SNO-Hb
- S-nitrosated hemoglobin
- GSNO
- S-nitrosoglutathione.
REFERENCES
- 1. Lancaster J. R., Jr. (1997) A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide 1, 18–30 [DOI] [PubMed] [Google Scholar]
- 2. Lancaster J. R., Jr. (1996) Diffusion of free nitric oxide. Methods Enzymol. 268, 31–50 [DOI] [PubMed] [Google Scholar]
- 3. Lancaster J. R., Jr. (1994) Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 91, 8137–8141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lancaster J. R., Jr., Gaston B. (2004) NO and nitrosothiols, spatial confinement and free diffusion. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L465–466 [DOI] [PubMed] [Google Scholar]
- 5. Eich R. F., Li T., Lemon D. D., Doherty D. H., Curry S. R., Aitken J. F., Mathews A. J., Johnson K. A., Smith R. D., Phillips G. N., Jr., Olson J. S. (1996) Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 35, 6976–6983 [DOI] [PubMed] [Google Scholar]
- 6. Keilin D., Hartree E. F. (1937) Reaction of nitric oxide with hemoglobin and methemoglobin. Nature 139, 548–548 [Google Scholar]
- 7. Chien J. C. (1969) Reactions of nitric oxide with methemoglobin. J. Am. Chem. Soc. 91, 2166–2168 [DOI] [PubMed] [Google Scholar]
- 8. Hoshino M., Maeda M., Konishi R., Seki H., Ford P. C. (1996) Studies on the reaction mechanism for reductive nitrosylation of ferrihemoproteins in buffer solutions. J. Am. Chem. Soc. 118, 5702–5707 [Google Scholar]
- 9. Fernandez B. O., Ford P. C. (2003) Nitrite catalyzes ferriheme protein reductive nitrosylation. J. Am. Chem. Soc. 125, 10510–10511 [DOI] [PubMed] [Google Scholar]
- 10. Shiva S., Wang X., Ringwood L. A., Xu X., Yuditskaya S., Annavajjhala V., Miyajima H., Hogg N., Harris Z. L., Gladwin M. T. (2006) Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat. Chem. Biol. 2, 486–493 [DOI] [PubMed] [Google Scholar]
- 11. Luchsinger B. P., Rich E. N., Gow A. J., Williams E. M., Stamler J. S., Singel D. J. (2003) Routes to S-nitrosohemoglobin formation with heme redox and preferential reactivity in the β subunits. Proc. Natl. Acad. Sci. U.S.A. 100, 461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nagababu E., Ramasamy S., Rifkind J. M. (2007) Intermediates detected by visible spectroscopy during the reaction of nitrite with deoxyhemoglobin. The effect of nitrite concentration and diphosphoglycerate. Biochemistry 46, 11650–11659 [DOI] [PubMed] [Google Scholar]
- 13. Herold S., Rock G. (2003) Reactions of deoxy-, oxy-, and methemoglobin with nitrogen monoxide. Mechanistic studies of the S-nitrosothiol formation under different mixing conditions. J. Biol. Chem. 278, 6623–6634 [DOI] [PubMed] [Google Scholar]
- 14. Angelo M., Singel D. J., Stamler J. S. (2006) An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc. Natl. Acad. Sci. U.S.A. 103, 8366–8371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Basu S., Grubina R., Huang J., Conradie J., Huang Z., Jeffers A., Jiang A., He X., Azarov I., Seibert R., Mehta A., Patel R., King S. B., Hogg N., Ghosh A., Gladwin M. T., Kim-Shapiro D. B. (2007) Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat. Chem. Biol. 3, 785–794 [DOI] [PubMed] [Google Scholar]
- 16. Roche C. J., Friedman J. M. (2010) NO reactions with sol-gel and solution phase samples of the ferric nitrite derivative of HbA. Nitric Oxide 22, 180–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Navati M. S., Friedman J. M. (2009) Reactivity of glass-embedded met hemoglobin derivatives toward external NO. Implications for nitrite-mediated production of bioactive NO. J. Am. Chem. Soc. 131, 12273–12279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gladwin M. T., Ognibene F. P., Pannell L. K., Nichols J. S., Pease-Fye M. E., Shelhamer J. H., Schechter A. N. (2000) Relative role of heme nitrosylation and β-cysteine 93 nitrosation in the transport and metabolism of nitric oxide by hemoglobin in the human circulation. Proc. Natl. Acad. Sci. U.S.A. 97, 9943–9948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gladwin M. T., Grubina R., Doyle M. P. (2009) The new chemical biology of nitrite reactions with hemoglobin. R-state catalysis, oxidative denitrosylation, and nitrite reductase/anhydrase. Acc. Chem. Res. 42, 157–167 [DOI] [PubMed] [Google Scholar]
- 20. Doyle M. P., Pickering R. A., DeWeert T. M., Hoekstra J. W., Pater D. (1981) Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J. Biol. Chem. 256, 12393–12398 [PubMed] [Google Scholar]
- 21. Huang K. T., Keszler A., Patel N., Patel R. P., Gladwin M. T., Kim-Shapiro D. B., Hogg N. (2005) The reaction between nitrite and deoxyhemoglobin. Reassessment of reaction kinetics and stoichiometry. J. Biol. Chem. 280, 31126–31131 [DOI] [PubMed] [Google Scholar]
- 22. Huang Z., Shiva S., Kim-Shapiro D. B., Patel R. P., Ringwood L. A., Irby C. E., Huang K. T., Ho C., Hogg N., Schechter A. N., Gladwin M. T. (2005) Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J. Clin. Invest. 115, 2099–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salhany J. M. (2008) Kinetics of reaction of nitrite with deoxy hemoglobin after rapid deoxygenation or predeoxygenation by dithionite measured in solution and bound to the cytoplasmic domain of band 3 (SLC4A1). Biochemistry 47, 6059–6072 [DOI] [PubMed] [Google Scholar]
- 24. Nagababu E., Ramasamy S., Abernethy D. R., Rifkind J. M. (2003) Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. J. Biol. Chem. 278, 46349–46356 [DOI] [PubMed] [Google Scholar]
- 25. Brooks J. (1937) The action of nitrite on Haemoglobin in the absence of oxygen. Proc. R. Soc. Lond. Ser. B Biol. Sci. 123, 368–382 [Google Scholar]
- 26. Ascenzi P., Bocedi A., Antonini G., Bolognesi M., Fasano M. (2007) Reductive nitrosylation and peroxynitrite-mediated oxidation of heme-hemopexin. FEBS J. 274, 551–562 [DOI] [PubMed] [Google Scholar]
- 27. Ascenzi P., Cao Y., di Masi A., Gullotta F., De Sanctis G., Fanali G., Fasano M., Coletta M. (2010) Reductive nitrosylation of ferric human serum heme-albumin. FEBS J. 277, 2474–2485 [DOI] [PubMed] [Google Scholar]
- 28. Ascenzi P., di Masi A., Gullotta F., Mattu M., Ciaccio C., Coletta M. (2010) Reductive nitrosylation of ferric cyanide horse heart myoglobin is limited by cyanide dissociation. Biochem. Biophys. Res. Commun. 393, 196–200 [DOI] [PubMed] [Google Scholar]
- 29. Geraci G., Parkhurst L. J., Gibson Q. H. (1969) Preparation and properties of α- and β-chains from human hemoglobin. J. Biol. Chem. 244, 4664–4667 [PubMed] [Google Scholar]
- 30. Parkhurst K. M., Parkhurst L. J. (1992) Rapid preparation of native α- and β-chains of human hemoglobin. Int. J. Biochem. 24, 993–998 [DOI] [PubMed] [Google Scholar]
- 31. Fang K., Ragsdale N. V., Carey R. M., MacDonald T., Gaston B. (1998) Reductive assays for S-nitrosothiols. Implications for measurements in biological systems. Biochem. Biophys. Res. Commun. 252, 535–540 [DOI] [PubMed] [Google Scholar]
- 32. Basu S., Wang X., Gladwin M. T., Kim-Shapiro D. B. (2008) Chemiluminescent detection of S-nitrosated proteins. Comparison of tri-iodide, copper/CO/cysteine, and modified copper/cysteine methods. Methods Enzymol. 440, 137–156 [DOI] [PubMed] [Google Scholar]
- 33. Chance B. (1952) Effect of pH upon the reaction kinetics of the enzyme-substrate compounds of catalase. J. Biol. Chem. 194, 471–481 [PubMed] [Google Scholar]
- 34. Keilin D., Nicholls P. (1958) Reactions of catalase with hydrogen peroxide and hydrogen donors. Biochim. Biophys. Acta 29, 302–307 [DOI] [PubMed] [Google Scholar]
- 35. Ford P. C. (2010) Reactions of NO and nitrite with heme models and proteins. Inorg. Chem. 49, 6226–6239 [DOI] [PubMed] [Google Scholar]
- 36. Sharma V. S., Traylor T. G., Gardiner R., Mizukami H. (1987) Reaction of nitric oxide with heme proteins and model compounds of hemoglobin. Biochemistry 26, 3837–3843 [DOI] [PubMed] [Google Scholar]
- 37. Saavedra J. E., Southan G. J., Davies K. M., Lundell A., Markou C., Hanson S. R., Adrie C., Hurford W. E., Zapol W. M., Keefer L. K. (1996) Localizing antithrombotic and vasodilatory activity with a novel, ultrafast nitric oxide donor. J. Med. Chem. 39, 4361–4365 [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y., Hogg N. (2002) Mixing artifacts from the bolus addition of nitric oxide to oxymyoglobin. Implications for S-nitrosothiol formation. Free Radic. Biol. Med. 32, 1212–1219 [DOI] [PubMed] [Google Scholar]
- 39. Han T. H., Hyduke D. R., Vaughn M. W., Fukuto J. M., Liao J. C. (2002) Nitric oxide reaction with red blood cells and hemoglobin under heterogeneous conditions. Proc. Natl. Acad. Sci. U.S.A. 99, 7763–7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Daff S. (2004) An appraisal of multiple NADPH binding-site models proposed for cytochrome P450 reductase, NO synthase, and related diflavin reductase systems. Biochemistry 43, 3929–3932 [DOI] [PubMed] [Google Scholar]
- 41. Malatesta F. (2005) The study of bimolecular reactions under non-pseudo-first order conditions. Biophys. Chem. 116, 251–256 [DOI] [PubMed] [Google Scholar]
- 42. Griffon N., Baudin V., Dieryck W., Dumoulin A., Pagnier J., Poyart C., Marden M. C. (1998) Tetramer-dimer equilibrium of oxyhemoglobin mutants determined from autoxidation rates. Protein Sci. 7, 673–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. White S. L. (1975) The molecular dissociation of ferrihemoglobin derivatives. J. Biol. Chem. 250, 1263–1268 [PubMed] [Google Scholar]
- 44. White S. L. (1976) Titration of the carboxyhemoglobin tetramer-dimer equilibrium by inositol hexaphosphate. J. Biol. Chem. 251, 4763–4769 [PubMed] [Google Scholar]
- 45. Imai K., Yonetani H. (1977) The hemoglobin-oxygen equilibrium associated with subunit dissociation. Biochim. Biophys. Acta 490, 164–170 [DOI] [PubMed] [Google Scholar]
- 46. Gray R. D. (1974) The effect of 2,3-diphosphoglycerate on the tetramer-dimer equilibrium of liganded hemoglobin. J. Biol. Chem. 249, 2879–2885 [PubMed] [Google Scholar]
- 47. Gray R. D. (1980) The effect of H+, inositol hexaphosphate, and Zn(II) on the tetramer-dimer equilibrium of liganded hemoglobin. J. Biol. Chem. 255, 1812–1818 [PubMed] [Google Scholar]
- 48. Antonini E., Brunori M. (1971) in Hemoglobin and Myoglobin in Their Reactions with Ligands (Neuberger A., Tatum E. L., eds) pp. 110–119, North-Holland Publishing Co., Amsterdam [Google Scholar]
- 49. Hensley P., Moffat K., Edelstein S. J. (1975) Influence of inositol hexaphosphate binding on subunit dissociation in methemoglobin. J. Biol. Chem. 250, 9391–9396 [PubMed] [Google Scholar]
- 50. Wink D. A., Darbyshire J. F., Nims R. W., Saavedra J. E., Ford P. C. (1993) Reactions of the bioregulatory agent nitric oxide in oxygenated aqueous media. Determination of the kinetics for oxidation and nitrosation by intermediates generated in the NO/O2 reaction. Chem. Res. Toxicol. 6, 23–27 [DOI] [PubMed] [Google Scholar]
- 51. Batie C. J., Kamin H. (1986) Association of ferredoxin-NADP+ reductase with NADP(H) specificity and oxidation-reduction properties. J. Biol. Chem. 261, 11214–11223 [PubMed] [Google Scholar]
- 52. Philo J. S., Lary J. W., Schuster T. M. (1988) Quaternary interactions in hemoglobin β-subunit tetramers kinetics of ligand binding and self-assembly. J. Biol. Chem. 263, 682–689 [PubMed] [Google Scholar]
- 53. Rifkind J. M., Nagababu E., Ramasamy S. (2011) The quaternary hemoglobin conformation regulates the formation of the nitrite-induced bioactive intermediate and the dissociation of nitric oxide from this intermediate. Nitric Oxide-Biol. Chem. 24, 102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Olson J. S. (1976) Effects of p-hydroxymercuribenzoate binding on the visible absorption spectrum of methemoglobin. J. Biol. Chem. 251, 441–446 [PubMed] [Google Scholar]
- 55. Olson J. S. (1976) Binding of inositol hexaphosphate to human methemoglobin. J. Biol. Chem. 251, 447–458 [PubMed] [Google Scholar]
- 56. Gibson Q. H., Gray R. D. (1970) The reaction of inositol hexaphosphate with hemoglobin. Biochem. Biophys. Res. Commun. 41, 415–420 [DOI] [PubMed] [Google Scholar]
- 57. Gray R. D., Gibson Q. H. (1971) The effect of inositol hexaphosphate on the kinetics of CO and O2 binding by human hemoglobin. J. Biol. Chem. 246, 7168–7174 [PubMed] [Google Scholar]
- 58. Cosby K., Partovi K. S., Crawford J. H., Patel R. P., Reiter C. D., Martyr S., Yang B. K., Waclawiw M. A., Zalos G., Xu X., Huang K. T., Shields H., Kim-Shapiro D. B., Schechter A. N., Cannon R. O., 3rd, Gladwin M. T. (2003) Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 9, 1498–1505 [DOI] [PubMed] [Google Scholar]
- 59. Nagababu E., Ramasamy S., Rifkind J. M. (2006) S-Nitrosohemoglobin: a mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide 15, 20–29 [DOI] [PubMed] [Google Scholar]
- 60. Keszler A., Piknova B., Schechter A. N., Hogg N. (2008) The reaction between nitrite and oxyhemoglobin. A mechanistic study. J. Biol. Chem. 283, 9615–9622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lundberg J. O., Gladwin M. T., Ahluwalia A., Benjamin N., Bryan N. S., Butler A., Cabrales P., Fago A., Feelisch M., Ford P. C., Freeman B. A., Frenneaux M., Friedman J., Kelm M., Kevil C. G., Kim-Shapiro D. B., Kozlov A. V., Lancaster J. R., Jr., Lefer D. J., McColl K., McCurry K., Patel R. P., Petersson J., Rassaf T., Reutov V. P., Richter-Addo G. B., Schechter A., Shiva S., Tsuchiya K., van Faassen E. E., Webb A. J., Zuckerbraun B. S., Zweier J. L., Weitzberg E. (2009) Nitrate and nitrite in biology, nutrition, and therapeutics. Nat. Chem. Biol. 5, 865–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lauer T., Preik M., Rassaf T., Strauer B. E., Deussen A., Feelisch M., Kelm M. (2001) Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc. Natl. Acad. Sci. U.S.A. 98, 12814–12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kleinbongard P., Dejam A., Lauer T., Rassaf T., Schindler A., Picker O., Scheeren T., Gödecke A., Schrader J., Schulz R., Heusch G., Schaub G. A., Bryan N. S., Feelisch M., Kelm M. (2003) Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic. Biol. Med. 35, 790–796 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.