

Background: SUR1 mutations generate hyperactive KATP channels that cause neonatal diabetes. The conformations underlying hyperactivity are not understood.
Results: Mutations outside the NBDs increase the affinity for nucleotides determined under non-hydrolytic equilibrium conditions.
Conclusion: An increased affinity for nucleotides at NBD2 of SUR1 can cause neonatal diabetes.
Significance: Nucleotide binding, without hydrolysis, appears sufficient to switch SUR1 to stimulatory conformations.
Keywords: ABC Transporter, Allosteric Regulation, Diabetes, Nucleotide, Potassium Channels, ABCC8, KATP Channel, SUR1, Neonatal Diabetes, Sulfonylurea
Abstract
KATP channels, (SUR1/Kir6.2)4 (sulfonylurea receptor type 1/potassium inward rectifier type 6.2) respond to the metabolic state of pancreatic β-cells, modulating membrane potential and insulin exocytosis. Mutations in both subunits cause neonatal diabetes by overactivating the pore. Hyperactive channels fail to close appropriately with increased glucose metabolism; thus, β-cell hyperpolarization limits insulin release. KATP channels are inhibited by ATP binding to the Kir6.2 pore and stimulated, via an uncertain mechanism, by magnesium nucleotides at SUR1. Glibenclamide (GBC), a sulfonylurea, was used as a conformational probe to compare nucleotide action on wild type versus Q1178R and R1182Q SUR1 mutants. GBC binds with high affinity to aporeceptors, presumably in the inward facing ATP-binding cassette configuration; MgATP reduces binding affinity via a shift to the outward facing conformation. To determine nucleotide affinities under equilibrium, non-hydrolytic conditions, Mg2+ was eliminated. A four-state equilibrium model describes the allosteric linkage. The KD for ATP4− is ∼1 versus 12 mm, Q1178R versus wild type, respectively. The linkage constant is ∼10, implying that outward facing conformations bind GBC with a lower affinity, 9–10 nm for Q1178R. Thus, nucleotides cannot completely inhibit GBC binding. Binding of channel openers is reported to require ATP hydrolysis, but diazoxide, a SUR1-selective agonist, concentration-dependently augments ATP4− action. An eight-state model describes linkage between diazoxide and ATP4− binding; diazoxide markedly increases the affinity of Q1178R for ATP4− and ATP4− augments diazoxide binding. NBD2, but not NBD1, has a higher affinity for ATP (and ADP) in mutant versus wild type (with or without Mg2+). Thus, the mutants spend more time in nucleotide-bound conformations, with reduced affinity for GBC, that activate the pore.
Introduction
Neuroendocrine ATP-sensitive K+ channels, (SUR1/Kir6.2)4, couple membrane electrical activity with cell metabolism in neurons and many endocrine cells, including pancreatic islet α- and β-cells (see Ref. 1 for a review). Loss of channel activity due to mutations in ABCC8 (gene encoding SUR1) or KCNJ11 (gene encoding Kir6.2) results in the excessive insulin release characteristic of hyperinsulinemic hypoglycemia, whereas gain-of-activity mutations that impair nucleotide regulation are a cause of neonatal diabetes (see Ref. 2 for a review). Neonatal diabetes mutations altering SUR1 “hyperactivate” the pore, thus increasing channel open probability (Po) which leads to membrane hyperpolarization, reduced activity of voltage-gated calcium channels, and hence less exocytosis of insulin (3). Patients with neonatal diabetes due to mutations in SUR1 are treatable with glibenclamide (GBC),2 a sulfonylurea commonly prescribed for type 2 diabetes; however, higher doses are typically needed to maintain glycemic control (4).
SUR1 has a canonical ABC transport protein “core” consisting of two nucleotide-binding domains (NBDs) and two six-helix bundles or transmembrane domains (TMDs). The SURs have an additional amino-terminal, five-helix bundle, TMD0, which interacts with Kir6.2 (5). The binding of ATP to ABC proteins results in the head-to-tail dimerization of the NBDs to produce two nucleotide-binding sites within the dimer interface. NBD dimerization reconfigures the TMDs from inward to outward facing conformations (6–11) (see Ref. 12 for a review). The binding of ATP, NBD dimerization, subsequent hydrolysis, and release of ADP·Pi produce the cyclic conformational changes that affect substrate transport across the cell membrane (see Refs. 12 and 13 for a review). SUR1 most closely resembles ABC exporters in which substrate binding is switched from high to low affinity upon binding of ATP. SUR1, like several other ABC proteins, has asymmetric NBDs (14–16) (see 17 for a review). The catalytic glutamate of NBD1 is substituted by an aspartate; affinity-labeling studies indicate that nucleotide dissociation constants for NBD1 are in the low micromolar range in the absence of Mg2+ (18). Mg2+ potentiates lower affinity nucleotide binding to NBD2 (19), where hydrolysis is reported to occur (19). Mg2+ is essential for ATP hydrolysis by SUR1 (20).
The transport properties, if any, of SURs are not known, but nucleotides do affect the binding of channel modulators, including sulfonylurea antagonists and agonist/openers, such as diazoxide, in a reciprocal manner. MgATP has a negative allosteric action on the binding of GBC to SUR1 (21–23), whereas the high affinity action of channel agonists is potentiated strongly by MgATP (24, 25). The data are consistent with the nucleotide dependence of transport substrate binding in ABC exporters and indicate that channel modulators interact differently with the enzymatic intermediates of SUR1.
The mechanism(s) underlying opening of the Kir pore by SUR1 are poorly understood; thus, the neonatal diabetes mutations may provide mechanistic insight into which conformations prompt channel openings. Here we focus on two mutations, Q1178R and R1182Q, located in the 15th helix (transmembrane helix) of the second transmembrane domain (TMD2). These substitutions are in a cluster of mutations that cause either neonatal diabetes (Q1178R, R1182Q, and A1184E) or hyperinsulinism (C1174F and S1185A). This cluster is spatially distinct from the NBDs, but Kir6.2/SUR1Q1178R channels display a strongly enhanced stimulation by MgATP (26). The results imply that MgATP shifts the distribution of SUR conformations or enzymatic intermediates toward those that stimulate openings of Kir6.2. The conventional view is that posthydrolytic, ADP-bound intermediates of SUR1 stimulate channel openings (i.e. that hydrolysis is essential for stimulation of KATP channel openings by SUR1) (27).
Using GBC as a reporter to probe nucleotide-driven changes in hyperactivating SUR1 mutants provides a means to better delineate the stimulatory conformation(s) and determine the molecular basis for channel overstimulation. ATP effectively reduced GBC binding in both wild and mutant receptors, presumably by switching from high affinity, inward facing to lower affinity, outward facing conformations. Eliminating Mg2+, a required enzymatic cofactor (20), showed that hydrolysis is not required; ATP4− concentration-dependently reduces the affinity for GBC. The elimination of Mg2+ allowed evaluation of the changes in nucleotide affinity due to the mutations.
In support of the hypothesis that ATP4− does switch SUR1 into a stimulatory conformation, we find that an agonist, diazoxide, stabilizes receptor intermediates with a reduced affinity for GBC in the presence but not the absence of ATP4−. The switching action of ATP4− requires that NBD2 be intact and functional; amino acid substitutions that affect nucleotide binding at NBD2 strongly diminish the allosteric action of ATP4− on SUR1Q1178R. The results imply that outward facing conformations with dimerized NBDs bind GBC and diazoxide with low and high affinity, respectively, and that the enhanced stimulatory action of Q1178R and R1182Q is due to their increased affinity for ATP and ADP. The data suggest that nucleotide-bound, outward facing conformations of SUR1 stimulate the channel, regardless of hydrolysis.
EXPERIMENTAL PROCEDURES
Cloning and Expression of WT and Mutant SUR1
The Abcc8/SUR1 hamster cDNA was subcloned under the control of the strong, methanol-inducible promoter AOX1 into the pSGP18 vector (28), a derivative of pPICZ (Invitrogen), by the ligation-independent method (29). Mutations and an amino-terminal His8 tag were introduced using standard site-directed mutagenesis methods and were confirmed by sequencing. The plasmids were transformed into Pichia pastoris strain KM71H by electroporation following standard procedures (Invitrogen). Transformants were selected on yeast peptone dextrose plates containing 1 mg/ml Zeocin. Transformants were cultured for 24 h in 10 ml of buffered minimal glycerol and then resuspended and cultured in buffered minimal methanol for an additional 24 h to induce protein expression. Membranes were isolated as described previously (30, 31) and then photolabeled with 1–3 nm [125I]azidoglibenclamide (32) and analyzed by SDS-PAGE and autoradiography to verify the presence of functional SUR1.
Large Scale P. pastoris Culture and Preparation of Microsomes
Overnight starter cultures (25 ml) were used to inoculate 1 liter of buffered minimal glycerol and grown to A600 = 20–30 (∼24 h) using the recommended conditions (Invitrogen). The cells were resuspended in buffered minimal methanol and cultured for 24 h, yielding, on average, 10–12 g of wet cells/liter. Cells were harvested and lysed, and microsomes were prepared as described previously (33).
GBC Saturation Experiments
Membranes containing WT or mutant SUR1 (∼150 pm) were suspended in a Mg2+-free physiological salt solution (139 mm NaCl, 5 mm KCl, 1 mm EDTA, 50 mm HEPES, pH 7.4) with increasing concentrations of 3H-labeled GBC for 30 min at 37 °C and then analyzed by rapid filtration, as described previously (21) to determine total binding. Nonspecific binding was determined in the presence of 1 μm unlabeled GBC (typically 10–15% of total binding). Specific GBC binding is defined as follows,
where Bmax is the total amount of receptor, G is the concentration of free 3H-labeled GBC in the reaction, KG is the equilibrium dissociation constant of GBC, and “nonspecific” is the amount of nonspecific binding.
[3H]GBC Binding Inhibition Experiments
Reaction conditions were similar to saturation experiments, except that 3H-labeled GBC was held fixed at 1 nm, and the reaction included the indicated concentrations of nucleotide and/or diazoxide. Experiments with MgATP included a creatine phosphokinase-based ATP-regenerating system to maintain a constant concentration of ATP over the 30-min incubation (34). The stability of ATP levels was verified using luciferase assays (Sigma; see supplemental material). MgADP-containing experiments included 10 mm AMP to inhibit endogenous adenylate kinases to reduce ATP production. Mg2+-containing experiments did not include EDTA. Nonspecific binding was determined in the presence of 1 μm unlabeled GBC and was typically 10–15% of total binding. The results are plotted as follows,
where X represents the reagent whose effect is being assayed (e.g. with or without ATP4−).
Homologous Competition Experiments
To quantify the allosteric relationship between GBC and ATP (at NBD2) on SUR1, a series of homologous competition experiments were performed at different ATP concentrations to yield an observed equilibrium dissociation constant for GBC, Kobs, for each given [ATP]. Binding assays were done in triplicate at two or three fixed concentrations of 3H-labeled GBC with increasing concentrations, 0.03 nm to 3 μm, of unlabeled GBC. The displacement data at a given concentration of ATP were fit globally in GraphPad (GraphPad Software, Inc.) using the equation,
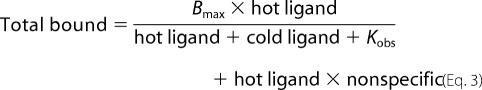 |
where “total bound” is the total amount of receptor occupied by 3H-labeled GBC (in dpm), Bmax is the total amount of receptors in the reaction, “hot ligand” is the concentration of 3H-labeled GBC, and “cold ligand” is the concentration of unlabeled GBC. Kobs is the observed equilibrium dissociation constant for GBC at a given [ATP], and “nonspecific” is the amount of nonspecific binding (in dpm). Reaction conditions were as described for the GBC binding reactions above.
Allosteric Analysis
The details of the analysis using four- and eight-state models are given in the supplemental material.
Photolabeling
∼20 μg of membrane protein was resuspended in 10 μl of TEE buffer (40 mm Tris, 100 μm EGTA, 1 mm EDTA) and 1 μm 8-N3-[γ-32P]ATP (Affinity Photoprobes, Lexington, KY) along with the indicated amounts of unlabeled nucleotide, incubated for 15 min on ice, and then irradiated on ice for 2 min at 254 nm and a distance of ∼7 cm (similar results obtained at 312 nm; data not shown). The samples were rapidly diluted in 800 μl of ice-cold TEE and pelleted at 200,000 × g for 30 min to remove free nucleotide. The membrane pellets were resuspended in Laemmli sample buffer and analyzed by SDS-PAGE and autoradiography. Autoradiograms were scanned and then analyzed using ImageJ software.
Statistics
The means ± S.E. are plotted; the number of replicate experiments varies, but n ≥ 3 in all cases. The t statistics and p values for fitting parameters to nonlinear models was done using Mathematica (Wolfram Research Inc., Champaign, IL).
RESULTS
GBC binding was used as a probe to compare nucleotide induced conformational changes in WT SUR1 versus two hyperactivating SUR1 mutants that produce neonatal diabetes. SUR1 is an ABC protein with ATPase activity (20). Fig. 1 gives a schematic diagram, a minimal model, representing the changes from inward to outward facing conformations of the ABC “core” of SUR1 during a nucleotide binding and hydrolysis cycle. This figure is based on work with several ABC proteins (6, 7, 35). The TMD0 domain, which couples SUR1 to the Kir6.2 pore is shown (5). ATP binds to both NBD1 and NBD2, but the SUR1 NBDs are not symmetric, and hydrolysis, in the intact receptor, is assumed to occur predominantly at NBD2 (19, 36). The sulfonylurea binding pocket is complex (37, 38), but residues in TMD2, spatially separate from the NBDs, affect binding (39, 40) (see Ref. 38 for a review). The addition of nucleotides reduces the apparent affinity for GBC, but the distinction between binding and hydrolysis is unresolved (21–23). We hypothesize that the inward facing, nucleotide-free conformation(s) of SUR1 has the highest affinity for sulfonylureas, whereas dimerization of the NBDs with switching to the outward facing configuration lowers the affinity. Although there are multiple estimates of the affinity of various sulfonylureas for apo-SUR1 and for mixtures of nucleotide-liganded states of SUR1 under steady-state conditions, there are none for the fully liganded receptor. Table 1 and Table S1 compare the dissociation constants, determined from saturation binding data, for the WT, Q1178R, and R1182Q aporeceptors (i.e. without nucleotides).
FIGURE 1.

A–D, a simplified enzymatic scheme correlating the binding of glibenclamide to distinct intermediates in the SUR1 hydrolysis cycle. GBC binds with nanomolar affinity to inward facing states (A and B). The binding of MgATP at NBD2 dimerizes the NBDs and reorients the TMDs to an outward facing conformation. C and D, hydrolysis at NBD2 and product release completes the cycle. MgATP has a negative allosteric coupling to GBC binding. This implies that the binding and/or hydrolysis of nucleotide at NBD2 is coupled to conformational changes that lower the affinity for GBC. The MgADP-bound posthydrolytic intermediate (D) is argued to be the conformation that activates the KIR pore, and hydrolysis has been reported to be necessary for the binding of diazoxide (Dz), a channel agonist. As shown here, we hypothesize that nucleotide binding is sufficient to bring about a conformational change that reciprocally affects the interactions with GBC and diazoxide. The TMD0 domain, which serves to couple SUR1 to Kir6.2, is added for completeness. E, neonatal diabetes (Q1178R in red and R1182Q in green) and hyperinsulinemia (C1174F in blue) causing mutations are clustered on transmembrane helix 15 (yellow), which feeds into NBD1 (light gray). Ser-1237 (purple) on the adjacent TM16 and Thr-1286/Met-1290 (orange) on TM17 have been shown to be important for the binding of sulfonylureas and openers (benzopyran and cyanoguanidine type), respectively (39–44) (see Ref. 38 for review). Panel E was prepared using DeepView and PovRay software.
TABLE 1.
KG of GBC for WT and neonatal diabetes SUR1
The dissociation constant, KG, of GBC for WT and neonatal diabetes SUR1 was determined in nucleotide-free conditions with 1 mm EDTA. Values are means ± S.E.
| SUR1 | KG |
|---|---|
| nm | |
| WT | 0.25 ± 0.02 |
| Q1178R | 1.0 ± 0.1 |
| R1182Q | 0.50 ± .15 |
Several channel agonists, including P1075 (N-cyano-N′-(1,1-dimethylpropyl)-N″-3-pyridylguanidine), which have a higher affinity for the closely related SUR2 receptors, bind at a more distal site(s) in TMD2 (41–44) (see Ref. 38 for a review), and their binding is potentiated by MgATP (24, 25, 42). Studies using chimeras of SUR1 and SUR2 (43) imply that diazoxide binds at a distinct site, which includes TMD1 and NBD1. Fig. 1E locates the cluster of hyperinsulinemia and neonatal diabetes mutations, referenced above, within a homology model of the carboxyl-terminal half of the SUR1 ABC core based on the bacterial ABC exporter, SAV1866 (7).
Q1178R and R1182Q Neonatal Diabetes Mutations Potentiate Negative Allosteric Action of MgATP on GBC Binding to SUR1
To assess quantitatively the effect of MgATP on GBC binding, membranes were incubated with increasing ATP concentrations maintained constant by a regenerating system (34); the level of ATP was constant over the 30-min time period of the incubation, as determined using luciferase assays (supplemental Fig. S1). Fig. 2A shows that under steady-state hydrolysis conditions, GBC binding to SUR1Q1178R and SUR1R1182Q is more sensitive to MgATP than WT SUR1. The IC50 values for MgATP are ∼850, 37, and 19 μm for the WT, R1182Q, and Q1178R receptors, respectively, an increase in the apparent affinity of SUR1Q1178R for ATP of ∼45-fold. In terms of an enzyme-substrate model, these differences could reflect changes in ATP binding and/or rates of hydrolysis but clearly show that the two mutant receptors spend more time in nucleotide-bound, outward facing conformations with a reduced affinity for GBC that can stimulate openings of Kir6.2 (26).
FIGURE 2.

Mutations in TMD2 increase the negative allosteric action of MgATP on GBC binding and increase the affinity for ATP4−. A, the curves through the data are logistic equations with IC50 values of 849 ± 195, 37 ± 13, and 19 ± 7 μm for WT, R1182Q, and Q1178R, respectively. The Hill coefficients (amplitudes in parentheses) are 0.78 ± 0.08 (0.8), 0.67 ± 0.08 (0.86), and 0.53 ± 0.04 (0.89). MgATP levels were kept constant using a regenerating system. B, the curves through the ATP4− data were calculated using a four-state allosteric model (inset and supplemental material). The model specifies the interactions of the SUR1 receptor (R) with GBC (G) and ATP4− (T) with dissociation constants KG and KT, respectively. β is an allosteric constant. The model assumes that NBD1 is occupied and that ATP binding to NBD2 induces a conformational change in the receptor, which reduces the affinity for GBC. The allosteric and ATP4− dissociation constants (β/KD) (μm) are 14/12,200, 6.8/1250, and 9.3/1000 for the WT, R1182Q, and Q1178R receptors, respectively. The dissociation constants for GBC are given in Table 1. Values are means ± S.E. (error bars); n = 4–6.
Q1178R and R1182Q Receptors Have Higher Affinity for ATP4− than WT
All ABC proteins have Walker-type nucleotide binding sites whose physiologic substrate is MgATP. Mg2+ binds to the β-phosphate of ATP and is a cofactor required for hydrolysis of ATP by SUR1 (20). To assess ATP binding without hydrolysis (i.e. under equilibrium rather than steady-state conditions), the changes in GBC binding induced by ATP without Mg2+ in the presence of 1 mm EDTA were determined). At pH 7.4, there is a mixture of ATP3− and ATP4−, but ATP4− is the predominant species, >90% calculated following Alberty (45). We use ATP4− as a descriptor and shorten HADP3− to ADP3−. Fig. 2B shows that ATP4− can reduce GBC binding in both WT and mutant receptors, presumably by switching SUR1 to lower affinity conformations. In terms of an enzyme-substrate model where hydrolysis is blocked, the results show that the mutant receptors have at least a 10-fold greater affinity for ATP4−, with Q1178R having a somewhat higher affinity than R1182Q (Tables 2 and S2). Comparison of Fig. 2A versus Fig. 2B implies that depleting Mg2+ increases the IC50 values of both WT and mutant receptors by at least 100-fold.
TABLE 2.
KT and β values for WT and neonatal diabetes SUR1 are shown ± S.E. (p values in parentheses)
| SUR1 | β | KT |
|---|---|---|
| μm | ||
| WT | 14 ± 10 (0.2) | 12,200 ± 4030 (0.02) |
| R1182Q | 6.8 ± 0.9 (<0.001) | 1250 ± 258 (0.003) |
| Q1178R | 9.3 ± 1.8 (<0.001) | 1000 ± 179 (<0.001) |
| Q1178R (Kobs method) | 10.5 ± 0.8 (0.005) | 846 ± 142 (0.03) |
Simplified Allosteric Model
Previous studies using affinity labeling with 8-N3-[32P]ATP show that NBD1 has a dissociation constant for NBD1 in the low micromolar range in the absence of Mg2+ (18), confirmed below for WT and Q1178R receptors. We hypothesize that the binding of a second ATP4− at NBD2 induces NBD dimerization and switches SUR1, mutant or WT, to conformations with a lower affinity for GBC. The four-state allosteric equilibrium model (inset in Fig. 2B and supplemental material) describes the linkage between occupation of the GBC binding site and ATP4− binding at NBD2. The derivation of the binding equations and methods for estimation of the binding parameters are given in the supplemental material. The curves through the SUR1Q1178R data points in Fig. 2B were calculated using the four-state model and KD values of 1 nm and 1000 μm for GBC and ATP4−, respectively, with an allosteric constant, β = 9.3. At saturating concentrations of ATP4−, the estimated affinity of the outward facing conformation for GBC, ∼9.3 nm, is given by βKG. This places a “floor” under the binding equilibrium. Thus, neither ATP4− nor MgATP will completely displace GBC from the receptor. Similarly, GBC binding reduces the apparent affinity of the receptors for ATP4−. The parameters for WT and R1182Q receptors are given in Table 2. The results show that the mutant receptors have at least a 10-fold greater affinity for nucleotide.
A second approach was used to measure the linkage between GBC and ATP4− binding to SUR1Q1178R and to estimate the ATP4− dissociation constant and β independently. The binding equation for the four-state model can be expressed as a simple binding isotherm (see supplemental material),
 |
where Kobs, the apparent dissociation constant at a specified concentration of ATP4−, is given by the following:
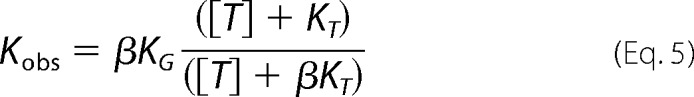 |
Kobs values at increasing concentrations of ATP4− were determined by homologous competition experiments (Fig. 3, A–D) and used to estimate KT and β. The results, β = 10.5 and KT = 846 μm (Table 2), agree with the values determined above.
FIGURE 3.

Evaluation of a four-state allosteric model linking the ATP and GBC binding sites in SUR1Q1178R. A–D, homologous displacement experiments, described under “Experimental Procedures,” were used to estimate Kobs at increasing concentrations of ATP4−. The displacement curves are based on the best fit values of Kobs at each [ATP4−] (shown in parentheses): specifically, 1.1 ± 0.1 nm (0), 5.5 ± 0.8 nm (7.5 mm), 7.0 ± 0.9 nm (17.5 mm), and 8.5 ± 1.3 nm (30 mm). E, plot of the Kobs data versus [ATP4−]. The curve is given by the equation, Kobs = βKG × ([ATP] + KT)/([ATP] + βKT), using the parameters KG = 1 nm, KT = 846 μm, and β = 10.5. The derivation of the Kobs equation and estimation of the parameters is given in the supplemental material. Values are the mean ± S.E. (error bars).
SUR1Q1178R Retains High Affinity ATP4− Binding at NBD1
The four-state model assumes that NBD1 is occupied and that binding of ATP4− at NBD2 causes dimerization and a shift to a lower affinity conformation. To determine if the Q1178R mutation affects binding at NBD1, WT and SUR1Q1178R receptors were labeled with 8-N3-[γ-32P]ATP plus increasing unlabeled ATP4−. Fig. 4 shows that both receptors are labeled; SUR1Q1178R has a 4-fold higher IC50 versus WT (∼40 versus 10 μm). The results indicate the Q1178R mutation somewhat reduces the apparent affinity of NBD1 for ATP4−, but NBD1 will be nearly saturated in both WT and SUR1Q1178R before there is a significant effect of ATP4− on GBC binding (compare Figs. 2B and 4).
FIGURE 4.

Q1178R retains a high affinity for ATP at NBD1. Membranes were photolabeled with 8-N3-[γ-32P]ATP (1 μm) in 1 mm EDTA. Labeled proteins were solubilized and separated as described under “Experimental Procedures.” Autoradiograms were prepared and quantified as described. A, the curves are best fits to a logistic equation. The IC50 for Q1178R is 39 ± 8 μm. The IC50 for the averaged values of WT and R1182Q is 9.8 ± 0.3 μm; the individual values are 10.1 ± 0.5 μm (WT) and 9.2 ± 0.8 μm (R1182Q). Values are means ± S.E. (error bars), n = 3. B, example autoradiograms comparing the labeling of WT and Q1178R receptors.
Diazoxide, KATP Channel Agonist, Stabilizes Low Affinity Conformation
To investigate further the idea that ATP4− binding is switching SUR1Q1178R to a conformation that stimulates channel activity, we tested whether diazoxide, a common KATP channel opener assumed to act by stabilizing a stimulatory state, can interact with SUR1Q1178R-ATP4− complexes. Diazoxide has been reported to require MgATP to stimulate KATP channels effectively (24, 46) and to interact with WT SUR1 with moderate affinity (24). Our previous interpretation has been that hydrolysis of MgATP is necessary for diazoxide binding (24) (i.e. that channel openers interact mainly with posthydrolytic conformations of SUR). Fig. 5 shows there is a clear linkage between ATP4− binding and the action of diazoxide in the absence of Mg2+ (i.e. under non-hydrolysis conditions).
FIGURE 5.

Diazoxide stabilizes the low affinity ATP4− bound state in SUR1Q1178R. A (inset), an eight-state allosteric model relating the binding of ATP4− (T), glibenclamide (G), and diazoxide (Z) to SUR1Q1178R (R) with dissociation constants, KT, KG, and KZ, respectively. α, β, and γ are allosteric constants. A, diazoxide reduces the binding of GBC to SUR1Q1178R in the presence of 2 mm ATP4− but has no effect in the absence of nucleotides. B, the reciprocal experiment demonstrates that 100 μm diazoxide potentiates the action of ATP4−. Values are means ± S.E. (error bars), n = 4–6.
An eight-state model that describes the linkage between the diazoxide, GBC, and nucleotide binding sites under equilibrium, non-hydrolysis conditions is given in Fig. 5. A derivation of the binding isotherm for this model is given in the supplemental material. Fig. 5A shows that diazoxide concentration-dependently reduces GBC binding to SUR1Q1178R in the presence of constant ATP4− (2 mm) but has no measurable effect without nucleotide. The final diazoxide concentration is limited by the solubility of the compound. Fig. 5B shows that adding diazoxide (100 μm) produces a left shift consistent with potentiation of the negative allosteric action of ATP4− on GBC binding to SUR1Q1178R. Overall, the data imply that ATP4− binds to both NBD1 and NBD2 in SUR1Q1178R, stabilizing a state with low affinity for glibenclamide and high affinity for diazoxide.
The parameters for the eight-state model were estimated as described in the supplemental material. Briefly, values of α = 0.0082, γ = 1.3, and KZ = 6.6 mm were obtained by fitting the eight-state binding isotherm to the combined data in Fig. 5, A and B, as described in the supplemental material.
The eight-state model provides semiquantitative insight into the linkage between the ATP, sulfonylurea, and diazoxide binding sites in SUR1Q1178R. Diazoxide binds very weakly to apo-SUR1, with a dissociation constant (KZ) of ∼6.6 mm, but more tightly to the ATP4−-bound conformation, αKZ ∼54 μm, an approximately 120-fold increase in affinity, 6600 μm/54 μm. Similarly, the diazoxide-bound form of the receptor has a significantly increased affinity, ∼120-fold, for ATP4−, αKT ∼8.2 μm versus KT ∼1000 μm. This is apparent from the left shift in the displacement curve when diazoxide (100 μm) is present (Fig. 5B).
Nucleotide Binding at NBD2 Is Necessary for Conformational Switching
The low affinity for ATP4− precludes using an 8-N3-32P analog to demonstrate nucleotide binding to NBD2. Therefore, based on studies with other ABC proteins, we mutated residues expected to reduce the affinity of NBD2 for ATP4− (47) (see Ref. 48 for a review) and assessed their effect on the ATP4− dependence of GBC binding and on affinity labeling of NBD1. Substitution of alanine for tyrosine at position 1354 (SUR1Y1354A), a substitution that in other ABC proteins lowers the affinity for ATP markedly, reduces the apparent affinity of SUR1Q1178R for ATP4− more than 100-fold (Fig. 6A). The Y1354A substitution in NBD2 leaves the apparent affinity for 8-N3-[32P]ATP at NBD1 essentially unchanged, 8 versus 10 μm (Figs. 4 and 6B). The results indicate that both NBDs bind ATP4− and are necessary for conformational switching and confirm that the affinity labeling is at NBD1.
FIGURE 6.

Functional binding at NBD2 is required for the ATP4− allosteric effect on SUR1Q1178R. A, the negative allosteric coupling between ATP4− and GBC binding to SUR1Q1178R is reduced significantly by a second mutation, Y1354A, in NBD2. The WT and Q1178R data and curves are taken from Fig. 2B. B, the Y1354A mutation does not significantly affect 8-N3-[γ-32P]ATP (1 μm) binding at NBD1. The IC50 values are 6 ± 0.9 and 10 ± 0.7 μm for SUR1Q1178R/Y1354A and WT SUR1, respectively. Values are means ± S.E. (error bars), n = 3.
Q1178R Receptor Has Higher Affinity for MgADP and ADP3−
MgADP stimulates KATP channel activity (49–53). This stimulation is usually assumed to imply that the posthydrolytic conformations of SUR1 stimulate KATP channel openings (e.g. see Ref. 27). We determined whether ADP will switch the conformation of SUR1Q1178R, the mutant with the higher affinity for ATP, and WT receptors. These experiments were carried out in the presence of 10 mm AMP to inhibit partially the generation of ATP by endogenous adenylate kinases (54). ATP was measured in these experiments using luciferase. Fig. 7 shows that MgADP has a stronger negative allosteric action on SUR1Q1178R than on WT receptors, IC50 values of 30 versus 200 μm, respectively. Additionally, the final displacements were greater for the mutant. Note that these are steady-state conditions, whereas the measured ATP values in these experiments are below 80 nm at ADP concentrations of ≤100 μm (see supplemental material); above this value, ATP binding and hydrolysis will make an increasing contribution and may determine the final plateau values. Interestingly, comparison with the MgATP data in Fig. 2A shows that MgADP switches both WT and SUR1Q1178R to the low affinity conformation more efficiently than MgATP.
FIGURE 7.

ADP can displace GBC from WT and Q1178R receptors. A logistic equation was used to estimate the IC50 values for displacement of GBC by MgADP. The IC50 values are 146 ± 47 and 24 ± 3 μm for WT and Q1178R, respectively. Mg2+ strongly potentiates ADP binding; ADP3− has a small, but significant, effect on Q1178R above 10 mm, but no detectable effect on WT at 100 mm. Values are means ± S.E. (error bars), n = 4–6.
To determine whether SUR1Q1178R has a higher affinity for ADP, GBC binding was carried out without Mg2+ with 1 mm EDTA present. At the highest concentration (100 mm), ADP3− reduced binding to SUR1Q1178R ∼35% but had no measurable effect on WT receptors. The data show the mutant receptor binds ADP3− weakly, albeit more tightly than WT. Although the ADP3− displacements are very far from saturation, Mg2+ must potentiate ADP binding by at least 10,000-fold. The results show that the occupation of the NBDs of SUR1Q1178R by MgADP switches or maintains the receptor in outward facing conformations with a reduced affinity for GBC. This implies that product dissociation, not hydrolysis per se, is required to return SUR1 to inward facing conformations with high affinity for GBC.
DISCUSSION
Previous studies demonstrated a negative allosteric effect of magnesium nucleotides on sulfonylurea receptors and established a concentration-dependent reduction in the binding of GBC to SURs with added MgATP and MgADP (21–23). These early studies provided no structural interpretation for these allosteric effects. Additionally, the data were usually interpreted to imply that hydrolysis was critical, if not required, particularly for the nucleotide effects on agonist binding (24). Here we relate the allosteric changes to the known dimerization of NBDs and switching of other ABC proteins from inward to outward facing conformations upon nucleotide binding. The results are consistent with the shifts in transport substrate affinity observed in other ABC transporters during a transport cycle (12) and imply that the apo- and single ATP-liganded, inward facing states have an approximately 10-fold greater affinity for GBC than outward facing conformations. Hydrolysis is not necessary for this switch; ATP4− induces the affinity shift in both mutant and WT receptors, albeit higher nucleotide concentrations are required for WT receptors, reflecting their lower affinity for ATP4−. Mg2+ strongly potentiates ATP and ADP binding to both WT and the two mutant receptors.
Depleting Mg2+ minimizes or eliminates ATP hydrolysis (20), allowing determination of the binding parameters under equilibrium versus steady-state hydrolysis conditions. The utility of the mutant receptors is that their increased affinity for nucleotides brings the ligand interactions into a range where they can be analyzed effectively. A four-state model was used to evaluate the allosteric linkage between ATP4− and GBC binding. The use of a simplified equilibrium model with binding of a single ATP is justified by the finding that the Q1178R substitution has only minor effects on the binding of ATP4− to NBD1. Therefore, NBD1 will be nearly saturated at ATP4− concentrations well below the IC50ATP for displacement of GBC, in which case the occupation of NBD2 induces the conformational switch. This idea is supported by the observation that a Y1354A substitution in NBD2, expected to reduce the affinity for nucleotides, strongly diminishes the action of ATP on GBC binding.
Although the affinities of the wild type and mutant receptors for ATP4− at NBD1 are similar as judged by affinity labeling, SUR1Q1178R binds ATP4− at NBD2 more tightly than WT. Although the displacement of GBC by ATP4− does not reach saturation by 100 mm, we used the four-state model to estimate, approximately, the KD for ATP4− at NBD2. The curve through the WT data in Fig. 2B is given by β ∼14, KG = 0.25 nm, and KT ∼12.2 mm, implying that ATP4− binding to WT SUR1 is at least 10-fold weaker versus SUR1Q1178R. The same argument holds for the R1182Q substitution (see Table 2). In an enzyme-substrate model, the results imply that SUR1Q1178R and SUR1R1182Q have a greater affinity for both ATP4− and MgATP. However, when Mg2+ is present, hydrolysis can occur, and the reduction in GBC binding can reflect both substrate interactions and hydrolysis.
Assessing the binding of ADP is more difficult. Pichia membrane preparations have endogenous adenylate kinase activity, which generates ATP and is only effectively suppressed by AMP at lower concentrations of ADP (see supplemental material). Under the conditions used, SUR1Q1178R apparently binds MgADP more tightly than WT receptors. As with ATP binding, Mg2+ markedly increases the apparent affinity for ADP in keeping with the Mg2+ coordination of the β-phosphate in several ABC structures. Depleting Mg2+ reduces the affinity of SUR1Q1178R for ADP3− by at least 4 orders of magnitude. The action of ADP3− is clearly not saturated in our experiments (∼35% displacement at 100 mm ADP3−; Fig. 7); thus, this estimate is provisional. ADP3− (100 mm) had no significant effect on GBC binding to WT receptors, implying that the affinity is quite weak. In an enzyme-substrate model, the results show that dissociation of ADP, not hydrolysis per se, is necessary for SUR1 to switch back to conformations with high affinity for GBC. In other words, the continued occupation of NBD2 by ADP maintains SUR1 in outward facing conformations with reduced affinity for sulfonylureas, which are presumably able to stimulate openings of Kir6.2.
Our results imply that the hyperactivation of Kir6.2 by SUR1Q1178R (26), and presumably by SUR1R1182Q, is a consequence of the greater affinity of the mutant receptors for nucleotides. To test the hypothesis that ATP4− switches SUR1 into conformations able to stimulate Kir6.2, we assessed the linkage between the binding of ATP4− and diazoxide, a known agonist of SUR1/Kir6.2 KATP channels. ATP4− stabilizes conformations of SUR1Q1178R with a higher affinity for diazoxide, and diazoxide potentiates the inhibitory effect of ATP4−. Analysis using an eight-state equilibrium model suggests that the SUR1Q1178R-ATP4− complex binds diazoxide ∼120-fold more tightly that the aporeceptor (KZ ∼6600 μm versus αKZ ∼54 μm, using values for KZ and α estimated by globally fitting data sets obtained by varying either [diazoxide] or [ATP4−]). Similarly, diazoxide-SUR1Q1178R binds ATP4− ∼120-fold more tightly than SUR1Q1178R. These results differ from earlier studies (24), which implied that KATP channel agonists, like diazoxide, bind with appreciable affinity only to posthydrolytic conformations of SUR1 (i.e. states with ADP bound to NBD2). Our results show that under non-hydrolytic conditions (i.e. without Mg2+) there is a strong, allosteric linkage between the ATP, diazoxide, and GBC binding sites. The results are consistent with the hypothesis that ATP binding and consequent NBD dimerization are sufficient to switch SUR1 to conformations that interact with diazoxide and stimulate the opening of the Kir6.2 pore. By analogy with other ABC proteins, we hypothesize that the stimulatory conformation is the outward facing state.
The negative allosteric linkage between ATP and sulfonylurea binding is broadly consistent with the reported need for higher doses of sulfonylureas to treat neonatal diabetes (4) (see Ref. 2 for a review). Assuming that pancreatic β-cells have an intracellular [ATP]c in the millimolar range, the results in Fig. 2A suggest that a greater percentage of the mutant receptors will be in conformations with a reduced affinity for sulfonylureas versus WT receptors.
Supplementary Material
This work was supported by American Diabetes Association Grant ADA 1–10-BS21 (to J. B.).

This article contains supplemental Tables S1 and S2 and Fig. S1.
- ABC
- ATP-binding cassette
- NBD
- nucleotide-binding domain
- GBC
- glibenclamide
- TMD
- transmembrane domain
- SUR
- sulfonylurea receptor
- 8-N3-[32P]ATP
- 8-azidoadenosine 5′-triphosphate.
REFERENCES
- 1. Aguilar-Bryan L., Bryan J. (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20, 101–135 [DOI] [PubMed] [Google Scholar]
- 2. Aguilar-Bryan L., Bryan J. (2008) Neonatal diabetes mellitus. Endocr. Rev. 29, 265–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Babenko A. P., Polak M., Cavé H., Busiah K., Czernichow P., Scharfmann R., Bryan J., Aguilar-Bryan L., Vaxillaire M., Froguel P. (2006) Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 355, 456–466 [DOI] [PubMed] [Google Scholar]
- 4. Rafiq M., Flanagan S. E., Patch A. M., Shields B. M., Ellard S., Hattersley A. T. (2008) Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care 31, 204–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Babenko A. P., Bryan J. (2003) SUR domains that associate with and gate KATP pores define a novel gatekeeper. J. Biol. Chem. 278, 41577–41580 [DOI] [PubMed] [Google Scholar]
- 6. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 8. Kreimer D. I., Chai K. P., Ferro-Luzzi Ames G. (2000) Nonequivalence of the nucleotide-binding subunits of an ABC transporter, the histidine permease, and conformational changes in the membrane complex. Biochemistry 39, 14183–14195 [DOI] [PubMed] [Google Scholar]
- 9. Manciu L., Chang X. B., Buyse F., Hou Y. X., Gustot A., Riordan J. R., Ruysschaert J. M. (2003) Intermediate structural states involved in MRP1-mediated drug transport. Role of glutathione. J. Biol. Chem. 278, 3347–3356 [DOI] [PubMed] [Google Scholar]
- 10. Mannering D. E., Sharma S., Davidson A. L. (2001) Demonstration of conformational changes associated with activation of the maltose transport complex. J. Biol. Chem. 276, 12362–12368 [DOI] [PubMed] [Google Scholar]
- 11. Sonveaux N., Vigano C., Shapiro A. B., Ling V., Ruysschaert J. M. (1999) Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. A tryptophan fluorescence quenching analysis. J. Biol. Chem. 274, 17649–17654 [DOI] [PubMed] [Google Scholar]
- 12. Higgins C. F., Linton K. J. (2004) The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11, 918–926 [DOI] [PubMed] [Google Scholar]
- 13. Rees D. C., Johnson E., Lewinson O. (2009) ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 10, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lubelski J., van Merkerk R., Konings W. N., Driessen A. J. (2006) Nucleotide-binding sites of the heterodimeric LmrCD ABC-multidrug transporter of Lactococcus lactis are asymmetric. Biochemistry 45, 648–656 [DOI] [PubMed] [Google Scholar]
- 15. Procko E., Ferrin-O'Connell I., Ng S. L., Gaudet R. (2006) Distinct structural and functional properties of the ATPase sites in an asymmetric ABC transporter. Mol. Cell 24, 51–62 [DOI] [PubMed] [Google Scholar]
- 16. Qin L., Zheng J., Grant C. E., Jia Z., Cole S. P., Deeley R. G. (2008) Residues responsible for the asymmetric function of the nucleotide binding domains of multidrug resistance protein 1. Biochemistry 47, 13952–13965 [DOI] [PubMed] [Google Scholar]
- 17. Procko E., O'Mara M. L., Bennett W. F., Tieleman D. P., Gaudet R. (2009) The mechanism of ABC transporters. General lessons from structural and functional studies of an antigenic peptide transporter. FASEB J. 23, 1287–1302 [DOI] [PubMed] [Google Scholar]
- 18. Matsuo M., Tanabe K., Kioka N., Amachi T., Ueda K. (2000) Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J. Biol. Chem. 275, 28757–28763 [DOI] [PubMed] [Google Scholar]
- 19. Matsuo M., Kioka N., Amachi T., Ueda K. (1999) ATP binding properties of the nucleotide-binding folds of SUR1. J. Biol. Chem. 274, 37479–37482 [DOI] [PubMed] [Google Scholar]
- 20. de Wet H., Mikhailov M. V., Fotinou C., Dreger M., Craig T. J., Vénien-Bryan C., Ashcroft F. M. (2007) Studies of the ATPase activity of the ABC protein SUR1. FEBS J. 274, 3532–3544 [DOI] [PubMed] [Google Scholar]
- 21. Hambrock A., Löffler-Walz C., Quast U. (2002) Glibenclamide binding to sulphonylurea receptor subtypes. Dependence on adenine nucleotides. Br. J. Pharmacol. 136, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwanstecher M., Löser S., Rietze I., Panten U. (1991) Phosphate and thiophosphate group donating adenine and guanine nucleotides inhibit glibenclamide binding to membranes from pancreatic islets. Naunyn Schmiedebergs Arch Pharmacol. 343, 83–89 [DOI] [PubMed] [Google Scholar]
- 23. Schwanstecher M., Löser S., Brandt C., Scheffer K., Rosenberger F., Panten U. (1992) Adenine nucleotide-induced inhibition of binding of sulphonylureas to their receptor in pancreatic islets. Br J. Pharmacol. 105, 531–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwanstecher M., Sieverding C., Dörschner H., Gross I., Aguilar-Bryan L., Schwanstecher C., Bryan J. (1998) Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J. 17, 5529–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hambrock A., Löffler-Walz C., Kurachi Y., Quast U. (1998) Mg2+ and ATP dependence of KATP channel modulator binding to the recombinant sulphonylurea receptor, SUR2B. Br J. Pharmacol. 125, 577–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Babenko A. P. (2008) A novel ABCC8 (SUR1)-dependent mechanism of metabolism-excitation uncoupling. J. Biol. Chem. 283, 8778–8782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zingman L. V., Alekseev A. E., Bienengraeber M., Hodgson D., Karger A. B., Dzeja P. P., Terzic A. (2001) Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 31, 233–245 [DOI] [PubMed] [Google Scholar]
- 28. Chloupková M., Pickert A., Lee J. Y., Souza S., Trinh Y. T., Connelly S. M., Dumont M. E., Dean M., Urbatsch I. L. (2007) Expression of 25 human ABC transporters in the yeast Pichia pastoris and characterization of the purified ABCC3 ATPase activity. Biochemistry 46, 7992–8003 [DOI] [PubMed] [Google Scholar]
- 29. Doyle S. A. (2005) High-throughput cloning for proteomics research. Methods Mol. Biol. 310, 107–113 [DOI] [PubMed] [Google Scholar]
- 30. Lerner-Marmarosh N., Gimi K., Urbatsch I. L., Gros P., Senior A. E. (1999) Large scale purification of detergent-soluble P-glycoprotein from Pichia pastoris cells and characterization of nucleotide binding properties of wild-type, Walker A, and Walker B mutant proteins. J. Biol. Chem. 274, 34711–34718 [DOI] [PubMed] [Google Scholar]
- 31. Urbatsch I. L., Beaudet L., Carrier I., Gros P. (1998) Mutations in either nucleotide-binding site of P-glycoprotein (Mdr3) prevent vanadate trapping of nucleotide at both sites. Biochemistry 37, 4592–4602 [DOI] [PubMed] [Google Scholar]
- 32. Schwanstecher M., Löser S., Chudziak F., Bachmann C., Panten U. (1994) Photoaffinity labeling of the cerebral sulfonylurea receptor using a novel radioiodinated azidoglibenclamide analogue. J. Neurochem. 63, 698–708 [DOI] [PubMed] [Google Scholar]
- 33. Wang Z., Stalcup L. D., Harvey B. J., Weber J., Chloupkova M., Dumont M. E., Dean M., Urbatsch I. L. (2006) Purification and ATP hydrolysis of the putative cholesterol transporters ABCG5 and ABCG8. Biochemistry 45, 9929–9939 [DOI] [PubMed] [Google Scholar]
- 34. Löffler-Walz C., Quast U. (1998) Binding of KATP channel modulators in rat cardiac membranes. Br J. Pharmacol. 123, 1395–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dawson R. J., Locher K. P. (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935–938 [DOI] [PubMed] [Google Scholar]
- 36. Ueda K., Inagaki N., Seino S. (1997) MgADP antagonism to Mg2+-independent ATP binding of the sulfonylurea receptor SUR1. J. Biol. Chem. 272, 22983–22986 [DOI] [PubMed] [Google Scholar]
- 37. Bryan J., Vila-Carriles W. H., Zhao G., Babenko A. P., Aguilar-Bryan L. (2004) Toward linking structure with function in ATP-sensitive K+ channels. Diabetes 53, Suppl. 3, S104–S112 [DOI] [PubMed] [Google Scholar]
- 38. Bryan J., Crane A., Vila-Carriles W. H., Babenko A. P., Aguilar-Bryan L. (2005) Insulin secretagogues, sulfonylurea receptors and KATP channels. Curr. Pharm. Des. 11, 2699–2716 [DOI] [PubMed] [Google Scholar]
- 39. Ashfield R., Gribble F. M., Ashcroft S. J., Ashcroft F. M. (1999) Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes 48, 1341–1347 [DOI] [PubMed] [Google Scholar]
- 40. Babenko A. P., Gonzalez G., Bryan J. (1999) The tolbutamide site of SUR1 and a mechanism for its functional coupling to KATP channel closure. FEBS Lett. 459, 367–376 [DOI] [PubMed] [Google Scholar]
- 41. Hambrock A., Kayar T., Stumpp D., Osswald H. (2004) Effect of two amino acids in TM17 of Sulfonylurea receptor SUR1 on the binding of ATP-sensitive K+ channel modulators. Diabetes 53, Suppl. 3, S128–S134 [DOI] [PubMed] [Google Scholar]
- 42. Schwanstecher M., Brandt C., Behrends S., Schaupp U., Panten U. (1992) Effect of MgATP on pinacidil-induced displacement of glibenclamide from the sulphonylurea receptor in a pancreatic beta-cell line and rat cerebral cortex. Br. J. Pharmacol. 106, 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Babenko A. P., Gonzalez G., Bryan J. (2000) Pharmaco-topology of sulfonylurea receptors. Separate domains of the regulatory subunits of KATP channel isoforms are required for selective interaction with K+ channel openers. J. Biol. Chem. 275, 717–720 [DOI] [PubMed] [Google Scholar]
- 44. Moreau C., Jacquet H., Prost A. L., D'hahan N., Vivaudou M. (2000) The molecular basis of the specificity of action of KATP channel openers. EMBO J. 19, 6644–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alberty R. A. (2003) Thermodynamics of Biochemical Reactions, pp. 5–11, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 46. Shyng S., Ferrigni T., Nichols C. G. (1997) Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol. 110, 643–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim I. W., Peng X. H., Sauna Z. E., FitzGerald P. C., Xia D., Müller M., Nandigama K., Ambudkar S. V. (2006) The conserved tyrosine residues 401 and 1044 in ATP sites of human P-glycoprotein are critical for ATP binding and hydrolysis. Evidence for a conserved subdomain, the A-loop in the ATP-binding cassette. Biochemistry 45, 7605–7616 [DOI] [PubMed] [Google Scholar]
- 48. Ambudkar S. V., Kim I. W., Xia D., Sauna Z. E. (2006) The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for ATP binding. FEBS Lett. 580, 1049–1055 [DOI] [PubMed] [Google Scholar]
- 49. Dunne M. J., Petersen O. H. (1986) Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Lett. 208, 59–62 [DOI] [PubMed] [Google Scholar]
- 50. Findlay I. (1988) Effects of ADP upon the ATP-sensitive K+ channel in rat ventricular myocytes. J. Membr. Biol. 101, 83–92 [DOI] [PubMed] [Google Scholar]
- 51. Kakei M., Kelly R. P., Ashcroft S. J., Ashcroft F. M. (1986) The ATP sensitivity of K+ channels in rat pancreatic B-cells is modulated by ADP. FEBS Lett. 208, 63–66 [DOI] [PubMed] [Google Scholar]
- 52. Lederer W. J., Nichols C. G. (1989) Nucleotide modulation of the activity of rat heart ATP-sensitive K+ channels in isolated membrane patches. J. Physiol. 419, 193–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Misler S., Falke L. C., Gillis K., McDaniel M. L. (1986) A metabolite-regulated potassium channel in rat pancreatic B cells. Proc. Natl. Acad. Sci. U.S.A. 83, 7119–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rosano T. G., Clayson K. J., Strandjord P. E. (1976) Evaluation of adenosine 5′-monophosphate and fluoride as adenylate kinase inhibitors in the creatine kinase assay. Clin. Chem. 22, 1078–1083 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.