

Abstract
It is commonly accepted that cancer is linked to inflammation. The possible mechanisms by which inflammation can contribute to carcinogenesis include induction of genomic instability, alterations in epigenetic events and subsequent inappropriate gene expression, enhanced proliferation of initiated cells, resistance to apoptosis, aggressive tumor neovascularization, invasion through tumor-associated basement membrane and metastasis. Inflammation also affects immune surveillance and responses to therapy. In this review, we overview the current understanding of different aspects of thyroid cancer and inflammation. Several studies have strongly suggested an increased risk of PTC in patients with Hashimoto's thyroiditis (HT), the most common autoimmune disease in thyroid cancer. Furthermore, an intense immune infiltrate is often associated with papillary thyroid carcinoma (PTC), and might play a critical role in the regulation of carcinogenesis and in carcinoma progression. The characterization of the most relevant inflammatory pathways of cancer-related inflammation (CRI) is instrumental for the identification of new target molecules that could lead to improved diagnosis and treatment.
Keywords: Cancer-related inflammation, thyroid cancer, inflammatory cell, chemokine
Introduction
Cancer is a complex disease involving numerous tempo-spatial changes in cell physiology. In an important review published in January 2000, Hanahan and Weinberg suggested that six essential alterations in cell physiology could underlie malignant cell growth. These six alterations occur in cancer cells independently of the originating tissue type and were described as the hallmarks of nearly all cancers. They included: self-sufficiency in growth signals, insensitivity to growth inhibitory signals, evasion of programmed cell death, limitless replicative potential, sustained angiogenesis, and ability to invade tissues and metastasize [1]. These hallmarks are usually quoted as the starting ground for new anticancer strategies, but recently new major features related to the probability of developing cancer have emerged [2]. Hanahan and Weinberg proposed other critical features to be functionally important for the development of cancer. The first involves the main mechanisms of cellular metabolism, which are reprogrammed supporting the growth and the proliferation of cancer cells, and thus favoring the progression of cancer. The second involves active evasion by cancer cells from attack and elimination by immune system [3]. The exploitation of immune mechanisms and evasion of immune surveillance are skills that cancer cells should acquire on their way to giving rise to a tumor [3,4]. An efficient immune response can eliminate cancer cells; however, the chronic activation of inflammatory cells present within or, more frequently, surrounding the neoplasia can sustain cancer cell proliferation.
Hanahan and Weinberg defined other two hallmarks of cancer as acquired functional capabilities that allow cancer cells to survive, proliferate, and disseminate: genome instability and tumor-promoting inflammation [3]. The loss of genomic “caretakers” or “guardians”, involved in sensing and repairing DNA damage, was proposed to explain the increased mutability of tumor cells [5,6]. The loss of these caretaker systems would allow genomic instability thus enabling pre-malignant cells to reach the six essential hallmarks of cancer.
In the last 10 years, it has also become increasingly clear that the inflammatory state of premalignant and malignant lesions play decisive roles in tumor initiation, promotion and progression. In particular, several lines of evidence suggest a strong association between chronic inflammation and increased susceptibility to neoplastic transformation and cancer development. In some studies, chronic infection leading to unresolved inflammation has been considered an important factor contributing to cellular transformation, tumorigenesis, and tumor progression [7].
The inflammation-cancer connection is not restricted to increased risk for a subset of tumors. An inflammatory component is present in the microenvironment of most neoplastic tissues, including those not causally related to an obvious inflammatory process [8]. The persistent release of inflammatory molecules in tumor site may affect tumor progression in a variety of ways, for instance by increasing tumor cell proliferation and resistance to apoptosis, by promoting angiogenesis and stroma remodeling, and by inhibiting the establishment of a protective antitumor immunity [9]. Recently, it has been proposed that two interrelated pathways link inflammation and cancer. The first pathway (intrinsic) is triggered by oncogenic events that initiate cancer. These events cause the transcription of inflammation-related genes in the neoplastic cells. The second pathway (extrinsic) is triggered by tumor-infiltrating inflammatory cells that increase cancer incidence or progression [8]. Cancer-associated inflammation includes leukocyte infiltration, prominently tumor-associated macrophages (TAM); expression of some cytokines and chemokines and tissue remodeling and angiogenesis. Consistent with this hypothesis, the use of anti-inflammatory therapies protects against several cancer types. Based upon these observations, it has been proposed that cancer-related inflammation (CRI) might be considered as the “seventh hallmark” of cancer [8]. Thyroid cancer obeys to these rules. First, an inflammatory component, which includes different leukocyte types, is frequently observed in thyroid tumors and the papillary histotype of thyroid cancer (PTC) is often associated with organ-specific autoimmune diseases, such as Hashimoto’s thyroiditis and Grave’s disease [10]. Second, RET/PTC, RAS (V12) or BRAF (V600E), the most common oncogenes found in human PTC, trigger an inflammatory transcriptional program in thyrocytes [10].
Thyroid cancer genetic alterations
Thyroid cancer is the most common cancer of the endocrine system and can derive from both the follicular and the parafollicular cells. The most common carcinomas deriving from thyroid follicular cells are: the well-differentiated thyroid carcinomas (WDTC), including the papillary carcinoma (PTC), and the follicular carcinoma (FTC); the poorly differentiated carcinomas, (PDC), histologically between the undifferentiated and the differentiated tumors; the undifferentiated or anaplastic carcinomas (ATC) [11]. Differentiated tumors (papillary or follicular) are highly treatable and usually curable. Undifferentiated tumors (poorly differentiated and anaplastic) are much less common, aggressive, metastasize early, and have a much poorer prognosis.
Finally, medullary thyroid carcinomas (MTC) derive from parafollicular C-cells. These carcinomas can occur as sporadic lesions or in the context of inherited neoplastic diseases, such as multiple endocrine neoplasia type 2A (MEN2A), type 2B (MEN2B) or familial medullary thyroid carcinoma (FMTC).
Papillary thyroid cancer (PTC) represents approximately 80% of all thyroid malignancies. The incidence of PTC is associated with radiation exposure; a considerable increase in this tumour type incidence has been observed after Chernobyl accident in 1986 [12]. Four genetic lesions, that are mutually exclusive, are associated with PTC. They include chromosomal aberrations targeting the RET or TRKA tyrosine kinase receptors and point mutations in RAS or BRAF genes. These genes encode for activators of the mitogen-activated protein kinase (MAPK) cascade. RET encodes for the tyrosine kinase receptor of growth factors belonging to the GDNF (Glial cell-Derived Neurotrophic Factor) family. In PTCs, RET tyrosine kinase domain is fused with the N-terminal region of constitutively expressed, heterologous genes, such as H4/CCD6 (in RET/PTC1) or RFG/NCOA4 (in RET/PTC3) [13]. In RET/PTC rearrangements, fusion with protein partners, possessing protein-protein interaction domains, provides RET/PTC proteins with coiled-coil domains, thereby resulting in ligand-independent activation of c-Ret tyrosine kinase activity [14]. RET/PTC oncogenes are detected with a high frequency in clinically-silent small PTC, confirming that they can be early events in thyroid tumorigenesis [15]. Similar rearrangements of the high affinity receptor for NGF (Nerve Growth Factor), TRKA, can be also found, albeit at a low prevalence in human PTC [16]. Activating point mutations in RAS small GTPases are found roughly in 10% of PTC, mainly in those belonging to the follicular variant (PTC-FV) [17]. Point mutations in BRAF are the most common genetic lesions found in PTC [18-22]. BRAF is a member of the RAF family of serine/threonine kinases and it is a component of the RAF-MEK-ERK signaling module. Activation of the RAF proteins is mediated through binding of RAS in its GTP-bound state.
Follicular thyroid cancer (FTC) accounts for 10-15% of thyroid cancers and its incidence is increased in areas of dietary iodine deficiency [23]. FTCs are single nodules that may be well circumscribed (minimally invasive), or widely infiltrative (with infiltration of adjacent thyroid tissue and blood vessels). In this carcinoma the presence of RAS mutations is quite common. Moreover it has been shown that a quite high proportion of FTCs carries the PAX8/PPARγ rearrangement [24]. The resulting fusion protein has dominant negative activity on wild type PPARγ [25].
Poorly differentiated carcinoma (PDTC) and Anaplastic thyroid carcinoma (ATC) account for 2-5% of thyroid malignancies; this carcinoma is highly malignant and markedly invasive and is composed of undifferentiated cells retaining markers of epithelial lineage. It can derive from pre-existing WDTC. RAS and BRAF point-mutations are prevalent in PDC and ATC [20,24,26,27]. Finally, p53 mutations are often found in ATC [28,29].
Medullary thyroid carcinoma (MTC) accounts for about 5% of all thyroid cancers; MTC can be sporadic or it could be one of the lesions that characterize the autosomal dominant MEN 2 syndromes (MEN2A, MEN2B and FMTC). MEN 2 syndromes are caused by germline point mutations that convert RET into a dominant oncogene [14].
In more than 80% of cases, MEN2B is caused by the Met918Thr substitution in the kinase domain of the receptor. In MEN2A and most FMTC patients, mutations affect one cysteine of the extracellular cysteine-rich domain of RET that can change to different residues.
About 40% of sporadic MTC cases harbor point mutations in RET [30].
Thyroid cancer related inflammation
The extrinsic pathway
The tumor mass is undoubtedly a multifaceted show, where different cell types, including neoplastic cells, fibroblasts, endothelial, and immune-competent cells communicate with each other by means of direct contact or cytokine and chemokine production and act in autocrine and paracrine manners to control and shape tumor growth. It is the expression of various immune mediators and modulators as well as the abundance and activation state of different cell types in the tumor microenvironment that dictate in which direction the balance is tipped and whether inflammation-promotes tumor growth or anti-tumor immunity will ensue [31,32].
The presence of leukocytes within tumors, observed in the 19th century by Rudolf Virchow, provided the first indication of a possible link between inflammation and cancer. A role for inflammation in tumorigenesis is now generally accepted, and it has become evident that an inflammatory microenvironment is an essential component of all tumors, including those which a direct causal relationship with inflammation is not yet proven [33].
Several reports indicate the presence of immune-inflammatory cell infiltrate also in thyroid cancer. Macrophages and immature dendritic cells accumulate in PTCs, both in tumoral stroma and at the invasive front [34,35]. The prevalence of lymphocytic infiltrate is generally significantly higher in patients with PTC than in those with benign thyroid lesions [36], indicating that the presence of these cells might favour cancer development. However, some authors indicate that the presence of chronic lymphocytic thyroiditis in patients with PTC correlates with an improved prognosis [37]. Moreover, PDCs and ATCs are characterized by a strongly reduced dendritic cell infiltrate with respect to PTCs, thus suggesting a protective role for these cell types in thyroid cancer [38]. Ryder and colleagues also demonstrated that PDTCs and ATCs displayed an increased density of TAM with respect to PTC and FTC and that TAM infiltration positively correlated with capsule invasion, extratiroidal extension and poor prognosis [35]. High levels of TAM are often correlated with a bad prognosis and are generally considered protumorigenic, and recent studies have also highlighted a link between their abundance and the process of metastasis [39,40]. These data suggest that TAM may favor the malignant progression of thyroid cancer.
During the last years it has been shown that mast cells (MC) also play an important role in neoplastic diseases. We showed that human PTCs, but not normal thyroid tissue, display an intense mast cell infiltrate and that its intensity positively correlates with the invasive behavior of thyroid carcinomas [41]. We found that thyroid carcinoma cell cultures are a potent chemoattractant for mast cells. This effect required thyroid carcinoma-cell-derived VEGF-A, because it was inhibited by an anti-VEGF-A antibody. Consistently, mast cells injected in the tail vein of immunodeficient mice are recruited to thyroid carcinoma cell xenografts. Interestingly, when mast cells and thyroid cancer cells were co-injected subcutaneously into nude mice, mast cells survived and proliferated suggesting that mediators released in situ can induce these effects [41]. Moreover, when thyroid cancer cells are treated with conditioned media from mast cells, they show higher proliferation rate, survival capacity and invasive ability. Accordingly, local co-injection of mast cells and thyroid cancer cells accelerated the growth of thyroid carcinoma xenograft in athymic mice. This effect was mediated by increased proliferation of thyroid cancer cells and vascularization and was reverted by sodium cromoglycate (Cromolyn), a specific MC degranulation inhibitor [41]. These data suggest that mast cells exert a protumorigenic effect on thyroid cancer cells (Figure 1).
Figure 1.
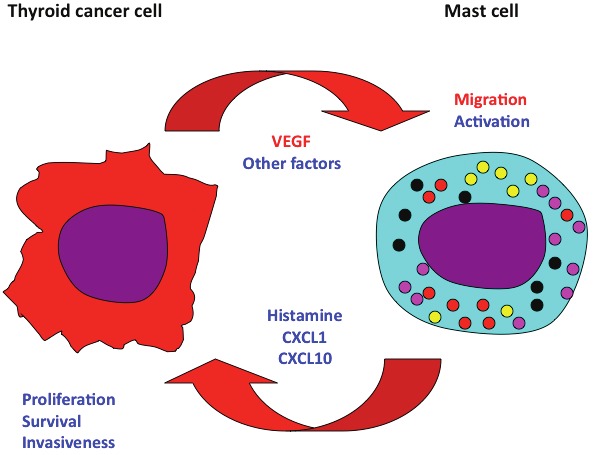
The complex relationship between mast cell and thyroid cancer cell. Mast cells exert a protumorigenic effect in thyroid cancer. They are recruited in tumor site by tumor-derived VEGF-A (and possibly by other chemoattractants), and are activated by cancer cell-secreted unknown factors. Activated mast cells secrete histamine, and the chemokines CXCL1 and CXCL10, that induce thyroid cancer cell proliferation, survival and invasion.
The presence of inflammatory cells has been also evaluated in transgenic mice expressing RET/PTC3 in the thyroid gland by means of a tissue specific promoter. The RET/PTC3 mice develop thyroid hyperplasia and solid tumor variants of papillary carcinoma. These PTC-like lesions were often characterized by a leukocytic infiltrate, composed mainly by macrophages [42]. Tumor incidence and burden of RET/PTC3 mice were influenced by the genetic background of the animals. In fact, when RET/PTC3 was expressed in C57BL/6 mice, tumors were significantly larger than those occurring in the C3H/HeJ animals. Cytokine expression was much higher in large tumors, suggesting that these molecules play a role in tumor growth. Since their discovery, the chemokine system has been strongly connected with cancer biology. In the last decade the knowledge in chemokine functions has expanded and now includes the promotion of the angiogenic switch and direct effects on tumor cells survival, proliferation and dissemination [43]. Probably, a different mixture of cytokine secreted by the C57BL/6 mice with respect to the C3H/HeJ animals could determinate a different polarization of the T CD4+ cells that is responsible for such effect. These data suggest that RET/PTC3 oncoprotein drives the recruitment of immune cells into tumor site, and is able to induce cytokine production that are involved in tumor progression.
In another set of experiments, Puffnock and Rothstein further clarified the role of the RET/PTC3 oncoprotein in the recruitment of immune cells. They studied tumor growth by using RET/PTC3 and a mutant isoform, RET/PTC3 Y1062F, that is defective in the activation of the most relevant RET-mediated signaling pathways [44]. Tumors were induced by injecting RET/PTC3- and RET/PTC3 Y1062F-expressing cells in syngeneic mice. RET/PTC3 tumors were significantly larger than RET/PTC3 mutant tumors. CD4+ and CD8+ T cell infiltrate density was comparable in both tumor groups. However, RET/PTC3, but not RET/PTC3 Y1062F tumors, displayed a remarkable leukocytic infiltrate characterized by CD11b+, Gr1+ myeloid cells, previously described as innate suppressive inflammatory cells [45,46]. These data show that RET/PTC3-positive thyroid cancer, as other cancer types, is capable of inducing escape from the immune response through the recruitment of CD11b+, Gr1+ cells. Whether other mechanisms of immune evasion are also operating in thyroid cancer is still unknown. Interestingly, the BRAF-MAPK signaling pathway has been shown to induce the synthesis and the secretion of immunosuppressive cytokines in melanomas [47]. These factors include IL-10, VEGF and IL6, cytokines that are also produced by thyroid cancer cells [42,44,48,50-53].
The extrinsic component of CRI in thyroid cancer is often represented by a coexisting autoimmune thyroid disease (AITD). AITD includes various clinical forms of autoimmune thyroiditis, such as classical Hashimoto’s thyroiditis (HT) and Graves’ disease (GD). HT is the most frequent chronic autoimmune disease diagnosed in the Western world and develops as a result of the interaction between predisposing genetic factors and environmental triggers. It is morphologically characterized by an inflammatory infiltrate and diffuse fibrosis which typically leads to progressive thyrocyte depletion. This loss of thyrocyte capacity results in impaired thyroid hormone production and clinical hypothyroidism. Sensitization of autoreactive CD4+ T-helper cells to thyroid antigens appears to be the initiating event, but multiple immunological mechanisms might contribute to thyrocyte death in HT. This disease is also characterized by proliferating nodules as well as cytological alterations and nuclear modifications similar to those of the papillary carcinomas [54]. The thyroid follicular cells in HT may have chromosomal defects, such as the RET/PTC1 rearrangement, the hallmark of many papillary thyroid carcinomas. Several other evidences suggest a role for RET/PTC in the association between thyroiditis and cancer. In fact, patients exposed to radiation from the Chernobyl nuclear power plant disaster often develop not only RET/PTC-induced papillary tumors but also an associated autoimmune thyroiditis [12]. Accordingly, transgenic mice engineered to express RET/PTC develop papillary carcinomas and chronic thyroiditis [55]. Finally, Wirtschafter and colleagues have detected RET/PTC expression in about 90% of the HT they have analyzed [56]. These data are, however, partially in contrast with other reports. Nikiforova and colleagues showed that RET/PTC rearrangements were detected only PTCs not associated with HT [57]. Rhoden and colleagues reported that only few follicular cells, expressing very low levels of RET/PTC, were detected in Hashimoto’s thyroiditis, thus suggesting that RET/PTC expression does not necessarily predicts the development of a PTC in patients with thyroiditis [58].
Two different models have been hypothesized to explain the correlation between Hashimoto’s thyroiditis and RET/PTC. The first one suggests that inflammation might facilitate the RET/PTC rearrangement. According to this hypothesis, free radical production, cytokine secretion, cellular proliferation as well as other phenomena correlated with inflammation might predispose to the rearrangement in follicular cells. It has been also show that the rate of mutations is much higher in inflamed than in normal tissues [8].
Chronic inflammation is linked to oxidative stress, which can cause DNA damage and thus contribute to the accumulation of cancer-initiating genetic alterations in cells. It is generally assumed that normal human epithelial cells do not tolerate oncogene expression, because excessive growth signals induce DNA replication stress, which induces oncogene-mediated senescence or apoptosis [59-61]. Thus, evasion from apoptosis or senescence is required for neoplastic transformation and can occur through additional genetic lesions. In support of this concept, different groups have shown that the ectopic expression of the RET/PTC oncogene in a normal continuous rat thyroid cell line (PC Cl3) induces apoptosis [62,63]. This might be due to the strong RET-mediated mitogenic stimuli. In thyroid cancer, in vitro and in vivo studies demonstrated that oncogene-induced senescence represents a barrier to the progress of thyroid tumors. In particular, the overexpression of main oncogenes activated in PTCs, BRAF, RET/PTC, TRK, and H-RAS are able to induce cellular senescence in human primary thyrocytes, promoting growth arrest, changes in cell morphology and chromatin modifications [64]. It is possible that cytokines and chemokines released by the inflammatory tumoral stroma by stimulating of thyroid cells in which oncogenic activation of RET, RAS, BRAF, randomly occurs, can confer resistance to oncogene-induced apoptosis. Proinflammatory cytokines and chemokines secreted by inflammatory cells can also regulate the senescent growth arrest of cancer cells and trigger an innate immune response that results in clearance of senescent lesions [65]. More recently, Guerra and colleagues suggested that one of the mechanisms by which pancreatitis-induced inflammation contributes to the progression of Pancreatic ductal adenocarcinoma is by eliminating the senescence barrier [66].
Another hypothesis suggests that RET/PTC rearrangements can promote recruitment of inflammatory cells. Accordingly, RET/PTC induces the synthesis of many inflammatory proteins in epithelial thyroid cells and a severe inflammatory response is observed in TG-RET/PTC transgenic mice in which RET/PTC expression is confined to the thyroid gland [44,53,65,67]. RET/PTC transgenic mice develop indeed papillary thyroid carcinomas and chronic thyroiditis; however, RET/PTC itself is not sufficient to induce a complete Hashimoto’s thyroiditis in these mice since this disease is characterized not only by lymphocytic infiltration, but by a humoral autoimmune reaction with the production of autoantibodies against thyroid antigens, and by the formation of lymphoid follicles in the thyroid parenchyma. Whether these features are present or not in TG-RET/PTC transgenic animals is still to be defined. In conclusion, we favour the hypothesis that AITD creates a protumorigenic microenvironment, in which the RET/PTC rearrangement is tolerated. The rearrangement itself than contributes to maintain the inflammatory reaction.
Intrinsic pathway
The intrinsic pathway of CRI is driven by the most frequent genetic alterations associated with thyroid cancer, such as RET and TRK rearrangements and BRAF and RAS mutations. In human PTCs, somatic chromosomal rearrangements lead to fusion of the 3’-terminal sequence of RET, which encodes the tyrosine kinase domain, with the 5’-terminal sequences of heterologous genes. The consequence of these rearrangements is the constitutive activation of RET, due to constitutive dimerization-oligomerization of the oncoprotein induced by the different RET-fused genes. Gain-of-function mutations of RET are involved in sporadic and familial cell-derived medullary thyroid carcinoma, including multiple endocrine neoplasia 2A (MEN2A), MEN2B and familial medullary thyroid carcinoma (FMTC) [69].
RET activation is achieved by constitutive dimerization of RET through the replacement of an extracellular cysteine that creates an unpaired residue, which can dimerize with another mutant RET molecule via an illegitimate disulfide bond resulting in constitutive activation of the signaling pathway [70]. Mutations that affect the intracellular domain, instead, induce an activation of the kinase in the absence of dimerization, presumably through a modification of the kinase structure. For instance, the MEN2B M918T point mutation induces a change in substrate specificity with respect to MEN2A mutants, which is reflected in the different capability of MEN2B mutants to phosphorylate endogenous tyrosines and signaling adaptors. These differences can account, at least in part, for the phenotypic differences between the two syndromes. Indeed, although both MEN2A and MEN2B are characterized by the presence of pheochromocytomas and MTCs, MEN2B MTCs occur earlier and are more aggressive. Furthermore, MEN2B patients also present skeletal abnormalities and ganglioneuromas [71].
Both point mutations and genetic rearrangements cause constitutive activation of the tyrosine kinase activity of RET in the absence of ligands. RET activation results in phosphorylation of key docking tyrosines that bind to several intracellular adaptor proteins such as SRC (at Y981), PLCγ (at Y1015), SHC, FRS2, IRS1. Among the tyrosines phosphorylated after RET activation, one of them, Y1062 has been shown to be important for RET-mediated biological activity in thyroid cells. The RET Y1062 phosphotyrosine serves as a docking site for multiple adaptors. It primarily supports SHC binding to recruit either the GRB2-SOS complex leading to MAPK activation or GRB2-GAB1 to stimulate PI3K/AKT signaling. GRB2 also binds directly to phospho-Y1096, where it preferentially activates PI3K/AKT signaling. By substituting Y1062 of RET with a phenylalanine, both in the context of a wild-type and of an oncogenically activated receptor, it is possible to abrogate RET-mediated signal transduction and biological activities [72]. We and others have shown that Ret is capable of activating the MAPK pathway in a Y1062-RAS- and BRAF-dependent manner [18,68]. Several groups also showed that RET-induced transcriptional activity depends almost entirely on the integrity of Y1062 residue and on the activation of the RAS/BRAF/MAPK pathway.
Russel and colleagues showed that the expression of RET/PTC3 isoform in PC Cl3 cells induced an increase in NFkB DNA-binding activity and a consequent increase in proinflammatory cytokine secretion. CXCL1/Groα, CCL2/mcp-1 and GM-CSF were up-regulated upon RET/PTC3 expression, and this increase depended on the integrity of residue 1062 of RET [73]. GDNF stimulation of neuroectodermal tumor cell line SK-N-MC, ectopically expressing the human wild-type RET, induced the production of high levels of IL8. This cytokine has also been found in TT human medullary thyroid carcinoma cell line carrying the RET/MEN2A oncogene and in the TPC1 human papillary thyroid carcinoma cell line carrying the RET/PTC1 oncogene [74]. IL8 is a pro-inflammatory, mitogenic and proangiogenic chemokine that contributes to several human cancer [73]. We used an oligonucleotide-based DNA microarray (Affymetrix) to search for RET/PTC-induced genes. To this aim, we used PC Cl3 cells. These cells are characterized by the typical differentiation parameters including thyroid epithelial morphology and dependence on TSH for growth. They were stably transfected with the three main oncogenes activated in human PTC: RET/PTC3, RASV12 and BRAFV600E. We compared oncogene-expressing cells with the parental ones, and found that RET/PTC3 induced a complex pattern of gene expression, which was completely dependent upon the integrity of the Y1062. HRAS (V12) or BRAF (V600E) oncoprotein modified the expression of several genes, and 50% of them were common between the three oncoprotein. Moreover, assays in vitro with siRNA and pharmacological inhibitors confirmed that most of these gene changes depended on the ERK pathway [68]. Global genome expression analysis of PC Cl3 transformed cells suggested also that the RET/PTC3 oncogene could induce a proinflammatory transcriptional program. This included several cytokines and chemokines such as osteopontin (OPN), VEGFA, CCL2, CXCL1 and CXCL10. Interestingly, CD44, CXCR2 and CXCR3, the receptors for respectively OPN, CXCL1 and CXCL10 were also expressed by transformed cells. Russell and colleagues found that RET/PTC3 caused the expression of proinflammatory proteins such as IL-1α, IL1β, IL6, and IL24 [42,50]. Similar results were obtained by Puxeddu and colleagues, who identified prostaglandin E2 (PGE2), microsomal prostaglandin E synthase1 (mPGES1), cycloxigenase2 (COX2), and several other genes involved in immune response and inflammation as RET/PTC3 induced genes [53,76]. We also found expression of CXCR4, a chemokine receptor whose ligand is ubiquitously expressed [77]. These results were confirmed by Borrello and coworkers by using human primary thyroid cells transduced with the RET/PTC oncogene [78]. The proinflammatory properties of RET/PTC in thyroid might have a dual effect: on one side, molecules such as OPN, CXCL1, CXCL10, CCL2, GM-CSF can influence immune response to the tumor by recruiting and functionally regulating immune cells. For instance, the transplantation of RET/PTC3 expressing thyrocytes into syngenic mice in vivo induced an intense macrophage infiltrate and neovascularization followed by cell death [73]. On the other side, secreted cytokines and chemokines, such as OPN, CXCL1, CXCL10, and IL24 can act as autocrine growth and survival factors for thyroid tumor cells, which express the cognate receptors on their plasma membrane [50,68,79]. These data are also corroborated by studies conducted on human thyroid tumors. Indeed, it has been shown that human thyroid samples express most of the inflammatory genes (CXCR4, CD44, OPN, CXCL1, CXCL10 and SDF-1) that were found in cell culture [68,77,78,80,81]. By searching for cytokines involved in the resistance of thyroid cancer cells to chemotherapeutic agents, Stassi and colleagues identified IL4 and IL10. These cytokines are typically secreted by T CD4+ cells polarized toward a TH2 phenotype. TH2 cytokines induce humoral immunity and promote thyrocyte survival in AITD by upregulating the levels of antiapoptotic proteins, such as Bcl2 and Bcl-XL [48]. Moreover, the authors demonstrated that IL4 and IL10 also induce resistance of thyroid cancer cells to FAS/FASL-mediated apoptosis. This effect is mediated by the IL4 and IL10-induction of two anti-apoptotic proteins, namely cFLIP and PED/PEA15 [49].
Conclusions
The inflammation-cancer link can be view as consisting of two pathways: an intrinsic pathway driven by genetic alterations that cause both inflammation and neoplasia and an extrinsic pathway, where inflammatory conditions promote cancer development. Key orchestrators of the inflammation-mediated tumor progression are transcription factors, cytokines, chemokines and infiltrating leukocytes [33].
It has become increasingly clear that a leukocyte infiltrate, varying in size, composition and distribution, is present in the majority of tumors and is involved in carcinogenesis, tumor growth, invasion and metastasis [9,82]. Nevertheless the role of these cells is complex; numerous studies highlighted the pro-tumoral activity of inflammation, while other evidences demonstrated that inflammation can support antitumor functions favoring protection against cancer progression. This paradox may be explained by the specific circuits expressed within the tumor microenvironment and by the abundance and activation state of different cell types in the tumor site.
Inflammatory infiltrates have also been found in human thyroid tumors. Macrophages and immature dendritic cells accumulate in PTCs, both in tumoral stroma and at the invasive front. Moreover we found that human thyroid carcinomas feature a remarkable mast cell infiltrate whose intensity correlates with the invasive phenotype [41]. Consistently, tumor-infiltrating mast cells are often associated with a bad prognosis in many cancers, suggesting a pro-tumorigenic role of these cells [83].
The oncogenes activated in thyroid carcinomas, RET/PTC, RAS and BRAF, triggering the MAPK cascade, can induce a cell-autonomous proinflammatory transcriptional program in thyrocytes, which mainly includes cytokines, chemokines and their receptors.
Chemokine receptors and their ligands are key orchestrators of leukocytes trafficking in homeostatic conditions as well as during inflammation and cancer. They are downstream of genetic events that cause neoplastic transformation and are abundantly expressed in chronic inflammatory conditions. Further, several tumors may use these molecules of the immune system also for growth, survival and metastasis [9,84]. In thyroid cancer, these molecules are able to act either by autocrine and paracrine mechanisms to sustain tumor cell proliferation, survival and invasiveness. Besides, by acting in a paracrine and, possibly, in an endocrine manner, they induce a remodeling of tumoral stroma by recruiting inflammatory, immune, endothelial and bone marrow-derived cells. Consistently, we found that mast cells are able to migrate toward thyroid cancer cell conditioned media, and this effect is due to thyroid cancer cell-derived VEGF-A. Based upon these observations, we favour the concept that, at least in the conclamed phase, thyroid cancer growth and progression are positively influenced by two major inflammatory components, one dependent on the cells that are present into cancer stroma, the other dependent on the activation, in epithelial cancer cells, of specific oncoprotein-mediated signaling.
For these reasons, not only oncoprotein, but the “cancer-related inflammation” represents an important target for innovative diagnostic and therapeutic strategies in thyroid cancers. The CRI can be targeted in several different ways: 1) inhibition of signal transducers and transcription factors that mediate survival and growth of malignant cells in response to inflammatory cytokines; 2) sequestration of chemokines and cytokines that recruit and sustain inflammatory cells in the tumor microenvironment; 3) depletion of immune and inflammatory cells that promote tumor development and progression, while sparing cell types and effector functions that support protective immune responses; 4) selective inhibition of tumor promoting cytokines without an effect on expression of antitumorigenic cytokines. In support of this, we have shown that a mast cell inhibitor, sodium cromoglycate (Cromolyn), strongly reduces the growth of thyroid cancer xenografts in immunodeficient mice. In conclusion, appraisal of the immune hallmarks of cancer, and of the possible countermeasures, opens the doors not only to widespread cancer immunoprevention but also to innovative and more efficacious cancer immunotherapies.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Lazebnik Y. What are the hallmarks of cancer? Nat Rev Cancer. 2010;10:232–233. doi: 10.1038/nrc2827. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–741. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Szent-Gyorgyi A. The living state and cancer. Proc Natl Acad sci USA. 1977;74:2844–2847. doi: 10.1073/pnas.74.7.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth DB, Gellert M. New guardians of the genome. Nature. 2000;404:823–825. doi: 10.1038/35009180. [DOI] [PubMed] [Google Scholar]
- 7.Swann JB, Vesely MD, Silva A, Sharkey J, Schreiber RD, Smyth MJ. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. PNAS. 2008;105:652–656. doi: 10.1073/pnas.0708594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related infalmmation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 9.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 10.Guarino V, Castellone MD, Avilla E, Melillo RM. Thyroid cancer and inflammation. Mol Cell Endocrinol. 2010;321:94–102. doi: 10.1016/j.mce.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Rosai J. Poorly differentiated thyroid carcinoma: introduction to the issue, its landmarks, and clinical impact. Endocr Pathol. 2004;15:293–296. doi: 10.1385/ep:15:4:293. [DOI] [PubMed] [Google Scholar]
- 12.Williams D. Cancer after nuclear fallout: lessons from the Chernobyl accident. Nat Rev Cancer. 2002;2:543–549. doi: 10.1038/nrc845. [DOI] [PubMed] [Google Scholar]
- 13.Manie S, Santoro M, Fusco A, Billaud M. The RET receptor: function in development and dysfunction in congenital malformation. Trends Genet. 2001;17:580–589. doi: 10.1016/s0168-9525(01)02420-9. [DOI] [PubMed] [Google Scholar]
- 14.Santoro M, Grieco M, Melillo RM, Fusco A, Vecchio G. Molecular defects in thyroid carcinomas: role of the RET oncogene in thyroid neoplastic transformation. Eur J Endocrinol. 1995;133:513–522. doi: 10.1530/eje.0.1330513. [DOI] [PubMed] [Google Scholar]
- 15.Fusco A, Chiappetta G, Hui P, Garcia-Rostan G, Golden L, Kinder BK, Dillon DA, Giuliano A, Cirafici AM, Santoro M, Rosai J, Tallini G. Assessment of RET/PTC oncogene activation and clonality in thyroid nodules with incomplete morphological evidence of papillary carcinoma: a search for the early precursors of papillary cancer. Am J Pathol. 2002;160:2157–2167. doi: 10.1016/S0002-9440(10)61164-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alberti L, Carniti C, Miranda C, Roccato E, Pierotti MA. RET and NTRK1 proto-oncogenes in human diseases. J Cell Physiol. 2003;195:168–186. doi: 10.1002/jcp.10252. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Z, Gandhi M, Nikiforova MN, Fischer AH, Nikiforov YE. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma. An unusually high prevalence of ras mutations. Am J Clin Pathol. 2003;120:71–77. doi: 10.1309/ND8D-9LAJ-TRCT-G6QD. [DOI] [PubMed] [Google Scholar]
- 18.Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signalling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–1457. [PubMed] [Google Scholar]
- 19.Xu X, Quiros RM, Gattuso P, Ain KB, Prinz RA. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res. 2003;63:4561–4567. [PubMed] [Google Scholar]
- 20.Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, Maximo V, Botelho T, Seruca R, Sobrinho-Simoes M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. 2003;22:4578–4580. doi: 10.1038/sj.onc.1206706. [DOI] [PubMed] [Google Scholar]
- 21.Fukushima T, Suzuki S, Mashiko M, Ohtake T, Endo Y, Takebayashi Y, Sekikawa K, Hagiwara K, Takenoshita S. BRAF mutations in papillary carcinomas of the thyroid. Oncogene. 2003;22:6455–6457. doi: 10.1038/sj.onc.1206739. [DOI] [PubMed] [Google Scholar]
- 22.Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, Beller U, Westra WH, Ladenson PW, Sidransky D. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst. 2003;95:625–627. doi: 10.1093/jnci/95.8.625. [DOI] [PubMed] [Google Scholar]
- 23.Williams D. The epidemiology of thyroid cancer. Ann Radiol (Paris) 1977;20:722–724. [PubMed] [Google Scholar]
- 24.Nikiforova MN, Kimura ET, Gandhi M, Biddinger PW, Knauf JA, Basolo F, Zhu Z, Giannini R, Salvatore G, Fusco A, Santoro M, Fagin JA, Nikiforov YE. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab. 2003;88:5399–5404. doi: 10.1210/jc.2003-030838. [DOI] [PubMed] [Google Scholar]
- 25.Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, Fletcher JA. PAX8 PPARgamma1 fusion oncogene in human thyroid carcinoma. Science. 2000;289:1357–1360. doi: 10.1126/science.289.5483.1357. [DOI] [PubMed] [Google Scholar]
- 26.García-Rostán G, Costa AM, Pereira-Castro I, Salvatore G, Hernandez R, Hermsem MJ, Herrero A, Fusco A, Cameselle-Teijeiro J, Santoro M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005;65:10199–10207. doi: 10.1158/0008-5472.CAN-04-4259. [DOI] [PubMed] [Google Scholar]
- 27.Begum S, Rosenbaum E, Henrique R, Cohen Y, Sidransky D, Westra WH. BRAF mutations in anaplastic thyroid carcinoma: implications for tumor origin, diagnosis and treatment. Mod Pathol. 2004;17:1359–1363. doi: 10.1038/modpathol.3800198. [DOI] [PubMed] [Google Scholar]
- 28.Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, Pierotti MA. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J Clin Invest. 1993;91:1753–1760. doi: 10.1172/JCI116385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest. 1993;91:179–184. doi: 10.1172/JCI116168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leboulleux S, Baudin E, Travagli JP, Schlumberger M. Medullary thyroid carcinoma. Clin Endocrinol (Oxf) 2004;61:299–310. doi: 10.1111/j.1365-2265.2004.02037.x. [DOI] [PubMed] [Google Scholar]
- 31.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. doi: 10.1016/S0065-2776(06)90001-7. [DOI] [PubMed] [Google Scholar]
- 33.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 34.Scarpino S, Stoppacciaro A, Ballerini F, Marchesi M, Prat M, Stella MC, Sozzani S, Allavena P, Mantovani A, Ruco LP. Papillary carcinoma of the thyroid: hepatocyte growth factor (HGF) stimulates tumor cells to release chemokines active in recruiting dendritic cells. Am J Pathol. 2000;156:831–837. doi: 10.1016/S0002-9440(10)64951-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryder M, Ghossein RA, Ricarte-Filho JC, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer. 2008;15:1069–1074. doi: 10.1677/ERC-08-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okayasu I. The Relationship of Lymphocytic Thyroiditis to the Development of Thyroid Carcinoma. Endocr Pathol. 1997;8:225–230. doi: 10.1007/BF02738789. [DOI] [PubMed] [Google Scholar]
- 37.Kebebew E, Treseler PA, Ituarte PH, Clark OH. Coexisting chronic lymphocytic thyroiditis and papillary thyroid cancer revisited. World J Surg. 2001;25:632–637. doi: 10.1007/s002680020165. [DOI] [PubMed] [Google Scholar]
- 38.Ugolini C, Basolo F, Proietti A, Vitti P, Elisei R, Miccoli P, Toniolo A. Lymphocyte and immature dendritic cell infiltrates in differentiated, poorly differentiated, and undifferentiated thyroid carcinoma. Thyroid. 2007;17:389–393. doi: 10.1089/thy.2006.0306. [DOI] [PubMed] [Google Scholar]
- 39.Pollard JW. Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol. 2008;84:623–630. doi: 10.1189/jlb.1107762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008;267:204–215. doi: 10.1016/j.canlet.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 41.Melillo RM, Guarino V, Avilla E, Galdiero MR, Liotti F, Prevete N, Rossi FW, Basolo F, Ugolini C, de Paulis A, Santoro M, Marone G. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene. 2010;29:6203–6215. doi: 10.1038/onc.2010.348. [DOI] [PubMed] [Google Scholar]
- 42.Russell JP, Engiles JB, Rothstein JL. Proinflammatory mediators and genetic background in oncogene mediated tumor progression. J Immunol. 2004;172:4059–4067. doi: 10.4049/jimmunol.172.7.4059. [DOI] [PubMed] [Google Scholar]
- 43.Mukaida N, Baba T. Chemokines in tumor development and progression. Exp Cell Res. 2012;318:95–102. doi: 10.1016/j.yexcr.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Pufnock JS, Rothstein JL. Oncoprotein signaling mediates tumor-specific inflammation and enhances tumor progression. J Immunol. 2009;182:5498–5506. doi: 10.4049/jimmunol.0801284. [DOI] [PubMed] [Google Scholar]
- 45.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–179. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 46.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–1656. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stassi G, Todaro M, Zerilli M, Ricci-Vitiani L, Di Liberto D, Patti M, Florena A, Di Gaudio F, Di Gesù G, De Maria R. Thyroid cancer resistance to chemotherapeutic drugs via autocrine production of interleukin-4 and interleukin-10. Cancer Res. 2003;63:6784–6790. [PubMed] [Google Scholar]
- 49.Todaro M, Zerilli M, Ricci-Vitiani L, Bini M, Perez Alea M, Maria Florena A, Miceli L, Condorelli G, Bonventre S, Di Gesù G, De Maria R, Stassi G. Autocrine production of interleukin-4 and interleukin- 10 is required for survival and growth of thyroid cancer cells. Cancer Res. 2006;66:1491–1499. doi: 10.1158/0008-5472.CAN-05-2514. [DOI] [PubMed] [Google Scholar]
- 50.Shinohara S, Rothstein JL. Interleukin 24 is induced by the RET/PTC3 oncoprotein and is an autocrine growth factor for epithelial cells. Oncogene. 2004;23:7571–7579. doi: 10.1038/sj.onc.1207964. [DOI] [PubMed] [Google Scholar]
- 51.Belletti B, Ferraro P, Arra C, Baldassarre G, Bruni P, Staibano S, De Rosa G, Salvatore G, Fusco A, Persico MG, Viglietto G. Modulation of in vivo growth of thyroid tumor-derived cell lines by sense and antisense vascular endothelial growth factor gene. Oncogene. 1999;18:4860–4869. doi: 10.1038/sj.onc.1202869. [DOI] [PubMed] [Google Scholar]
- 52.Jo YS, Li S, Song JH, Kwon KH, Lee JC, Rha SY, Lee HJ, Sul JY, Kweon GR, Ro HK, Kim JM, Shong M. Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J Clin Endocrinol Metab. 2006;91:3667–3670. doi: 10.1210/jc.2005-2836. [DOI] [PubMed] [Google Scholar]
- 53.Puxeddu E, Knauf JA, Sartor MA, Mitsutake N, Smith EP, Medvedovic M, Tomlinson CR, Moretti S, Fagin JA. RET/PTC-induced gene expression in thyroid PCCL3 cells reveals early activation of genes involved in regulation of the immune response. Endocr Relat Cancer. 2005;12:319–334. doi: 10.1677/erc.1.00947. [DOI] [PubMed] [Google Scholar]
- 54.Weetman AP. Cellular immune responses in autoimmune thyroid disease. Clin Endocrinol (Oxf) 2004;61:405–413. doi: 10.1111/j.1365-2265.2004.02085.x. [DOI] [PubMed] [Google Scholar]
- 55.Powell DJ Jr, Russell J, Nibu K, Li G, Rhee E, Liao M, Goldstein M, Keane WM, Santoro M, Fusco A, Rothstein JL. The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res. 1998;58:5523–5528. [PubMed] [Google Scholar]
- 56.Wirtschafter A, Schmidt R, Rosen D, Kundu N, Santoro M, Fusco A, Multhaupt H, Atkins JP, Rosen MR, Keane WM, Rothstein JL. Expression of the RET/PTC fusion gene as a marker for papillary carcinoma in Hashimoto’s thyroiditis. Laryngoscope. 1997;107:95–100. doi: 10.1097/00005537-199701000-00019. [DOI] [PubMed] [Google Scholar]
- 57.Nikiforova MN, Caudill CM, Biddinger P, Nikiforov YE. Prevalence of RET/PTC rearrangements in Hashimoto's thyroiditis and papillary thyroid carcinomas. Int J Surg Pathol. 2002;10:15–22. doi: 10.1177/106689690201000104. [DOI] [PubMed] [Google Scholar]
- 58.Rhoden KJ, Unger K, Salvatore G, Yilmaz Y, Vovk V, Chiappetta G, Qumsiyeh MB, Rothstein JL, Fusco A, Santoro M, Zitzelsberger H, Tallini G. RET/papillary thyroid cancer rearrangement in nonneoplastic thyrocytes: follicular cells of Hashimoto's thyroiditis share low-level recombination events with a subset of papillary carcinoma. J Clin Endocrinol Metab. 2006;91:2414–2423. doi: 10.1210/jc.2006-0240. [DOI] [PubMed] [Google Scholar]
- 59.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 60.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 61.Evan GI, d'Adda di Fagagna F. Cellular senescence: hot or what? Curr Opin Genet. 2009;1:25–31. doi: 10.1016/j.gde.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 62.Castellone MD, Cirafici AM, De Vita G, De Falco V, Malorni L, Tallini G, Fagin JA, Fusco A, Melillo RM, Santoro M. Ras-mediated apoptosis of PC CL 3 rat thyroid cells induced by RET/PTC oncogenes. Oncogene. 2003;22:246–255. doi: 10.1038/sj.onc.1206112. [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Knauf JA, Basu S, Puxeddu E, Kuroda H, Santoro M, Fusco A, Fagin JA. Conditional expression of RET/PTC induces a weak oncogenic drive in thyroid PCCL3 cells and inhibits thyrotropin action at multiple levels. Mol Endocrinol. 2003;17:1425–1436. doi: 10.1210/me.2003-0041. [DOI] [PubMed] [Google Scholar]
- 64.Vizioli MG, Possik P, Tarantino E, Meissl K, Borrello MG, Miranda C, Anania MC, Pagliarini S, Seregni E, Pierrotti M, Pilotti S, Pepper D, Greco A. Evidence of oncogene-induced senescence in thyroid carcinogenesis. Endocrine-related Cancer. 2011;18:743–757. doi: 10.1530/ERC-11-0240. [DOI] [PubMed] [Google Scholar]
- 65.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 66.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernández-Porras I, Cañamero M, Rodriguez-Justo M, Serrano M, Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Powell DJ Jr, Eisenlohr LC, Rothstein JL. A thyroid tumor-specific antigen formed by the fusion of two self proteins. J Immunol. 2003;170:861–869. doi: 10.4049/jimmunol.170.2.861. [DOI] [PubMed] [Google Scholar]
- 68.Melillo RM, Castellone MD, Guarino V, De Falco V, Cirafici AM, Salvatore G, Caiazzo F, Basolo F, Giannini R, Kruhoffer M, Orntoft T, Fusco A, Santoro M. The RET/PTC-RAS-BRAF linear signalling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J Clin Invest. 2005;115:1068–1081. doi: 10.1172/JCI22758. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA Jr, Marx SJ. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- 70.Santoro M, Carlomagno F, Melillo RM, Fusco A. Dysfunction of the RET receptor in human cancer. Cell Mol Life Sci. 2004;61:2954–2964. doi: 10.1007/s00018-004-4276-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hansford JR, Mulligan LM. Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. J Med Genet. 2000;37:817–827. doi: 10.1136/jmg.37.11.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 2001;12:361–373. doi: 10.1016/s1359-6101(01)00012-0. [DOI] [PubMed] [Google Scholar]
- 73.Russell JP, Shinohara S, Melillo RM, Castellone MD, Santoro M, Rothstein JL. Tyrosine kinase oncoprotein, RET/PTC3, induces the secretion of myeloid growth and chemotactic factors. Oncogene. 2003;22:4569–4577. doi: 10.1038/sj.onc.1206759. [DOI] [PubMed] [Google Scholar]
- 74.Iwahashi N, Murakami H, Nimura Y, Takahashi M. Activation of RET tyrosine kinase regulates interleukin-8 production by multiple signaling pathways. Biochem Biophys Res Commun. 2002;294:642–649. doi: 10.1016/S0006-291X(02)00528-4. [DOI] [PubMed] [Google Scholar]
- 75.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 76.Puxeddu E, Mitsutake N, Knauf JA, Moretti S, Kim HW, Seta KA, Brockman D, Myatt L, Millhorn DE, Fagin JA. Microsomal prostaglandin E2 synthase-1 is induced by conditional expression of RET/PTC in thyroid PCCL3 cells through the activation of the MEK-ERK pathway. Biol Chem. 2003;278:52131–52138. doi: 10.1074/jbc.M306003200. [DOI] [PubMed] [Google Scholar]
- 77.Castellone MD, Guarino V, De Falco V, Carlomagno F, Basolo F, Faviana P, Kruhoffer M, Orntoft T, Russell JP, Rothstein JL, Fusco A, Santoro M, Melillo RM. Functional expression of the CXCR4 chemokine receptor is induced by RET/PTC oncogenes and is a common event in human papillary thyroid carcinomas. Oncogene. 2004;23:5958–5967. doi: 10.1038/sj.onc.1207790. [DOI] [PubMed] [Google Scholar]
- 78.Borrello MG, Alberti L, Fischer A, Degl'innocenti D, Ferrario C, Gariboldi M, Marchesi F, Allavena P, Greco A, Collini P, Pilotti S, Cassinelli G, Bressan P, Fugazzola L, Mantovani A, Pierotti MA. Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc Natl Acad Sci USA. 2005;102:14825–14830. doi: 10.1073/pnas.0503039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Castellone MD, Celetti A, Guarino V, Cirafici AM, Basolo F, Giannini R, Medico E, Kruhoffer M, Orntoft TF, Curcio F, Fusco A, Melillo RM, Santoro M. Autocrine stimulation by osteopontin plays a pivotal role in the expression of the mitogenic and invasive phenotype of RET/PTC-transformed thyroid cells. Oncogene. 2004;23:2188–2196. doi: 10.1038/sj.onc.1207322. [DOI] [PubMed] [Google Scholar]
- 80.Guarino V, Faviana P, Salvatore G, Cartellone MD, Cirafici AM, De Falco V, Celetti A, Giannini R, Basolo F, Melillo RM, Santoro M. Osteopontin is overexpressed in human papillary thyroid carcinomas and enhances thyroid carcinoma cell invasiveness. J Clin Endocrinol Metab. 2005;90:5270–5278. doi: 10.1210/jc.2005-0271. [DOI] [PubMed] [Google Scholar]
- 81.De Falco V, Guarino V, Avilla E, Castellone MD, Salerno P, Salvatore G, Faviana P, Basolo F, Santoro M, Melillo RM. Biological role and potential therapeutic targeting of the chemokine receptor CXCR4 in undifferentiated thyroid cancer. Cancer Res. 2007;67:11821–11829. doi: 10.1158/0008-5472.CAN-07-0899. [DOI] [PubMed] [Google Scholar]
- 82.Coussens LM, Werb Z. Inflammatory cells and cancer: think different. J Exp Med. 2001;193:23–26. doi: 10.1084/jem.193.6.f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khazaie K, Blatner NR, Khan MW, Gounari F, Gounaris E, Dennis K, Bonertz A, Tsai FN, Strouch MJ, Cheon E, Phillips JD, Beckhove P, Bentrem DJ. The significant role of mast cells in cancer. Cancer Metastasis. 2011;30:45–60. doi: 10.1007/s10555-011-9286-z. [DOI] [PubMed] [Google Scholar]
- 84.Allavena P, Germano G, Marchesi F, Mantovani A. Chemokines in cancer related inflammation. Exp Cell Res. 2011;317:664–673. doi: 10.1016/j.yexcr.2010.11.013. [DOI] [PubMed] [Google Scholar]