

Abstract
The transcription factor LSF (Late SV40 Factor), also known as TFCP2, belongs to the LSF/CP2 family related to Grainyhead family of proteins and is involved in many biological events, including regulation of cellular and viral promoters, cell cycle, DNA synthesis, cell survival and Alzheimer’s disease. Our recent studies establish an oncogenic role of LSF in Hepatocellular carcinoma (HCC). LSF overexpression is detected in human HCC cell lines and in more than 90% cases of human HCC patients, compared to normal hepatocytes and liver, and its expression level showed significant correlation with the stages and grades of the disease. Forced overexpression of LSF in less aggressive HCC cells resulted in highly aggressive, angiogenic and multi-organ metastatic tumors in nude mice. Conversely, inhibition of LSF significantly abrogated growth and metastasis of highly aggressive HCC cells in nude mice. Microarray studies revealed that as a transcription factor LSF modulated specific genes regulating invasion, angiogenesis, chemoresistance and senescence. LSF transcriptionally regulates thymidylate synthase (TS) gene, thus contributing to cell cycle regulation and chemoresistance. Our studies identify a network of proteins, including osteopontin (OPN), Matrix metalloproteinase-9 (MMP-9), c-Met and complement factor H (CFH), that are directly regulated by LSF and play important role in LSF-induced hepatocarcinogenesis. A high throughput screening identified small molecule inhibitors of LSF DNA binding and the prototype of these molecules, Factor Quinolinone inhibitor 1 (FQI1), profoundly inhibited cell viability and induced apoptosis in human HCC cells without exerting harmful effects to normal immortal human hepatocytes and primary mouse hepatocytes. In nude mice xenograft studies, FQI1 markedly inhibited growth of human HCC xenografts as well as angiogenesis without exerting any toxicity. These studies establish a key role of LSF in hepatocarcinogenesis and usher in a novel therapeutic avenue for HCC, an invariably fatal disease.
Keywords: Late SV40 Factor (LSF), hepatocellular carcinoma (HCC), osteopontin (OPN), matrix metalloproteinase-9 (MMP-9), c-Met, thymidylate synthase (TS), angiogenesis, metastasis, cell cycle regulation, small molecule inhibitors, FQI1
Hepatocellular carcinoma: the clinical scenario
Hepatocellular carcinoma (HCC) is a major global health problem and one of the five most common cancers worldwide [1,2]. Although overall incidence of cancers is decreasing, the incidence of HCC is increasing significantly and it is the third highest cause of cancer-related death globally [3]. In the US, the estimated new cases of HCC for 2011 were 26,190 out of which 19,590 were expected to die [3]. In Asian countries Hepatitis B virus (HBV) infection is the predominant cause of HCC while in Western countries Hepatitis C virus (HCV) infection and alcoholism play the major role [1,4]. Dietary toxin, such as aflatoxin, is a major cause of HCC in sub-Saharan Africa. These diverse etiologies along with enumerable modulations in gene expression, signaling pathways and mutations have posed a major impediment in understanding the pathophysiology of this complex disease and developing effective therapeutic strategies. HCC is a tumor with rapid growth and early vascular invasion so that most HCC patients present with advanced symptomatic tumors that are not amenable to surgical resection [5]. Even after surgical resection, the recurrence rate is very high [6]. The tumors are resistant to conventional chemotherapy and no systemic therapy is available for the metastatic disease [7-9]. The treatment options for HCC depend upon the stages and grades of the disease [10]. Surgical resection, radiofrequency ablation and liver transplantations are the treatments of choice for the localized disease [11,12]. However, surgical resection or transplantation is limited to very few cases since most HCC patients present with advanced symptomatic tumors with underlying cirrhotic changes. Transarterial chemoembolization (TACE) and systemic therapy with doxorubicin alone or a combination of cisplatin, interferon, doxorubicin and 5-fluorouracil (PIAF) are being used for advanced disease with moderate improvement in overall survival duration varying between 6.8 months to 8.6 months [9,12-16]. Sorafenib, an inhibitor of c-Raf and BRaf kinases as well as of vascular endothelial growth factor receptor (VEGFR) family, has been introduced in the clinics following Phase III clinical trials [17,18]. While the median survival for placebo-treated patients was approximately 7.9 months, sorafenib-treated patients survived 10.7 months [17]. Bevacizumab, a vascular endothelial growth factor (VEGF) pathway inhibitor, either alone or in combination with chemotherapy, also demonstrates very limited response [11,19]. In view of this dire scenario, understanding the molecular pathogenesis of HCC and developing targeted and effective treatments are mandatory to significantly increase the survival interval and ameliorate the sufferings of the patients.
The transcription factor Late SV40 Factor (LSF/TFCP2)
LSF is a ubiquitous transcription factor involved in many biological processes, including cell cycle, cell growth and development [20,21]. Using HeLa cells LSF was first identified as a transcriptional activator of the major late Simian Virus 40 promoter [22]. Simultaneous cloning of the cDNA as a transcriptional regulator of additional genes led to multiple nomenclature for LSF, including LBP-1 or UBP-1 (HIV long terminal repeat), CP2 (murine α-globin) and SEF-1 (murine serum amyloid A3) [20,23-28]. The gene encoding for LSF is identified as TFCP2. LSF belongs to an evolutionary conserved family of transcription factors consisting of LSF/CP2 subfamily and grainyhead (GRH) subfamily [29-31]. Human LSF subfamily includes LSF (chromosome 12q13), LBP-1a/b (chromosome 3) and LBP-9 (chromosome 2) [32,33]. The human LSF is a 502 amino acids protein with a predicted molecular mass of 57 kDa (Figure 1).
Figure 1.

Schematic structure of human LSF protein. The numbers indicate amino acid residues. NLS: nuclear localizationsignal.
As a transcription factor LSF binds DNA predominantly as a homotetramer [34,35]. The DNA binding region of LSF spans amino acid residues 67-260 and shows structural similarity to p53/p63/p73 family of proteins [21,36,37]. Two mutations in the DNA binding region, Q234L and K236E, prevent LSF DNA binding and generate a dominant negative inhibitor (LSFdn) [37,38]. The DNA recognition sequence of LSF spans 15 base pairs containing two half sites separate by a spacer. The consensus sequence is CNRG-N6-CNR(G/C) in which the first half site is more conserved than the second [39]. The C-terminal region of LSF mediates oligomerization, either dimerization or tetramerization. The region containing 326 to 389 amino acid residues function as the tetramerization domain and is structurally similar to the SAM protein-protein interaction domain [34]. Deletion mutation analysis revealed the region containing residues 448 to 502 as the dimerization domain which is structurally similar to ubiquitin-like fold domain [34,36]. The amino acid residues 189 to 239 mediate nuclear localization of LSF [40,41].
In addition to homotypic interaction, LSF interacts with other members of the LSF subfamily, such as LBP-1a/b [28], and also with unrelated proteins to generate novel DNA-binding complexes. LSF partners with NF-E4 in a particular stage of erythroid development leading to a stage- and tissue-specific transcriptional activity regulating expression of the β-globin gene cluster [42,43]. LSF interacts with transcriptional repressor YY1 to inhibit transcription from HIV LTR [44,45]. Additionally LSF has been identified in a switch recombination site-binding complex in B cells and the IL-4 promoter-binding complex in Jurkat cells, although the LSF-interacting partners in these complexes remain to be identified [40,46].
Regulation of transcription by LSF: Functional significance
Viral promoters
LSF functions both as a transcription activator and repressor and regulates a variety of viral and cellular promoters. LSF stimulates transcription at initiation sites L325 and L264 of the SV40 late promoter which are the major transcription sites utilized after DNA replication during the SV40 lytic cycle, indicating a potential role of LSF in early to late transcriptional switch in SV40 lytic cycle [22]. On the other hand LSF binds to sequences within the HIV-1 long terminal repeat (LTR) initiation region and recruits YY1 and histone deacetylase 1 (HDAC1) to the LTR, inhibiting transcription and thereby contributing to HIV persistence within resting CD4+ T cells [44,45]. Phosphorylation of LSF by ERK decreases and by p38MAPK increases binding of LSF to the HIV LTR and HIV was recovered from the resting CD4+ T cells of avire-mic, HIV-infected donors upon treatment of these cells with specific p38MAPK inhibitors indicating a potential role of these inhibitors in disrupting latent HIV infection [47].
Cellular promoters
Globin genes
LSF performs a lineage-specific function in erythroid cells regulating α, β and γ-globin gene expression. The human β-like globin genes (ε, Aγ, Gγ, δ and β) exist in a cluster and are expressed in a temporal and tissue-specific manner [42]. Expression of the ε-gene occurs until the twelfth week of gestation; the γ genes are expressed predominantly in the fetal liver until birth when a switch to adult δ and β gene expression occurs. The competitive silencing of the β-gene in the fetal developmental stage is mediated by the stage selector element (SSE) located in the -50 region of the γ-promoter. It was demonstrated that LSF binds as a heterodimer with the erythroid-specific factor NF-E4 to the SSE thus suppressing adult globin gene expression and allowing both ε and γ globin gene expression [42,43].
In mouse erythroid cells LSF binds as a hetero-tetramer to the adult α-globin promoter, inducing its expression [23,24,48]. It was shown that small ubiquitin-like-modifier (SUMO) E3 ligase PIAS1 facilitates nuclear translocation of LSF in a sumoylation-independent manner and a hexameric complex containing two units each of LSF, PIAS1 and another member of LSF family CP2b [49]. Whether similar mechanism of gene regulation also occurs for human α-globin remains to be determined.
Uroporphyrinogen III synthase: regulation of heme synthesis
Congenital erythropoietic porphyria (CEP), an autosomal recessive inborn error of heme biosynthesis, results from the markedly deficient activity of uroporphyrinogen III synthase (URO synthase). The erythroid-specific promoter of URO synthase gene is regulated by GATA1 and LSF and mutation in the GATA1 and LSF binding sites in the promoter region of human URO synthase gene has been identified in CEP patients [50]. Mutation in the LSF binding site resulted in profound inhibition in promoter activity and URO synthase enzymatic activity and patients harboring such mutation presented severe transfusion-dependent disease indicating the importance of LSF in regulating heme biosynthesis.
Glycogen synthase kinase3 β (GSK3β) and transferrin: link to Alzheimer’s disease (AD)
Alzheimer’s disease (AD) is characterized by the deposition of neuritic plaques which are predominantly composed of amyloid β (Aβ) peptides derived from amyloid precursor proteins (APP) [51]. APP family proteins also include two APP-like proteins, APLP1 and APLP2. It was reported that the intracellular domain (ICD) of APLP2 translocates to the nucleus where it interacts with LSF and promotes LSF-mediated transcription of glycogen synthase kinase3 β (GSK3β) [52]. The functional significance of this event in the pathogenesis of AD was documented by increased phosphorylation of tau (τ), a substrate of GSK3β the hyperphosphorylated forms of which are a component of neurofibrillary tangles, upon treatment with APLP2-ICD; protection by LiCl, a GSK3β inhibitor, from APLP2-ICD-induced cell killing in differentiated PC12 cells; and increased staining for GSK3β in AD patients [52].
Transferrin, an iron regulatory protein, is up-regulated in AD frontal cortex [53]. It was demonstrated that Aβ peptides induce transferrin expression by promoting binding of LSF to transferrin promoter [54]. Overexpression of LSF induced while shRNA-mediated knockdown of LSF downregulated both basal and Aβ-induced expression of transferrin. Abnormalities in iron distribution is observed in AD patients, thus LSF contributes to AD by multiple mechanisms. Interestingly epidemiologic studies have demonstrated an association between a non-coding polymorphism (G to A) in the 3'-untranslated region of LSF gene in AD patients with the A allele showing a protective effect [55-58].
Interleukin-4 (IL-4): allergic response
IL-4 promotes the differentiation of T helper 2 (Th2) subset of CD4+ T cells, thereby regulating allergic and humoral responses [59]. It was demonstrated that overexpression of LSF significantly augmented IL-4 promoter activity in human Jurkat T cells and binding of LSF to the IL-4 promoter was confirmed by the electrophoretic mobility shift assay [46]. Additionally, overexpression of LSF augmented, while a dominant negative mutant, lacking the DNA binding domain, inhibited IL-4 mRNA expression in murine Th2 clone D10 [46]. However, LSF did not induce promoter activity of IL-2, a Th1 cell-associated cytokine, indicating that LSF might be involved in regulating Th2 differentiation and function.
Acute phase proteins: link to inflammation
Following tissue injury or infection, proinflammatory cytokines, such as IL-1 or IL-6 released from macrophages or monocytes, trigger the release of acute phase proteins from the liver. The promoter region of the mouse gene encoding major acute-phase protein Serum Amyloid A3 (SAA3) contains an enhancer region to which CCAAT/enhancer-binding protein (C/EBP) and LSF bind [27,60,61]. Overexpression of LSF increased SAA3 promoter activity which was further enhanced by IL-1 treatment. It was shown that IL-1 treatment resulted in NF-κB activation and a putative NF-κB binding sequence was identified in the same region where LSF binds albeit with a significantly weak affinity for NF-κB [60]. Immunoprecipitation with anti-LSF antibody pulled down both LSF and the p65 subunit of NF-κB upon IL-1 treatment and the authors hypothesized that LSF might interact with NF-κB and facilitate cytokine-mediated activation of SAA3 gene by stabilizing NF-κB binding [60]. Thus LSF might be involved in acute inflammatory response. A major limitation of these studies is that they are restricted to SAA3 promoter analysis without showing changes in SAA3 mRNA or protein levels. Additionally, whether similar regulatory mechanism is also involved in human acute phase proteins has not been elucidated.
PAX6 and αA-crystallin: regulation of eye function
The transcription factor PAX6 is a master regulator of eye development and is regulated by two promoters P0 and P1, the latter directing PAX6 expression in the eye [62]. It was demonstrated that LSF plays a major role in regulating P1 promoter activity of human Pax6 gene [63]. Both LSF and Sp1 enhanced P1 promoter activity and co-immunoprecipitation studies documented a potential interaction between LSF and Sp1. Crystallins are water-soluble components of ocular lens and LSF binds to the promoter of chicken αA-crystallin gene in a lens specific manner and positively regulates promoter activity [35]. These two independent studies in two species suggest a potential role of LSF in regulating eye development. However, both these studies focused only on promoter activity analysis without showing changes at the mRNA or protein levels upon LSF inhibition.
LSF and immunoglobulin (Ig) class switch recombination
The expression of Ig isotypes is determined by Ig heavy chain class switch recombination. The upstream region of each heavy chain gene segment contains a switch (S) region where nonhomologous recombination occurs and it was demonstrated that LSF and its paralog LBP-1a binds to both Sμ and Sα region, the switch regions of human IgM and IgA heavy chain gene segments, but not to Sγ1, the switch region for IgG1 [40,64]. Overexpression of LSF dominant negative (LSFdn) inhibited endogenous LSF and LBP-1a binding and increased class switch recombination indicated by increased production of IgA, but not IgG1. However, LSFdn overexpression did not increase the levels of the sterile IgA transcripts that are also required for recombination to occur, indicating that the effect is mediated by a non-transcriptional mechanism. Class switch recombination requires open chromatin structure, and in vitro studies demonstrated that LSF interacts with histone deacetylases (HDAC) and the co-repressor Sin3A suggesting that LSF/LBP-1a might induce histone deacetylation resulting in inhibition of class switch recombination specifically to the Sα region [40,64]. Thus LSF might play a role in regulating the antibody-mediated immune response.
LSF and cell cycle regulation
LSF plays an important role in cell cycle regulation and is ubiquitously expressed in all cell types. LSF is essential for cell cycle progression at the G1/S transition after re-entry of quiescent cells into the cell cycle, predominantly through inducing thymidylate synthase (TS) gene, which encodes the rate limiting enzyme in the production of dTTP, required for DNA synthesis [38]. The de novo pathway to dTTP synthesis first requires the use of dUMP from the metabolism of either UDP or CDP. The dUMP is converted to dTMP by the action of TS [65]. The methyl group is donated by N5,N10-methylene tetrahydrofolate (THF). The unique property of the action of TS is that the THF is converted to dihydrofolate (DHF), the only such reaction yielding DHF from THF [66]. In order for the TS reaction to continue, THF must be regenerated from DHF. This is accomplished through the action of dihydrofolate reductase (DHFR) [67]. THF is then converted to N5,N10-THF via the action of serine hydroxymethyl transferase. Thus TS and DHFR play a crucial role in thymidine nucleotide biosynthesis and are required for S-phase. Imbalance of dNTPs, including lack of dTTPs, induces cell cycle arrest and apoptosis. The TS essential promoter region (-105 to -75) contains an LSF-recognition sequence where LSF binds upon mitogenic stimuli to induce TS expression [38]. Inhibition of LSF by LSFdn induces apoptosis in S phase in NIH3T3 mouse fibroblasts or DU145 prostate cancer cell which might be relieved by exogenous thymidine, thereby circumventing the requirement for TS activity [38]. The adapter protein Fe65 interacts with the β-amyloid precursor protein (APP), a central player in the pathogenesis of Alzheimer’s disease, which prevents nuclear translocation of Fe65 [68]. Overexpression of the Fe65 results in its nuclear accumulation and induces G1/S arrest in NIH3T3 cells which could be rescued by APP that prevents nuclear translocation of Fe65 [68]. Interestingly, in the nucleus Fe65 interacts with LSF and prevents LSF-mediated induction of TS and addition of exogenous thymidine counteracts the Fe65-mediated block of cell cycle progression [68].
The activity of LSF is regulated during G1 via phosphorylation by both ERK and Cyclin C/Cdk2 [69,70]. LSF is rapidly phosphorylated by ERK on Ser-291 upon mitogenic stimulation of multiple cell types [69]. LSF is then phosphorylated by Cyclin C/Cdk2 on Ser-309 in mouse fibroblasts, with maximal phosphorylation occurring in early G1, 1-2 h following mitogenic stimulation [70]. Phosphorylation at both Ser-291 and Ser-309 inhibits the transcriptional activity of LSF and both sites are dephosphorylated as cell progress into late G1, prior to activation of TS at the G1/S transition suggesting a time-delay mechanism of LSF activation preventing premature induction of LSF target genes. The dephosphorylation of LSF, required for TS induction, does not affect E2F target genes, such as Cyclin E1 and MCM3, indicating that LSF functions parallel to the Rb/E2F pathway in regulating expression of genes necessary for G1/S transition.
Recent studies have highlighted the role of Prolyl isomerase Pin1 in regulating dephosphorylation and activation of LSF [71]. Both ERK and Cdk2 are proline-directed kinases that target Ser/Thr residues immediately N-terminal to Pro residues, similar to Ser-291 and Ser-309 of LSF. One consequence of phosphorylation at proline-directed sites is association with prolyl isomerase, such as Pin1, which can reversibly catalyze cis-trans isomerization of the phospho-Ser/Thr-Pro peptide bond resulting in a conformational change. A series of coimmunoprecipitation and mutational studies demonstrated that Pin1 associates with three SP/TP motifs in LSF at residues Ser-291, Ser-309 and Thr-329 [71]. At early G1, LSF is phosphorylated at Ser-291 and Ser-309 sites by ERK and Cyclin C/Cdk2. Binding of Pin1 at Thr-329-Pro-330 motif in LSF is predicted to facilitate isomerization by Pin1 of the peptide bonds at the nearby phosphorylated SP motifs (Ser-291 and Ser-309) to the trans configuration. In late G1, the kinase/phosphatase balance is predicted to drive dephosphorylation by the phosphatase PP2A, coupled with a reversion of proline isomerization to the cis configuration, and increasing transcriptional activity of LSF. Indeed, overexpression of Pin1 increases LSF transcriptional activity that requires residue Thr-329 of LSF and both the WW and PPiase domains of Pin1 and correlates with hypophos-phorylation of LSF at Ser-291 and Ser-309. These findings are evident in mouse fibroblasts that respond to mitogenic stimuli. Whether similar level of regulation of LSF by phosphorylation/dephosphorylation events persists in HCC cells that have outgrown requirement of mitogens, remains to be determined. It might be possible that the superabundance of LSF in HCC cells results in persistent activation of LSF with subsequent promiscuous induction of genes that would not occur under physiological circumstances.
However, our studies document that LSF is required during S phase in human HCC cells as well. We have established stable clones expressing dominant negative LSF (LSFdn) and in human HCC cells QGY-7703 and demonstrated that these clones, LSFdn-8 and LSFdn-15, have a slower growth rate compared to control clones (Control-1 and Control-7, stable neomycin-resistant clones) [72]. To decipher the mechanism of reduced growth rate following LSF inhibition, we synchronized Control-1 and LSFdn-15 clones of QGY-7703 cells by double thymidine block and then allowed them to continue through the cell cycle by removal of the high concentration of thymidine that inhibits ribonucleotide reductase. At 0 h, cells in both groups were predominantly in early S-phase indicating the efficacy of double thymidine block (Figure 2). However, the Control-1 cells completed the S-phase and moved to G2/M phase significantly faster than the LSFdn-15 cells indicating that LSF is required for efficient passing through S phase during which DNA synthesis takes place.
Figure 2.
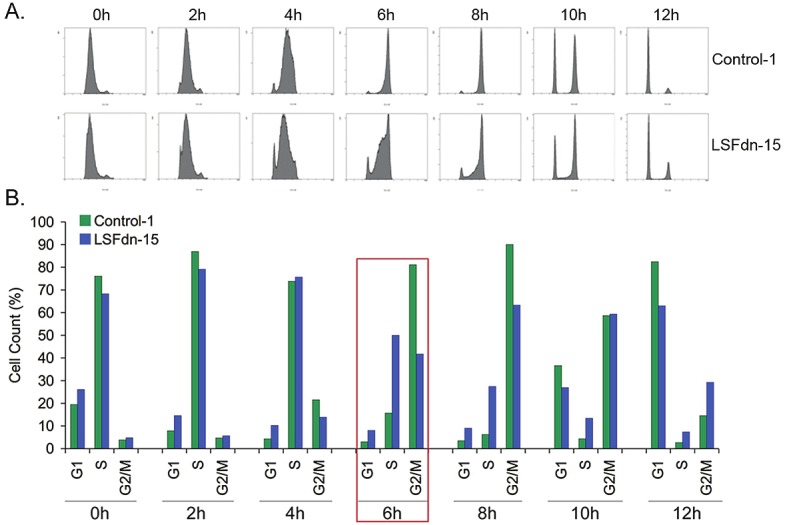
LSF inhibition causes slower cell cycle progression through S-phase. Control-1 and LSFdn-15 clones of QGY-7703 cells were synchronized by double thymidine block and then allowed to enter the cell cycle by thymidine removal. Samples were collected every two hours and cell cycle was analyzed by flow cytometry of propidium iodide-stained DNA. A. Histograms of the cell cycle profile. B. Graphical representation of the percentage of cells in eachphase of the cell cycle.
To identify the proteins that are critical for S-phase progression and are regulated by LSF we performed a dual color 2D gel electrophoresis, where Control-1 sample was labeled with Cy3 (green) and LSFdn-15 sample was labeled with Cy5 (red) (Figure 3). Overlay of the images identified proteins that are modulated by LSF. A green spot indicates a protein that is decreased in the LSFdn-15 sample indicating positive regulation by LSF. A red spot indicates a protein that is increased in the LSFdn-15 sample indicating negative regulation by LSF. A yellow spot indicates equal amount of protein in each sample indicating no change. The analysis was performed in synchronized cells by double thymidine block at 0 h. Seventy-one (71) differentially modulated spots were thus identified out of which 20 spots showed significant downregulation in the LSFdn-15 sample and their identity was determined by mass spectrometry. Very interestingly, we identified both TS and DHFR to be profoundly downregulated in the LSFdn-15 clone (Figure 3). DHFR is a novel LSF-regulated gene and the observation that LSF regulates two important genes controlling folate metabolism further establishes the importance of LSF in cell cycle regulation. Whether LSF transcriptionally regulates DHFR remains to be determined.
Figure 3.

Dual color 2D DIGE detecting protein expression in Control-1 and LSFdn-15 clones of QGY-7703 cells. Leftpanel shows single color and overlay images. Right panel shows peak intensities of Spot 20 and Spot 43. Spot 20and Spot 43 represent Dihydrofolate reductase (DHFR) and thymidylate synthase (TS), respectively. LSF and hepatocellular carcinoma (HCC).
LSF and hepatocellular carcinoma (HCC)
The oncogene Astrocyte elevated gene-1 (AEG-1) is overexpressed in >90% of human HCC patients and AEG-1 plays an important role in regulating development and progression of HCC [73]. Affymetrix microarray comparing global gene expression profiles between control clone and AEG-1-overexpressing clone of HepG3 cells identified LSF as an AEG-1 downstream gene [73]. LSF mRNA expression was ~15-fold higher in AEG-1-overexpressing clones compared to a control clone indicating a potential role of LSF in mediating the oncogenic functions of AEG-1. While in normal hepatocytes LSF protein expression was virtually undetected, its expression was robustly upregulated in human HCC cells, except HepG3 cells, which do not form tumors in nude mice [72]. These findings were extended by tissue microarrays containing 86 primary HCC, 23 metastatic HCC and 9 normal adjacent liver samples that were immunostained using anti-LSF antibody [72]. Little to no LSF immunostaining was detected in the 9 normal liver samples while significant LSF staining was observed in HCC samples. LSF expression was detected predominantly in the nucleus. Among the 109 HCC samples, only nine scored negative for LSF and the remaining 100 (91.7%) showed variable levels of LSF that could be significantly correlated with the stages and grades of the disease [72].
Amplification of chromosome band 12q13, the location of the LSF gene, has been reported in some cases of HCC [74,75]. Although fluorescence in-situ hybridization (FISH) analysis did not identify amplification of LSF gene, in 34 out of 50 HCC samples (68%), polyploidy of chromosome 12 was detected suggesting a potential mechanism of LSF overexpression in human HCC patients [72]. Using a small cohort of 25 HCC patients, a statistically significant correlation was observed between Notch-1-intracellular domain (Notch-1-ICD) and LSF protein expression levels [76]. Overexpression of Notch-1-ICD resulted in significant induction of LSF mRNA and protein in hepatic stellate cells (HSC), HEK-293 cells and human hepatocyte cell line L02 [76]. Interestingly, co-expression of Notch-1-ICD and Ha-Ras further augmented LSF expression. Inhibition of Notch signaling by a secretase inhibitor DAPT downregulated endogenous LSF expression in HepG2 cells [76]. These studies indicate that multiple mechanisms might contribute to LSF overexpression in human HCC. However, the transcription factor(s) mediating AEG-1 or Notch-1-induced upregulation of LSF remain to be determined.
To interrogate the role of LSF in HCC, gain- and loss-of function studies were performed using HepG3 cells, expressing low level of LSF, and QGY-7703 cells, an aggressive HCC cell line expressing high level of LSF [72]. Stable overexpression of LSF in HepG3 cells (LSF-1 and LSF-17 clones) resulted in significant increase in proliferation, colony formation, soft agar growth and Matrigel invasion while stable expression of a dominant negative mutant of LSF (LSFdn, a double amino acid substitution mutant of LSF initially named 234QL/236KE that is unable to bind DNA) in QGY-7703 cells (LSFdn-8 and LSFdn-15 clones) significantly abrogated the aforementioned phenotypes in vitro [72]. While control clones of HepG3 cells did not form any tumors, LSF-1 and LSF-17 clones formed large, aggressive tumors in nude mice xenograft assays [72]. Conversely, LSFdn-8 and LSFdn-15 clones formed significantly smaller tumors when compared to the control clones of QGY-7703 cells in nude mice. The angiogenesis marker CD31 and proliferation marker Ki-67 were significantly upregulated in LSF-1 and LSF-17 clones and significantly downregulated in LSFdn-8 and LSFdn-15 clones. In tail vein metastasis assays in nude mice, the LSF-17 clone formed multi-organ metastatic tumors while inhibition of LSF in LSFdn-15 clone profoundly abrogated its metastatic ability [72]. These findings establish LSF as a key player regulating aggressive progression of HCC.
LSF, osteopontin (OPN) and c-Met axis: a major contributor to HCC
To identify LSF target genes contributing to the aggressive carcinogenic phenotype, Affymetrix oligonucleotide microarray was performed between control HepG3 clone (Con-8) and HepG3 clone stably overexpressing LSF (Hep-LSF-17) [72]. With a 2.0 fold cut-off, expression levels of 125 genes were upregulated while those of 148 genes were downregulated upon overexpression of LSF. Twenty-one of these genes are directly involved in the process of tumorigenesis. The gene SPP1, encoding osteopontin (OPN), showed the most robust induction by LSF [72]. OPN levels can be used as a sensitive and specific marker in predicting disease progression in diverse cancers, including HCC, and OPN is known to promote every step in metastasis as well as growth of the primary tumor [77]. OPN mRNA and protein levels were markedly increased in LSF-overexpressing clones with corresponding significant decrease in LSFdn-overexpressing clones. An ~1kb human OPN promoter region was cloned and OPN promoter luciferase reporter showed significantly higher activity in LSF-overexpressing clones [72]. Chromatin immunoprecipitation (ChIP) assay confirmed LSF binding to OPN promoter establishing transcriptional regulation of OPN by LSF. Stable clones established in Hep-LSF-17 background and expressing OPN shRNA demonstrated significant inhibition of proliferation, colony formation, soft agar growth and Matrigel invasion in vitro; and of tumor growth and metastasis in vivo using nude mice xenograft studies and tail vein metastasis assays [72]. OPN functions through the αvβ3 integrin and CD44 receptors. Anti-αvβ3 integrin or anti-CD44 antibody significantly inhibited Matrigel invasion by Hep-LSF-17 cells and the combination of the two antibodies decreased the invasion further confirming that OPN working through its canonical receptors plays a key role in regulating LSF function [72].
Analysis of a human phospho-receptor tyrosine kinase (RTK) array using lysates from Con-8 and LSF-17 clones of HepG3 cells identified robust activation of c-Met in LSF-17 clone [78]. c-Met, a cell surface receptor for hepatocyte growth factor (HGF), conveys a unique combination of pro-migratory, anti-apoptotic and mitogenic signals [79]. When activated, c-Met initiates epithelial-mesenchymal transition (EMT) by facilitating cell scattering, thereby facilitating migration and invasive growth. That c-Met plays a pivotal role in HCC was confirmed by the observation that conditional overexpression of wildtype c-Met in hepatocytes of transgenic mice (an experimental condition that mimics the spontaneous amplification of the c-Met gene observed in human tumors) is sufficient to cause hepatocellular carcinoma that regresses following transgene inactivation [80]. The level of activated (phosphorylated) c-Met was significantly higher in LSF-1 and LSF-17 clones compared to the Control-8 clone of HepG3 cells [78]. Similarly, activated c-Met was significantly downregulated in LSFdn-8 and LSFdn-15 clones, compared to the Control-7 clone of QGY-7703 cells. The level of total c-Met did not change in either group. In HCC tissue microarray (TMA), LSF, OPN and c-Met levels were significantly higher compared to normal human liver and a significant statistical correlation was observed among LSF, OPN and phospho-c-Met levels [78]. The level of c-Met ligand HGF was found to be unaltered between Con-8 and LSF-17 clones and a search for an alternative mechanism of c-Met activation identified OPN as an activator of c-Met. Using a series of coimmunoprecipitation and receptor blocking studies it was demonstrated that secreted OPN, acting through CD44 receptors, induced phosphorylation and activation of c-Met [78].
Inhibition of c-Met by SU11274 abrogated matrigel invasion of LSF-17 and QGY-7703 cells and blocked OPN-induced activation of downstream signaling, such as Akt and ERK [78]. Stable clones established in the Hep-LSF-17 background and expressing c-Met shRNA demonstrated significant inhibition of proliferation and Matrigel invasion, and activation of Akt and ERK along with changes in EMT markers, such as upregulation of E-cadherin and downregulation of vimentin, in vitro. In nude mice xenograft assays these clones generated significantly smaller tumors with concomitant inhibition of tumor angiogenesis, as evidenced by CD31 staining, and proliferation, assayed by Ki-67 staining and the metastatic potential of these clones was virtually wiped out as evidenced by tail vein metastasis assay [78]. These findings indicate that the LSF-OPN-c-Met axis might play an important role in mediating hepatocarcinogenesis.
LSF and angiogenesis
The observation that LSF overexpression in HepG3 cells results in highly aggressive, metastatic tumors while LSFdn expression in QGY-7703 cells profoundly inhibits tumor growth and metastasis, prompted the hypothesis that LSF might modulate tumor angiogenesis, a hallmark of aggressive tumors. Using human vascular endothelial cells (HUVEC) differentiation assay and chicken chorioallantoic membrane (CAM) assay it was demonstrated that conditioned media (CM) from LSF-overexpressing cells augments while CM from LSFdn-expressing cells abrogates angiogenesis significantly [81]. To identify a master regulator of angiogenesis that might be transcriptionally regulated by LSF, ChIP-on-chip analysis was performed Using Roche Nimblegen’s ChIP-on-chip 2.1 M tiling array that allows identification of promoter regions to which a specific transcription factor binds [81]. The promoter region of matrix metalloproteinase-9 (MMP-9) exhibited the highest statistical significance in terms of LSF binding. MMP-9 mRNA and protein levels and enzymatic activity were significantly higher in LSF-overexpressing cells and conversely significantly lower in LSFdn-overexpressing cells. An ~2 kb region of human MMP-9 promoter was cloned to generate a luciferase reporter vector (MMP-9-Prom-luc) [81]. The activity of MMP-9-Prom-luc was significantly higher in LSF-overexpressing cells while it was significantly lower in LSFdn-overexpressing cells. Deletion mutation and ChIP assays identified two LSF binding sites in the human MMP-9 promoter between the regions-1306 to -746 when the translation start site was regarded as +1. Stable clones established in the Hep-LSF-17 background and expressing MMP-9 shRNA demonstrated significant inhibition of proliferation, colony formation, migration and Matrigel invasion in vitro, of angiogenesis by HUVEC differentiation and CAM assays, and of tumor growth, metastasis and angiogenesis in vivo using nude mice xenograft studies and tail vein metastasis assays [81]. These clones also showed reduced expression of angiogenic factors, such as vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8), indicating that MMP-9 might be a major player mediating LSF-induced angiogenic response. Angiogenesis is an invasive process that requires proteolysis of the extracellular matrix and proliferation and migration of endothelial cells [82]. As such, MMPs play an important role in tumor angiogenesis by facilitating ECM degradation, mainly degradation of collagen in the basement membrane [83]. MMP-9 is overexpressed in many tumors including HCC. MMP-9 is a major contributor to tumor-induced angiogenesis and a crucial factor triggering activation of quiescent vasculature [84,85]. MMP-9 degrades ECM allowing invasion of pericytes that stabilizes newly-formed capillaries, sustain blood flow, modulate vascular permeability and regulate the function of endothelial cells [86]. Inhibition of MMP-9 results in inhibition of expression of pro-angiogenic genes, such as VEGF and ICAM-1 [87]. It should be noted LSF specifically upregulates MMP-9, and not other MMPs such as MMP-2 and MT1-MMP, further stressing the importance of MMP-9 in LSF function [81].
Studies from mouse models provide interesting insights into the role of LSF/TFCP2 and LBP-1a/NF2d9, members of grainyhead family of transcription factors, in angiogenesis [88]. An LSF knock-out mouse is viable and has a normal life expectancy suggesting that LBP1-a might compensate for LSF function. However, LBP-1a knockout mice die at embryonic day 10.5 due to severe placental insufficiency because of an angiogenic defect in the mesodermal components of the labyrinthine layer and the yolk sac vasculature. These findings strongly suggest that this family of proteins is essential for angiogenesis. However, the underlying mechanism by which LBP-1a regulates angiogenesis is not clear. It is also not clear why LSF cannot compensate for LBP-1a deficiency, whereas LBP1-a can compensate for LSF, even though their tissue distribution is quite similar and they have a high degree of similarity in protein structure. Additionally, angiogenic factors, transcriptionally regulated by LBP-1a, have also not been identified. It might be possible that LSF has acquired more functional importance than LBP-1a in humans. Additionally, acquired tissue- and tumor-specific roles of LSF might also underlie its specialized function in regulating tumor angiogenesis. It should be noted that it is LSF, not LBP-1a, which is overexpressed in human HCC [81]. Another possibility is that the angiogenesis functions are attributable to the LBP-1b protein, which due to differential splicing contains additional 36 amino acids, and which is particularly important in erythroid-specific gene expression [49], but overexpression of LSF can allow it to substitute for LBP-1b. The super-abundance of LSF in human HCC cells might allow it to transcriptionally regulate OPN or MMP-9, which may not happen under physiological conditions. Analysis of the role of LSF in regulating MMP-9 in inflammation- and/or wound-associated angiogenesis will provide important insight into the role of this unique transcription factor in biological processes.
LSF and chemoresistance
Chemoresistance is a hallmark of aggressive cancers [89]. HCC is notoriously resistant to chemotherapy and a combination of multiple chemotherapeutics drugs, including cisplatin, doxorubicin and 5-fluorouracil (5-FU), along with interferon, barely affects survival of HCC patients [9]. 5-FU is converted intracellularly into 5’-fluoro-2’-deoxyuridine by thymidine phosphorylase with subsequent phosphorylation by thymidine kinase into the active metabolite 5-fluoro-2’-deoxyuridine 5’-monophosphate (FdUMP) [90]. FdUMP inhibits thymidylate synthase (TS) which reduces the thymidine pool and increases the uracil pool leading to the inhibition of DNA synthesis. 5-FU is converted into its inactive metabolite fluoro-5,6-dihydrouracil (FUH2) by dihydropyrimidine dehydrogenase (DPYD). TS and DPYD gene expression and/or activity are major determinants of the efficacy of 5-FU [91,92]. Since LSF transcriptionally regulates TS it might be presumed that LSF overexpression might contribute to 5-FU resistance. Interestingly, AEG-1 overexpression resulted in increase in both TS and DPYD with subsequent resistance to 5-FU [93]. Inhibition of LSF by LSFdn in AEG-1 overexpressing cells resulted in downregulation of TS and rendered these cells susceptible to 5-FU again indicating that by augmenting TS expression LSF also contributes to chemoresistance [93].
LSF and epithelial mesenchymal transition (EMT)
EMT, demonstrated by downregulation of epithelial markers and upregulation of mesenchymal markers, is a hallmark of advanced tumor cells required for increased motility during invasion and metastasis [94]. As a transcription factor Snail1 plays an important role in regulating EMT [95]. Studies using human pancreatic and colorectal carcinoma cell lines revealed a role of LSF in mediating Snail1-induced upregulation of EMT marker Fibronectin [96]. Fibronectins are a class of cell-secreted glycoproteins that modulate cell-substrate adhesion and regulate a diverse array of events, including cell attachment, spreading, migration, differentiation and oncogenic transformation [97]. Overexpression of Snail1 resulted in increased binding of LSF to Fibronectin 1 (FN1) promoter thereby upregulating FN1 expression [96]. Overexpression of LSFdn prevented Snail1-induced FN1 upregulation and was associated with reduced migration. The effect of LSF is specific to FN1 since other major regulators of EMT, such as LEF1 and ZEB1, were not modulated by LSF [96]. More importantly, induction of FN1 by TGF-β, another major regulator of EMT, was also inhibited by LSFdn indicating a general role of FN1 expression by LSF during EMT. Although Snail1 did not change LSF protein level, an increased nuclear accumulation of LSF was observed in Snail1-overexpressed cells [96]. However, the underlying mechanism remains to be determined. These findings suggest that even without changes in its expression level LSF might mediate EMT, an important determinant of aggressive cancers.
Small molecule inhibitors of LSF: potential therapeutics for HCC
The pivotal role of LSF in regulating hepatocarcinogenesis indicates that LSF inhibition might be an effective therapeutic approach to counteract this fatal malady. In an attempt to identify small molecule inhibitors of LSF-binding to DNA, 110,000 commercially available compounds were evaluated by a high throughput screening using a fluorescence polarization assay indicating an ability to diminish the DNA-binding activity of LSF [98]. A secondary electrophoretic mobility shift assay (EMSA) was performed to establish selectivity for LSF DNA-binding activity, eliminating compounds that also inhibited other transcription factors, such as, Sp1, Oct1 and E2F3 [98]. These dual assays identified 4-aryl-3,4-dihydroquinolin-2(1H)-ones as potent and specific inhibitors of in vitro LSF-DNA binding. A collection of compounds based on the dihydroquinolin- 2(1H)-one structure was prepared and the prototype of these compounds, factor quinolinone inhibitor 1 (FQI1), demonstrated 50% inhibition in LSF transactivation activity (IC50) at a concentration of 2.1 μM using a LSF-dependent luciferase reporter assay. FQI1 did not affect USF-dependent luciferase reporter activity [98]. As discussed above, the DNA binding region of LSF is predicted to be structurally similar to that of the p53 transcription factor family. To test whether FQI1 could also diminish p53 activity, cells were treated with nutlin-3, which inhibits MDM2, therefore enhancing p53 levels. The resulting transcriptional induction of p21 was unaffected by co-treatment with FQI1, further confirming specificity of FQI1 towards LSF [98].
To probe into the mechanism by which FQI1 inhibits LSF transactivation, an inducible stable cell line was generated in HEK-293 cells in which expression of an HA-tagged LSF is induced by the non-steroidal diphenylhydrazine compound, RSL1 and LSF binding to the promoter of POLA1, encoding the catalytic subunit of DNA polymerase α, was determined by ChIP assay [98]. RSL1 treatment resulted in increased binding of LSF to POLA1 promoter which was completely inhibited by co-treatment with FQI1. FQI1 treatment did not affect that level of RSL1-induced HA-LSF protein level indicating that FQI1 functions by inhibiting LSF DNA-binding rather than modulating LSF expression.
Separation of FQI1 isomers demonstrated that while the (R)-enantiomer of FQI1 [(R)-FQI1] showed significantly less activity, the (S)-enantiomer [(S)-FQI1] was more potent than FQI1 in inhibiting LSF transactivation activity [98]. The achiral quinolin-2(1H)-one, FQI2, which might adopt a similar conformation as (S)-FQI1, was more potent than FQI1, demonstrating functional activity at sub-micromolar concentrations. As a corollary, using a variety of cell lines, it was demonstrated that (R)-FQI1 is at least ten times less effective while (S)-FQI1 and FQI2 were four-fold more effective as FQI1 in inhibiting cell proliferation at a comparable dose that inhibits LSF transactivation. Structure-activity relationship (SAR) analysis comparing the GI50 values (concentrations that reduce cell proliferation by 50%) from all the FQI analogs containing combinations of peripheral structural variations revealed consistent inhibition of cell growth and LSF transcriptional activity in all cell lines. Highly correlated structure-activity relationships and specificity strongly suggests that the antiproliferative activity of the FQI compounds is the consequence of targeting a single, or multiple highly related, molecules.
To develop FQI as a potential therapeutic for HCC, the anti-proliferative activity of FQI1 and FQI2 was tested using QGY-7703 and Hep3B cells that overexpress LSF [98]. Both FQI1 and FQI2 inhibited viability of QGY-7703 cells profoundly at doses between 0.5 to 2 μM and by 48 h 90% of QGY-7703 cells showed apoptosis. The kinetics of the killing was delayed in Hep3B cells, in which by 96 h, 65% cells showed apoptosis. QGY-7703 cells are highly aggressive and express high level of LSF, compared to Hep3B cells. The findings of viability and apoptosis assays support an oncogene addiction phenomenon where QGY-7703 cells are more dependent on LSF for its survival and therefore are more susceptible to LSF inhibition. In human prostate cancer cells inhibition of LSF by dominant negative LSF leads to cell cycle arrest at S phase with subsequent apoptosis due to inhibition of expression of LSF target gene thymidylate synthase. However, incubation with thymidine to overcome cellular dependence on thymidylate synthase did not rescue QGY-7703 cells from FQI1-induced apoptosis, indicating inhibition of additional LSF target genes mediating FQI1-induced apoptosis. Importantly, FQI1 did not adversely affect viability of normal immortalized human hepatocytes (Hc3716-hTERT) and primary mouse hepatocytes in vitro further supporting the phenomenon of oncogene addiction in QGY-7703 cells.
To confirm in vivo efficacy of FQI1, subcutaneous xenografts were established using QGY-7703 cells in athymic nude mice and after the establishment of the tumors the animals were treated with FQI1 intraperitoneally at a dose of 1 mg/kg once every 3 days for a total of 5 times [98]. The mice were followed for another 2 weeks after the last treatment. FQI1 treatment inhibited tumor growth by ~90% with profound inhibition in tumor angiogenesis, as evidenced by CD31 staining; proliferation, assayed by Ki-67 staining; and expression of LSF target gene, such as OPN without affecting LSF expression. More importantly no general toxicity was observed in FQI1-treated animals with preservation of normal morphology and architecture of the internal organs. Liver enzymes and serum protein levels were unaffected upon FQI1 treatment demonstrating that FQI1 did not induce any liver damage. These studies indicate that FQI1 is safe and potently effective in inhibiting human HCC.
Conclusion
Our studies have identified novel targets of LSF, such as OPN, MMP-9 and c-Met, that play essential roles in hepatocarcinogenesis (Figure 4). OPN promotes tumorigenesis via a vast array of mechanisms, one of which is induction of MMP-9. While we demonstrate that LSF transcriptionally regulates MMP-9, the two most frequent transcription factors regulating MMP-9 transcription are NF-κB and AP-1. We have previously demonstrated that LSF itself can activate NF-κB and it also activates ERK signaling that might lead to AP-1 activation [72]. These properties of LSF might be attributed to OPN, a known activator of NF-κB and AP-1, and OPN-induced c-Met activation. As such there might be multiple mechanisms by which LSF induces MMP-9 expression. On the other hand, MMP-9 is known to cleave OPN into specific fragments which augment HCC cell invasion [99]. Thus MMP-9 and OPN work in concert to promote hepatocarcinogenesis. Affymetrix microarray studies identified complement factor H (CFH) as an LSF-inducible gene [72]. OPN can sequester complement factor H (CFH) to the surface of cancer cells that disables the formation of the membrane attack complex and subsequent complement-mediate lysis of cancer cells, thus favoring their escape from host immune defense during the process of metastasis [100]. Thus LSF functions as a master regulator of a network of proteins, involving OPN, MMP-9, c-Met, CFH and FN1, controlling tumor progression, angiogenesis and metastasis, and also cell cycle regulatory proteins, such as TS, and inhibition of LSF might be an effective strategy to counteract a fatal disease like HCC. The identification of FQI family of small molecules ushers in a novel therapeutic modality for HCC [98]. The initial characterization of FQI1 demonstrates its safety and efficacy in pre-clinical immune-compromised mouse models. More stringent studies using transgenic and immunecompetent animal models are warranted for future translation of FQI to clinics to promote disease-free survival in HCC patients.
Figure 4.

Model for molecular mechanism by which LSF promotes hepatocarcinogenesis. LSF transcriptionally upregulates osteopontin (OPN), matrix metalloproteinase-9 (MMP-9), complement factor H (CFH) and fibronectin 1 (FN1). Secreted OPN binds to CD44 receptor resulting in activation of c-Met and its downstream signaling Akt and ERK. Collectively these events contribute to proliferation, invasion, angiogenesis and metastasis. LSF upregulates thymidylate synthase (TS) thus contributing to cell cycle progression and chemoresistance.
Acknowledgement
The present study was supported in part by grants from the James S McDonnell Foundation and National Cancer Institute Grant R01 CA138540 (DS), the Samuel Waxman Cancer Research Foundation (SWCRF) Grant (DS and PBF) and NIH grant R01 CA134721 (PBF). DS is the Harrison Endowed Scholar in Cancer Research and a Blick scholar. PBF holds the Thelma Newmeyer Corman Chair in Cancer Research and is a SWCRF Investigator.
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 4.El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med. 2003;139:817–823. doi: 10.7326/0003-4819-139-10-200311180-00009. [DOI] [PubMed] [Google Scholar]
- 5.Pang RW, Joh JW, Johnson PJ, Monden M, Pawlik TM, Poon RT. Biology of hepatocellular carcinoma. Ann Surg Oncol. 2008;15:962–971. doi: 10.1245/s10434-007-9730-z. [DOI] [PubMed] [Google Scholar]
- 6.Poon RT, Fan ST, Lo CM, Ng IO, Liu CL, Lam CM, Wong J. Improving survival results after resection of hepatocellular carcinoma: a prospective study of 377 patients over 10 years. Ann Surg. 2001;234:63–70. doi: 10.1097/00000658-200107000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuchs CS, Clark JW, Ryan DP, Kulke MH, Kim H, Earle CC, Vincitore M, Mayer RJ, Stuart KE. A phase II trial of gemcitabine in patients with advanced hepatocellular carcinoma. Cancer. 2002;94:3186–3191. doi: 10.1002/cncr.10607. [DOI] [PubMed] [Google Scholar]
- 8.Lopez PM, Villanueva A, Llovet JM. Systematic review: evidence-based management of hepatocellular carcinoma--an updated analysis of randomized controlled trials. Aliment Pharmacol Ther. 2006;23:1535–1547. doi: 10.1111/j.1365-2036.2006.02932.x. [DOI] [PubMed] [Google Scholar]
- 9.Yeo W, Mok TS, Zee B, Leung TW, Lai PB, Lau WY, Koh J, Mo FK, Yu SC, Chan AT, Hui P, Ma B, Lam KC, Ho WM, Wong HT, Tang A, Johnson PJ. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97:1532–1538. doi: 10.1093/jnci/dji315. [DOI] [PubMed] [Google Scholar]
- 10.Llovet JM, Bru C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis. 1999;19:329–338. doi: 10.1055/s-2007-1007122. [DOI] [PubMed] [Google Scholar]
- 11.O'Neil BH, Venook AP. Hepatocellular carcinoma: the role of the North American GI Steering Committee Hepatobiliary Task Force and the advent of effective drug therapy. Oncologist. 2007;12:1425–1432. doi: 10.1634/theoncologist.12-12-1425. [DOI] [PubMed] [Google Scholar]
- 12.Georgiades CS, Hong K, Geschwind JF. Radiofrequency ablation and chemoembolization for hepatocellular carcinoma. Cancer J. 2008;14:117–122. doi: 10.1097/PPO.0b013e31816a0fac. [DOI] [PubMed] [Google Scholar]
- 13.Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology. 2003;37:429–442. doi: 10.1053/jhep.2003.50047. [DOI] [PubMed] [Google Scholar]
- 14.Nerenstone S, Friedman M. Medical treatment of hepatocellular carcinoma. Gastroenterol Clin North Am. 1987;16:603–612. [PubMed] [Google Scholar]
- 15.Leung TW, Patt YZ, Lau WY, Ho SK, Yu SC, Chan AT, Mok TS, Yeo W, Liew CT, Leung NW, Tang AM, Johnson PJ. Complete pathological remission is possible with systemic combination chemotherapy for inoperable hepatocellular carcinoma. Clin Cancer Res. 1999;5:1676–1681. [PubMed] [Google Scholar]
- 16.Patt YZ, Hoque A, Roh M, Ellis L, Lozano R, Carrasco CH, Charnsangavej C, Cleary K. Durable clinical and pathologic response of hepatocellular carcinoma to systemic and hepatic arterial administration of platinol, recombinant interferon alpha 2B, doxorubicin, and 5-fluorouracil: a communication. Am J Clin Oncol. 1999;22:209–213. doi: 10.1097/00000421-199904000-00024. [DOI] [PubMed] [Google Scholar]
- 17.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 18.Simpson D, Keating GM. Sorafenib: in hepatocellular carcinoma. Drugs. 2008;68:251–258. doi: 10.2165/00003495-200868020-00007. [DOI] [PubMed] [Google Scholar]
- 19.Zhu AX, Blaszkowsky LS, Ryan DP, Clark JW, Muzikansky A, Horgan K, Sheehan S, Hale KE, Enzinger PC, Bhargava P, Stuart K. Phase II study of gemcitabine and oxaliplatin in combination with bevacizumab in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2006;24:1898–1903. doi: 10.1200/JCO.2005.04.9130. [DOI] [PubMed] [Google Scholar]
- 20.Veljkovic J, Hansen U. Lineage-specific and ubiquitous biological roles of the mammalian transcription factor LSF. Gene. 2004;343:23–40. doi: 10.1016/j.gene.2004.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen U, Owens L, Saxena UH. Transcription factors LSF and E2Fs: tandem cyclists driving G0 to S? Cell Cycle. 2009;8:2146–2151. doi: 10.4161/cc.8.14.9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim CH, Heath C, Bertuch A, Hansen U. Specific stimulation of simian virus 40 late transcription in vitro by a cellular factor binding the simian virus 40 21-base-pair repeat promoter element. Proc Natl Acad Sci USA. 1987;84:6025–6029. doi: 10.1073/pnas.84.17.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnhart KM, Kim CG, Banerji SS, Sheffery M. Identification and characterization of multiple erythroid cell proteins that interact with the promoter of the murine alpha-globin gene. Mol Cell Biol. 1988;8:3215–3226. doi: 10.1128/mcb.8.8.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim CG, Barnhart KM, Sheffery M. Purification of multiple erythroid cell proteins that bind the promoter of the alpha-globin gene. Mol Cell Biol. 1988;8:4270–4281. doi: 10.1128/mcb.8.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones KA, Luciw PA, Duchange N. Structural arrangements of transcription control domains within the 5'-untranslated leader regions of the HIV-1 and HIV-2 promoters. Genes Dev. 1988;2:1101–1114. doi: 10.1101/gad.2.9.1101. [DOI] [PubMed] [Google Scholar]
- 26.Wu FK, Garcia JA, Harrich D, Gaynor RB. Purification of the human immunodeficiency virus type 1 enhancer and TAR binding proteins EBP-1 and UBP-1. Embo J. 1988;7:2117–2130. doi: 10.1002/j.1460-2075.1988.tb03051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang JH, Liao WS. Induction of the mouse serum amyloid A3 gene by cytokines requires both C/EBP family proteins and a novel constitutive nuclear factor. Mol Cell Biol. 1994;14:4475–4484. doi: 10.1128/mcb.14.7.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon JB, Li G, Roeder RG. Characterization of a family of related cellular transcription factors which can modulate human immunodeficiency virus type 1 transcription in vitro. Mol Cell Biol. 1994;14:1776–1785. doi: 10.1128/mcb.14.3.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venkatesan K, McManus HR, Mello CC, Smith TF, Hansen U. Functional conservation between members of an ancient duplicated transcription factor family, LSF/Grainyhead. Nucleic Acids Res. 2003;31:4304–4316. doi: 10.1093/nar/gkg644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilanowski T, Tuckfield A, Cerruti L, O'Connell S, Saint R, Parekh V, Tao J, Cunningham JM, Jane SM. A highly conserved novel family of mammalian developmental transcription factors related to Drosophila grainyhead. Mech Dev. 2002;114:37–50. doi: 10.1016/s0925-4773(02)00046-1. [DOI] [PubMed] [Google Scholar]
- 31.Traylor-Knowles N, Hansen U, Dubuc TQ, Martindale MQ, Kaufman L, Finnerty JR. The evolutionary diversification of LSF and Grainyhead transcription factors preceded the radiation of basal animal lineages. BMC Evol Biol. 2010;10:101. doi: 10.1186/1471-2148-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swendeman SL, Spielholz C, Jenkins NA, Gilbert DJ, Copeland NG, Sheffery M. Characterization of the genomic structure, chromosomal location, promoter, and development expression of the alpha-globin transcription factor CP2. J Biol Chem. 1994;269:11663–11671. [PubMed] [Google Scholar]
- 33.Cunningham JM, Vanin EF, Tran N, Valentine M, Jane SM. The human transcription factor CP2 (TFCP2), a component of the human gamma-globin stage selector protein, maps to chromosome region 12q13 and is within 250 kb of the NF-E2 gene. Genomics. 1995;30:398–399. [PubMed] [Google Scholar]
- 34.Shirra MK, Hansen U. LSF and NTF-1 share a conserved DNA recognition motif yet require different oligomerization states to form a stable protein-DNA complex. J Biol Chem. 1998;273:19260–19268. doi: 10.1074/jbc.273.30.19260. [DOI] [PubMed] [Google Scholar]
- 35.Murata T, Nitta M, Yasuda K. Transcription factor CP2 is essential for lens-specific expression of the chicken alphaA-crystallin gene. Genes Cells. 1998;3:443–457. doi: 10.1046/j.1365-2443.1998.00204.x. [DOI] [PubMed] [Google Scholar]
- 36.Kokoszynska K, Ostrowski J, Rychlewski L, Wyrwicz LS. The fold recognition of CP2 transcription factors gives new insights into the function and evolution of tumor suppressor protein p53. Cell Cycle. 2008;7:2907–2915. doi: 10.4161/cc.7.18.6680. [DOI] [PubMed] [Google Scholar]
- 37.Shirra MK, Zhu Q, Huang HC, Pallas D, Hansen U. One exon of the human LSF gene includes conserved regions involved in novel DNA-binding and dimerization motifs. Mol Cell Biol. 1994;14:5076–5087. doi: 10.1128/mcb.14.8.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powell CM, Rudge TL, Zhu Q, Johnson LF, Hansen U. Inhibition of the mammalian transcription factor LSF induces S-phase-dependent apoptosis by downregulating thymidylate synthase expression. Embo J. 2000;19:4665–4675. doi: 10.1093/emboj/19.17.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frith MC, Hansen U, Weng Z. Detection of cis-element clusters in higher eukaryotic DNA. Bioinformatics. 2001;17:878–889. doi: 10.1093/bioinformatics/17.10.878. [DOI] [PubMed] [Google Scholar]
- 40.Drouin EE, Schrader CE, Stavnezer J, Hansen U. The ubiquitously expressed DNA-binding protein late SV40 factor binds Ig switch regions and represses class switching to IgA. J Immunol. 2002;168:2847–2856. doi: 10.4049/jimmunol.168.6.2847. [DOI] [PubMed] [Google Scholar]
- 41.Zambrano N, Minopoli G, de Candia P, Russo T. The Fe65 adaptor protein interacts through its PID1 domain with the transcription factor CP2/LSF/LBP1. J Biol Chem. 1998;273:20128–20133. doi: 10.1074/jbc.273.32.20128. [DOI] [PubMed] [Google Scholar]
- 42.Jane SM, Nienhuis AW, Cunningham JM. Hemoglobin switching in man and chicken is mediated by a heteromeric complex between the ubiquitous transcription factor CP2 and a developmentally specific protein. Embo J. 1995;14:97–105. doi: 10.1002/j.1460-2075.1995.tb06979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou W, Clouston DR, Wang X, Cerruti L, Cunningham JM, Jane SM. Induction of human fetal globin gene expression by a novel erythroid factor, NF-E4. Mol Cell Biol. 2000;20:7662–7672. doi: 10.1128/mcb.20.20.7662-7672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74:6790–6799. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romerio F, Gabriel MN, Margolis DM. Repression of human immunodeficiency virus type 1 through the novel cooperation of human factors YY1 and LSF. J Virol. 1997;71:9375–9382. doi: 10.1128/jvi.71.12.9375-9382.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casolaro V, Keane-Myers AM, Swendeman SL, Steindler C, Zhong F, Sheffery M, Georas SN, Ono SJ. Identification and characterization of a critical CP2-binding element in the human interleukin-4 promoter. J Biol Chem. 2000;275:36605–36611. doi: 10.1074/jbc.M007086200. [DOI] [PubMed] [Google Scholar]
- 47.Ylisastigui L, Kaur R, Johnson H, Volker J, He G, Hansen U, Margolis D. Mitogen-activated protein kinases regulate LSF occupancy at the human immunodeficiency virus type 1 promoter. J Virol. 2005;79:5952–5962. doi: 10.1128/JVI.79.10.5952-5962.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim LC, Swendeman SL, Sheffery M. Molecular cloning of the alpha-globin transcription factor CP2. Mol Cell Biol. 1992;12:828–835. doi: 10.1128/mcb.12.2.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang HC, Chae JH, Jeon J, Kim W, Ha DH, Shin JH, Kim CG. PIAS1 regulates CP2c localization and active promoter complex formation in erythroid cell-specific alpha-globin expression. Nucleic Acids Res. 2010;38:5456–5471. doi: 10.1093/nar/gkq286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solis C, Aizencang GI, Astrin KH, Bishop DF, Desnick RJ. Uroporphyrinogen III synthase erythroid promoter mutations in adjacent GATA1 and CP2 elements cause congenital erythropoietic porphyria. J Clin Invest. 2001;107:753–762. doi: 10.1172/JCI10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang P, Yang G, Mosier DR, Chang P, Zaidi T, Gong YD, Zhao NM, Dominguez B, Lee KF, Gan WB, Zheng H. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. J Neurosci. 2005;25:1219–1225. doi: 10.1523/JNEUROSCI.4660-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Y, Kim HS, Joo Y, Choi Y, Chang KA, Park CH, Shin KY, Kim S, Cheon YH, Baik TK, Kim JH, Suh YH. Intracellular domains of amyloid precursor- like protein 2 interact with CP2 transcription factor in the nucleus and induce glycogen synthase kinase-3beta expression. Cell Death Differ. 2007;14:79–91. doi: 10.1038/sj.cdd.4401928. [DOI] [PubMed] [Google Scholar]
- 53.Loeffler DA, Connor JR, Juneau PL, Snyder BS, Kanaley L, DeMaggio AJ, Nguyen H, Brickman CM, LeWitt PA. Transferrin and iron in normal, Alzheimer's disease, and Parkinson's disease brain regions. J Neurochem. 1995;65:710–724. doi: 10.1046/j.1471-4159.1995.65020710.x. [DOI] [PubMed] [Google Scholar]
- 54.Jang SM, Kim JW, Kim CH, An JH, Kang EJ, Kim CG, Kim HJ, Choi KH. Control of transferrin expression by beta-amyloid through the CP2 transcription factor. FEBS J. 2010;277:4054–4065. doi: 10.1111/j.1742-4658.2010.07801.x. [DOI] [PubMed] [Google Scholar]
- 55.Lambert JC, Goumidi L, Vrieze FW, Frigard B, Harris JM, Cummings A, Coates J, Pasquier F, Cottel D, Gaillac M, St Clair D, Mann DM, Hardy J, Lendon CL, Amouyel P, Chartier-Harlin MC. The transcriptional factor LBP-1c/CP2/LSF gene on chromosome 12 is a genetic determinant of Alzheimer's disease. Hum Mol Genet. 2000;9:2275–2280. doi: 10.1093/oxfordjournals.hmg.a018918. [DOI] [PubMed] [Google Scholar]
- 56.Taylor AE, Yip A, Brayne C, Easton D, Evans JG, Xuereb J, Cairns N, Esiri MM, Rubinsztein DC. Genetic association of an LBP-1c/CP2/LSF gene polymorphism with late onset Alzheimer's disease. J Med Genet. 2001;38:232–233. doi: 10.1136/jmg.38.4.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luedecking-Zimmer E, DeKosky ST, Nebes R, Kamboh MI. Association of the 3' UTR transcription factor LBP-1c/CP2/LSF polymorphism with late-onset Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet. 2003;117B:114–117. doi: 10.1002/ajmg.b.10026. [DOI] [PubMed] [Google Scholar]
- 58.Bertram L, Parkinson M, McQueen MB, Mullin K, Hsiao M, Menon R, Moscarillo TJ, Blacker D, Tanzi RE. Further evidence for LBP-1c/CP2/LSF association in Alzheimer's disease families. J Med Genet. 2005;42:857–862. doi: 10.1136/jmg.2004.024596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boulay JL, Paul WE. The interleukin-4-related lymphokines and their binding to hematopoietin receptors. J Biol Chem. 1992;267:20525–20528. [PubMed] [Google Scholar]
- 60.Bing Z, Huang JH, Liao WS. NFkappa B interacts with serum amyloid A3 enhancer factor to synergistically activate mouse serum amyloid A3 gene transcription. J Biol Chem. 2000;275:31616–31623. doi: 10.1074/jbc.M005378200. [DOI] [PubMed] [Google Scholar]
- 61.Bing Z, Reddy SA, Ren Y, Qin J, Liao WS. Purification and characterization of the serum amyloid A3 enhancer factor. J Biol Chem. 1999;274:24649–24656. doi: 10.1074/jbc.274.35.24649. [DOI] [PubMed] [Google Scholar]
- 62.Kammandel B, Chowdhury K, Stoykova A, Aparicio S, Brenner S, Gruss P. Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev Biol. 1999;205:79–97. doi: 10.1006/dbio.1998.9128. [DOI] [PubMed] [Google Scholar]
- 63.Zheng JB, Zhou YH, Maity T, Liao WS, Saunders GF. Activation of the human PAX6 gene through the exon 1 enhancer by transcription factors SEF and Sp1. Nucleic Acids Res. 2001;29:4070–4078. doi: 10.1093/nar/29.19.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Repetny KJ, Zhong X, Holodick NE, Rothstein TL, Hansen U. Binding of LBP-1a to specific immunoglobulin switch regions in vivo correlates with specific repression of class switch recombination. Eur J Immunol. 2009;39:1387–1394. doi: 10.1002/eji.200838226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 66.Danenberg PV. Thymidylate synthetase - a target enzyme in cancer chemotherapy. Biochim Biophys Acta. 1977;473:73–92. doi: 10.1016/0304-419x(77)90001-4. [DOI] [PubMed] [Google Scholar]
- 67.Schweitzer BI, Dicker AP, Bertino JR. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990;4:2441–2452. doi: 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- 68.Bruni P, Minopoli G, Brancaccio T, Napolitano M, Faraonio R, Zambrano N, Hansen U, Russo T. Fe65, a ligand of the Alzheimer's beta-amyloid precursor protein, blocks cell cycle progression by down-regulating thymidylate synthase expression. J Biol Chem. 2002;277:35481–35488. doi: 10.1074/jbc.M205227200. [DOI] [PubMed] [Google Scholar]
- 69.Pagon Z, Volker J, Cooper GM, Hansen U. Mammalian transcription factor LSF is a target of ERK signaling. J Cell Biochem. 2003;89:733–746. doi: 10.1002/jcb.10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saxena UH, Powell CM, Fecko JK, Cacioppo R, Chou HS, Cooper GM, Hansen U. Phosphorylation by cyclin C/cyclin-dependent kinase 2 following mitogenic stimulation of murine fibroblasts Inhibits transcriptional activity of LSF during G1 progression. Mol Cell Biol. 2009;29:2335–2345. doi: 10.1128/MCB.00687-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saxena UH, Owens L, Graham JR, Cooper GM, Hansen U. Prolyl isomerase Pin1 regulates transcription factor LSF (TFCP2) by facilitating dephosphorylation at two serine-proline motifs. J Biol Chem. 2010;285:31139–31147. doi: 10.1074/jbc.M109.078808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoo BK, Emdad L, Gredler R, Fuller C, Dumur CI, Jones KH, Jackson-Cook C, Su ZZ, Chen D, Saxena UH, Hansen U, Fisher PB, Sarkar D. Transcription factor Late SV40 Factor (LSF) functions as an oncogene in hepatocellular carcinoma. Proc Natl Acad Sci USA. 2010;107:8357–8362. doi: 10.1073/pnas.1000374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, Mills AS, Waxman S, Fisher RA, Llovet JM, Fisher PB, Sarkar D. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J Clin Invest. 2009;119:465–477. doi: 10.1172/JCI36460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zondervan PE, Wink J, Alers JC, JN IJ, Schalm SW, de Man RA, van Dekken H. Molecular cytogenetic evaluation of virus-associated and non-viral hepatocellular carcinoma: analysis of 26 carcinomas and 12 concurrent dysplasias. J Pathol. 2000;192:207–215. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH690>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 75.Wilkens L, Bredt M, Flemming P, Kubicka S, Klempnauer J, Kreipe H. Cytogenetic aberrations in primary and recurrent fibrolamellar hepatocellular carcinoma detected by comparative genomic hybridization. Am J Clin Pathol. 2000;114:867–874. doi: 10.1309/BMTT-JBPD-D13H-1UVD. [DOI] [PubMed] [Google Scholar]
- 76.Fan RH, Li J, Wu N, Chen PS. Late SV40 factor: a key mediator of Notch signaling in human hepatocarcinogenesis. World J Gastroenterol. 2011;17:3420–3430. doi: 10.3748/wjg.v17.i29.3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bellahcene A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer. 2008;8:212–226. doi: 10.1038/nrc2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoo BK, Gredler R, Chen D, Santhekadur PK, Fisher PB, Sarkar D. c-Met activation through a novel pathway involving osteopontin mediates oncogenesis by the transcription factor LSF. J Hepatol. 2011;55:1317–1324. doi: 10.1016/j.jhep.2011.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 80.Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol. 2001;153:1023–1034. doi: 10.1083/jcb.153.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Santhekadur PK, Gredler R, Chen D, Siddiq A, Shen XN, Das SK, Emdad L, Fisher PB, Sarkar D. Late SV40 Factor (LSF) Enhances Angiogenesis by Transcriptionally Up-regulating Matrix Metalloproteinase-9 (MMP-9) J Biol Chem. 2012;287:3425–3432. doi: 10.1074/jbc.M111.298976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harper J, Moses MA. Molecular regulation of tumor angiogenesis: mechanisms and therapeutic implications. EXS. 2006:223–268. doi: 10.1007/3-7643-7378-4_10. [DOI] [PubMed] [Google Scholar]
- 83.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 84.Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9:267–285. doi: 10.1111/j.1582-4934.2005.tb00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chantrain CF, Henriet P, Jodele S, Emonard H, Feron O, Courtoy PJ, DeClerck YA, Marbaix E. Mechanisms of pericyte recruitment in tumour angiogenesis: a new role for metalloproteinases. Eur J Cancer. 2006;42:310–318. doi: 10.1016/j.ejca.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 87.Aalinkeel R, Nair MP, Sufrin G, Mahajan SD, Chadha KC, Chawda RP, Schwartz SA. Gene expression of angiogenic factors correlates with metastatic potential of prostate cancer cells. Cancer Res. 2004;64:5311–5321. doi: 10.1158/0008-5472.CAN-2506-2. [DOI] [PubMed] [Google Scholar]
- 88.Parekh V, McEwen A, Barbour V, Takahashi Y, Rehg JE, Jane SM, Cunningham JM. Defective extraembryonic angiogenesis in mice lacking LBP-1a, a member of the grainyhead family of transcription factors. Mol Cell Biol. 2004;24:7113–7129. doi: 10.1128/MCB.24.16.7113-7129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 90.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 91.Yoshinare K, Kubota T, Watanabe M, Wada N, Nishibori H, Hasegawa H, Kitajima M, Takechi T, Fukushima M. Gene expression in colorectal cancer and in vitro chemosensitivity to 5-fluorouracil: a study of 88 surgical specimens. Cancer Sci. 2003;94:633–638. doi: 10.1111/j.1349-7006.2003.tb01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oguri T, Achiwa H, Bessho Y, Muramatsu H, Maeda H, Niimi T, Sato S, Ueda R. The role of thymidylate synthase and dihydropyrimidine dehydrogenase in resistance to 5-fluorouracil in human lung cancer cells. Lung Cancer. 2005;49:345–351. doi: 10.1016/j.lungcan.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 93.Yoo BK, Gredler R, Vozhilla N, Su ZZ, Chen D, Forcier T, Shah K, Saxena U, Hansen U, Fisher PB, Sarkar D. Identification of genes conferring resistance to 5-fluorouracil. Proc Natl Acad Sci USA. 2009;106:12938–12943. doi: 10.1073/pnas.0901451106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 95.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 96.Porta-de-la-Riva M, Stanisavljevic J, Curto J, Franci C, Diaz VM, Garcia de Herreros A, Baulida J. TFCP2c/LSF/LBP-1c is required for Snail1-induced fibronectin gene expression. Biochem J. 2011;435:563–568. doi: 10.1042/BJ20102057. [DOI] [PubMed] [Google Scholar]
- 97.Yamada KM. Cell surface interactions with extracellular materials. Annu Rev Biochem. 1983;52:761–799. doi: 10.1146/annurev.bi.52.070183.003553. [DOI] [PubMed] [Google Scholar]
- 98.Grant TJ, Bishop JA, Christadore LM, Barot G, Chin HG, Woodson S, Kavouris J, Siddiq A, Gredler R, Shen X-N, Sherman J, Meehan T, Fitzgerald K, Pradhan S, Briggs LA, Andrews WH, Sarkar D, Schaus SE, Hansen U. Antiproliferative small molecule inhibitors of transcription factor LSF reveal oncogene addiction to LSF in hepatocellular carcinoma. Proc Natl Acad Sci USA. 2012;12:4503–4508. doi: 10.1073/pnas.1121601109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW. An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene. 2007;26:6361–6371. doi: 10.1038/sj.onc.1210463. [DOI] [PubMed] [Google Scholar]
- 100.Jain A, Karadag A, Fohr B, Fisher LW, Fedarko NS. Three SIBLINGs (small integrin-binding ligand, N-linked glycoproteins) enhance factor H's cofactor activity enabling MCP-like cellular evasion of complement-mediated attack. J Biol Chem. 2002;277:13700–13708. doi: 10.1074/jbc.M110757200. [DOI] [PubMed] [Google Scholar]