

Background: Podocyte dedifferentiation is the hallmark of many glomerular kidney diseases.
Results: Krüppel-like factor 15 (KLF15) increases podocyte differentiation; KLF15 null mice exhibit more injury in models of kidney disease.
Conclusion: KLF15 is a novel transcriptional regulator of podocyte differentiation; a loss of KLF15 increases the susceptibility to kidney injury.
Significance: Identification of KLF15 in mediating podocyte differentiation provides new insight into kidney disease.
Keywords: HIV-1, Kidney, Kruppel-like Factor (KLF), Nephrology, Podocytes, Focal Segmental Glomerulosclerosis (FSGS), Adriamycin, HIV-associated Nephropathy, Nephrin, Wilms Tumor-1
Abstract
Podocyte injury resulting from a loss of differentiation is the hallmark of many glomerular diseases. We previously showed that retinoic acid (RA) induces podocyte differentiation via stimulation of the cAMP pathway. However, many podocyte maturity markers lack binding sites for RA-response element or cAMP-response element (CREB) in their promoter regions. We hypothesized that transcription factors induced by RA and downstream of CREB mediate podocyte differentiation. We performed microarray gene expression studies in human podocytes treated with and without RA to identify differentially regulated genes. In comparison with known CREB target genes, we identified Krüppel-like factor 15 (KLF15), a kidney-enriched nuclear transcription factor, that has been previously shown to mediate cell differentiation. We confirmed that RA increased KLF15 expression in both murine and human podocytes. Overexpression of KLF15 stimulated expression of differentiation markers in both wild-type and HIV-1-infected podocytes. Also, KLF15 binding to the promoter regions of nephrin and podocin was increased in RA-treated podocytes. Although KLF15−/− mice at base line had minimal phenotype, lipopolysaccharide- or adriamycin-treated KLF15−/− mice had a significant increase in proteinuria and podocyte foot process effacement with a reduction in the expression of podocyte differentiation markers as compared with the wild-type treated mice. Finally, KLF15 expression was reduced in glomeruli isolated from HIV transgenic mice as well as in kidney biopsies from patients with HIV-associated nephropathy and idiopathic focal segmental glomerulosclerosis. These results indicate a critical role of KLF15 in mediating podocyte differentiation and in protecting podocytes against injury.
Introduction
In normal mature kidneys, podocytes are regarded as highly differentiated and growth-arrested cells with a limited capacity for replication. Podocyte injury manifests clinically as proteinuria and structurally as foot process effacement. Significant podocyte injury leads to proliferation, apoptosis, and a loss of podocyte differentiation markers. For example, in minimal change disease, nephrotic range proteinuria is associated with significant but reversible effacement of the foot process. However, in diabetic nephropathy, the podocyte number is reduced because of either cell detachment or apoptosis (1, 2). In collapsing focal segmental glomerulosclerosis (FSGS)3 of HIV-associated nephropathy (HIVAN), proliferation and dedifferentiation of podocytes are prominent features (3, 4). Specifically, the viral infection of the podocytes is responsible for podocyte proliferation and dedifferentiation in HIVAN (5–8). Because podocyte dedifferentiation is a major mechanism in the pathogenesis of podocyte injury, it is critical to identify targets for therapy that can prevent or reverse this disease process.
Retinoic acids (RA) are derivatives of vitamin A and have multiple cellular functions, including inhibition of proliferation, induction of cell differentiation, and inhibition of inflammation (9). In addition to their established benefits in the treatment of hematologic malignancies, RA also has renal protective effects in several experimental models of kidney disease (10–13), including HIV-1 transgenic mouse (Tg26), a model for HIVAN (14). Furthermore, RA also induces the expression of podocyte differentiation markers in vitro and in vivo (15). These studies provide a strong scientific basis supporting the use of RA in podocyte diseases. In fact, a phase II clinical trial examining the efficacy of RA for treatment of podocyte diseases, including minimal change disease, FSGS, or collapsing glomerulopathy, is ongoing (ClinicalTrials.gov Identifier NCT00098020). The mechanism by which RA protects against podocytes injury, however, has yet to be clearly characterized.
We previously described that RA can attenuate podocyte dedifferentiation in a cAMP-dependent manner via the activation of protein kinase A (PKA) and cAMP-response element-binding protein (CREB) (14). However, many markers of a differentiated podocyte, such as synaptopodin or nephrin, do not have CREB-binding sites in their promoter region. Thus, we hypothesized that transcription factors induced by RA and downstream of CREB could regulate the expression of podocyte differentiation markers. By comparing RA-regulated genes in podocytes with known CREB-targeted genes, we identified a nuclear transcription factor, Krüppel-like factor 15 (KLF15), that is a kidney-enriched factor known to promote adipocyte differentiation (16). KLFs are a subfamily of 17 DNA-binding transcriptional regulators that are involved in a broad range of cellular processes, including cell differentiation (17, 18). Comparative promoter analysis of podocyte slit diaphragm components revealed that KLF is one of four common transcriptional binding sites on many podocyte-specific genes (19), suggesting a potential role of KLF family proteins in regulating podocyte-specific functions. Here, we demonstrate that KLF15 is a key transcription factor for podocyte differentiation, and a loss of KLF15 increases the susceptibility of podocytes to injury.
EXPERIMENTAL PROCEDURES
Cell Culture
Conditionally immortalized murine and human podocytes were gifts from Dr. Peter Mundel (Massachusetts General Hospital, Boston) and Dr. Moin Saleem (University of Bristol, Southmead Hospital, Bristol, UK). Methods for podocyte cultivation, immortalization, and differentiation will be based on a previously described protocol (14). These cells proliferate under permissive conditions (γ-interferon at 33 °C) but differentiate under nonpermissive conditions (37 °C).
Infection of Murine and Human Podocytes with HIV-1 or Control Vector
The HIV constructs have been described previously (7). Expression of HIV-1 genes was confirmed by immunoblot analysis. Briefly, the HIV-1 gag/pol-deleted construct pNL4-3:d1443 was derived from the provirus pNL4-3. A fragment that contained the enhanced GFP gene (from pEGFP-C1; Clontech) was inserted at the SphI/MscI gag/pol deletion site. The expression of HIV-1 genes was confirmed by Western blot analysis. The HIV-1 gag/pol genes and the VSV.G envelope glycoprotein were provided in trans using pCMV R8.91 and pMD.G plasmids, respectively (gifts of Dr. Didier Trono, Salk Institute, La Jolla, CA). As a negative control, the virus was also produced from pHR-CMV-IRES2-GFP-ΔB, which contains the HIV-1 long term repeat and enhanced GFP. In all experiments, cells were grown at 37 °C on type I collagen-coated dishes for 10 days to inactivate the temperature-sensitive T antigen and to allow for differentiation. By Western blot, the T antigen was confirmed to be absent in these cells.
Retinoic Acid Treatment of Podocytes in Culture
Wild-type and HIV-1-infected murine podocytes were treated with atRA (1 μm) or control vehicle as described previously (20). Similarly, human podocytes infected with and without HIV-1 were treated with or without atRA (1 μm).
KLF15 Construct
A KLF15 cDNA clone purchased from Thermo Scientific (Huntsville, AL) was inserted into a gag-, pol-, and env-deficient lentivector construct, VVE/BBW (a gift of Dr. G Luca Gusella, Mount Sinai School of Medicine). Lentiviral particles were generated and used for infection of podocytes. Podocytes with stable KLF15 expression were selected using blasticidin. Cultured murine and human podocytes infected with the empty VVE/BBW lentivector served as controls.
Real Time PCR
Total RNA was extracted by using TRIzol (Invitrogen). First strand cDNA was prepared from total RNA (1.5 μg) using the SuperScriptTM III first strand synthesis kit (Invitrogen), and cDNA (1 μl) was amplified in triplicate using SYBR GreenER qPCR Supermix on an ABI PRISM 7900HT (Applied Biosystems, Foster City, CA). Pre-designed primer sets were obtained from Sigma for synaptopodin, nephrin, WT-1, podocin, GAPDH, and KLF15 (mouse F, 5′-GAGACCTTCTCGTCACCGAAA-3′, and R, 5′-GCTGGAGACATCGCTGTCCAT-3′), (human F, 5′-GTTGGGTATCTGGGTGATAGGC-3′, and R, TGAGAGTCGGGACTGGAACAG-3′). Light Cycler analysis software was used to determine crossing points using the second derivative method. Data were normalized to housekeeping genes (GAPDH) and presented as fold increase compared with RNA isolated from WT animals using the 2−ΔΔCT method.
Chromatin Immunoprecipitation Assay (ChIP)
The ChIP assay was performed using a kit from Upstate Biotechnologies, Inc. (Lake Placid, NY), as described previously (21). Briefly, 3 × 107 cultured murine podocytes per experimental condition were serum-starved for 16 h and then treated with either atRA (1μm) or control vehicle for 4 h. Cells were cross-linked with formaldehyde for 10 min, followed by the addition of 1/20 volume of 2.5 m glycine to quench unreacted formaldehyde. Cells were lysed using a series of non-SDS-containing buffers as described previously (21). Chromatin extracted from the lysed cells was sonicated using a Misonix 3000 sonicator with microtip to generate chromatin fragments of between 300 and 1000 bp. Immunoprecipitation of KLF15-cross-linked chromatin was carried out using M-280 Dynabeads (Invitrogen) with sheep anti-rabbit immunoglobulin G (IgG) preincubated with goat anti-KLF15 (Abcam) antibody. To control for nonspecific IgG binding, rabbit IgG (Sigma) was used. After incubation of chromatin with antibody-coupled Dynabeads, the beads were washed several times, and immunoprecipitated chromatin complexes were eluted from the beads. DNA-protein cross-links were reversed by incubation at 65 °C for 6 h, and then RNase A and protein kinase K were added sequentially to remove RNA and proteins. DNA was purified using the QIAquick® PCR purification kit (Qiagen) by following the manufacturer's instructions. Purified DNA was used for the analysis of the nephrin and podocin proximal promoter by real time PCR on an ABI PRISM 7900HT using SYBR GreenER qPCR supermix. PCR primers for murine nephrin and podocin promoters were derived from available sequence (GenBankTM accession numbers AAK38483 and AY050309, respectively) and were the following: nephrin F, 5′-CCCACGCACACAGGCCTAGC-3′, and R, 5′-TTCCTGAGTCTGCCCAGCCG; podocin F, 5′-AGAGAACCCCAAGGACACGTCA-3′, and R, 5′-AGCAGGCTCTGCTTAGGTGGCT-3′. The relative amplification of the promoter sequence of each gene was calculated using the 2−ΔΔCT method, and normalization was performed against the 1:100 diluted input of DNA.
Genotyping of Tg26 Mice
Mount Sinai School of Medicine Animal Institute Committee approved all animal studies, and the National Institutes of Health Guide for the Care and Use of Laboratory Animals was strictly followed. Derivation of a transgenic mouse line (Tg26 mice) that bears a defective HIV-1 provirus lacking gag-pol (Tg26) has been described (22). Tg26 mice are in the FVB/N background. Mice generated from the same litter of Tg26 mice were used as the controls in the studies. Genotyping by tail preparation and PCR was performed at 2 weeks of age as described previously (20).
Generation of KLF15−/− Mice
KLF15 gene was targeted in mice by homologous recombination using the targeting strategy with a KLF15-targeting construct as described previously (23). Generation of targeted C57BL/6 cell clones and germ line transmission of targeted construct was verified (23). KLF15−/− mice were viable, fertile, and born in the Mendelian ratios as expected in C57BL/6 background.
LPS Murine Model
Transient nephrotic syndrome with podocyte FP effacement was induced by LPS injection as described previously (24). Base-line urine was collected in wild-type and KLF15−/− mice (C57BL/6 at 8–9 weeks of age). All mice were administered low dose LPS (10 μg/g) intraperitoneally at 0 and 24 h. Urine was collected at 12-h increments with 1 ml of normal saline boluses intraperitoneally. Mice were sacrificed at 48 h.
Adriamycin Murine Model
In the adriamycin model, wild-type and KLF15−/− mice (C57BL/6 at 8–9 weeks of age) were administered adriamycin (20 mg/kg) intravenously by tail vein injection (25). Urine was collected weekly to assess for proteinuria, and mice were sacrificed 4 weeks post-treatment.
AtRA Treatment of Mice
Administration of atRA was performed as described previously (14). 12 h prior to treatment with LPS, mice were administered intraperitoneal injections of atRA at 16 mg/kg or vehicle alone (DMSO). Following the initial injection, two additional injections were given at 24-h intervals at the same dose. Mice were sacrificed at 48 h.
Measurement of Urine Albumin and Creatinine
Urine albumin was quantified by ELISA using a kit from Bethyl Laboratories, Inc. (Houston, TX). Urine creatinine levels were measured in the same samples using QuantiChromTM creatinine assay kit (DICT-500) (BioAssay Systems) according to the manufacturer's instruction. The urine albumin excretion rate was expressed as the ratio of albumin to creatinine.
Histopathology by Transmission Electron Microscopy
Mice were perfused with PBS and then immediately fixed in glutaraldehyde for EM. Sections were mounted on a copper grid and photographed under a Hitachi H7650 microscope. Briefly, negatives were digitized, and images with a final magnitude of approximately ×15, 000 were obtained. ImageJ 1.26t software (National Institutes of Health, rsb.info.nih.gov) was used to measure the length of the peripheral GBM, and the number of slit pores overlying this GBM length was counted. The arithmetic mean of the foot process width (WFP) was calculated as shown in Equation 1,
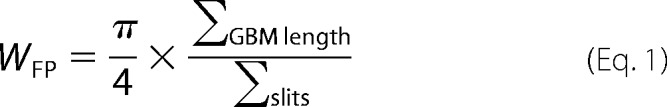 |
where Σslits indicates the total number of slits counted; ΣGBM length indicates the total GBM length measured in one glomerulus, and π/4 is the correction factor for the random orientation by which the foot processes were sectioned (26).
Isolation of Glomeruli from Mice for RNA Extraction
Mouse glomeruli were isolated as described (27). Briefly, animals were perfused with Hanks' buffered salt solution containing 2.5 mg/ml iron oxide and 1% bovine serum albumin. At the end of perfusion, kidneys were removed, decapsulated, minced into 1-mm3 pieces, and digested in Hanks' buffered salt solution containing 1 mg/ml collagenase A and 100 units/ml deoxyribonuclease I. Digested tissue was then passed through a 100-μm cell strainer and collected by centrifugation. The pellet was resuspended in 2 ml of Hanks' buffered salt solution, and glomeruli were collected using a magnet. The purity of glomerular was verified under microscopy. Total RNA was isolated from kidney glomeruli of mice using TRIzol (Invitrogen).
Isolation of Primary Podocytes
After glomerular isolation, primary podocytes were isolated as described previously (28). In brief, isolated glomeruli were initially cultured on collagen I-coated culture dishes in RPMI 1640 medium containing 10% fetal bovine serum (Cansera International, Canada) supplemented with 1% insulin/transferrin/selenium A liquid media supplement (Invitrogen) and 100 units/ml penicillin. Cultures were incubated in a 37 °C humidified incubator. Subculture of primary podocytes was performed after 5 days of culture of isolated glomeruli. Cellular outgrowths were detached with trypsin/EDTA solution (Sigma) and passed through a 25-mm sieve to remove the remaining glomerular cores. The filtered cells were cultured on collagen I-coated dishes and processed for RNA or protein preparation.
Western Blot
Glomeruli were lysed with a buffer containing 1% Triton, a protease inhibitor mixture and tyrosine and serine/threonine phosphorylation inhibitors. Lysates were subjected to immunoblot analysis using goat anti-KLF15 (Abcam), rabbit anti-synaptopodin (Sigma), rabbit anti-nephrin (a gift from Dr. Tomoko Takano), and rabbit anti-GAPDH antibodies (Sigma). Densitometry analysis for quantification was performed as described previously (29).
Immunofluorescence
Kidney sections from these mice were prepared in an identical fashion. Immunostaining was performed using rabbit anti-synaptopodin (Fitzgerald), rabbit anti-nephrin (a gift from Dr. Larry Holzman), and mouse anti-WT-1 antibodies (Santa Cruz Biotechnology). After washing, sections were incubated with a fluorophore-linked secondary antibody (Alexa Fluor 488 anti-rabbit IgG and Alexa Fluor 568 anti-mouse IgG from Invitrogen). After staining, slides were mounted in Aqua Poly/Mount (Polysciences Inc.) and photographed under an AxioVision IIe microscope with a digital camera.
Immunohistochemistry
Archival human biopsy specimens of healthy donor nephrectomies, HIVAN, and idiopathic FSGS for immunohistochemistry were collected at Columbia University under a protocol approved by its Institutional Review Board. Specimens were initially baked for 20 min in 55–60 °C oven and then processed as described previously below. Briefly formalin-fixed and paraffin-embedded sections were deparaffinized, and endogenous peroxidase was inactivated with H2O2. Sections were then blocked in 2% goat serum in phosphate-buffered saline (PBS) for 1 h at room temperature and then incubated with a rabbit anti-KLF15 antibody (1:1000, GenScript) at 4 °C overnight. The next day, sections were washed three times with PBS and then incubated with secondary antibody for 30 min. Positive staining was revealed by peroxidase-labeled streptavidin and diaminobenzidine substrate. The control included a section stained with only secondary antibody.
Quantification of Immunostaining
After sections were stained with anti-KLF15 antibody, negatives were digitized, and images with a final magnitude of approximately ×40 were obtained. ImageJ 1.26t software was used to measure the level of immunostaining in the glomeruli. First, the images were converted to 8-bit grayscale. Next, the glomerular region was selected for measurement of area and integrated density. Then, the background intensity was measured by selecting three distinct areas in the background with no staining. The corrected optical density (COD) was determined as shown in Equation 2,
where ID is the integrated density of the selected glomerular region; A is the area of the selected glomerular region, and MGV is the mean gray value of the background readings) (30).
Statistical Analysis
Data were expressed as mean ± S.D. The unpaired t test was used to analyze data between two groups. The analysis of variance followed by Bonferroni correction was used when more than two groups were present. All experiments were repeated at least three times, and representative experiments are shown. Statistical significance will be considered when at p < 0.05.
RESULTS
Identification of KLF15 as Potential Downstream Transcription Factor Mediating RA-induced Podocyte Differentiation
Our previous studies suggest that RA-induced podocyte differentiation occurs through activation of the cAMP/PKA/CREB pathway (14). However, many podocyte-specific genes lack CREB-binding sites. To identify genes downstream of CREB that may potentially mediate RA-induced podocyte differentiation, we performed GeneChip expression analysis on podocytes stimulated with either atRA (1 μm) or with dimethyl sulfoxide (DMSO) (control) at 6 h (n = 4). A total of 111 genes (90 up and 21 down) were selected based on t test with a false discovery rate of 0.1, indicating that 11 genes on the list may be picked at random. The functions of these genes include anti-oxidant, anti-inflammation, regulation of cell proliferation, apoptosis, and differentiation. We then compared this list of genes with CREB ChIP sequencing database deposited in Sequence Read Archive of NCBI (Series GSE17067; 20442865). From this comparison, we identified several important CREB-mediated genes in podocytes in response to atRA. These genes include 16 up-regulated genes (>1.5-fold) and 12 down-regulated genes (>1.5-fold) (Table 1). These genes are involved in various cellular processes, including cell differentiation, anti-inflammatory processes, and inhibition of reactive oxidant species generation. Interestingly, only two of the identified targets were directly implicated in cellular differentiation, glial cell-derived neurotrophic factor (GDNF), and KLF15. Previous studies have already demonstrated that GDNF plays a role in podocyte differentiation (31). KLF15, which was originally identified in kidneys (14), has been shown to promote differentiation of adipocytes under metabolic stress (16), but its role in podocyte differentiation was to date unknown. In addition, recent studies suggest that many of podocyte-specific genes share four common binding sites in their promoter regions with one of the most common binding domains being for Kruppel-like factor (19). Thus, we chose to further explore the role of KLF15 in regulation of podocyte differentiation.
TABLE 1.
Genes regulated by RA and targeted by CREB in mouse podocytes
Significant criteria are as follows: 1.5-fold change and 0.1 false discovery rate.
| Gene symbol | Description |
|---|---|
| Up-regulated genes | |
| GSTA4 | Glutathione S-transferase α4 |
| PER2 | Period homolog 2 |
| IFRD1 | Interferon-related developmental regulator 1 |
| HIVEP1 | Human immunodeficiency virus type I enhancer binding protein 1 |
| LIPE | Lipase, hormone-sensitive |
| METTL1 | Methyltransferase-like 1 |
| RFX1 | Regulatory factor X, 1 (influences HLA class II expression) |
| SNX5 | Sorting nexin 5 |
| PLAUR | Plasminogen activator, urokinase receptor |
| CLCN3 | Chloride channel 3 |
| SFXN4 | Sideroflexin 4 |
| DMD | Dystrophin |
| FOXC1 | Forkhead box C1 |
| KLF15 | Kruppel-like factor 15 |
| GDNF | Glial cell-derived neurotrophic factor |
| GABARAPL1 | GABAA receptor-associated protein like 1 |
| Down-regulated genes | |
| GATA3 | GATA-binding protein 3 |
| CCND2 | Cyclin D2 |
| ISGF3G | Signal transducer and activator of transcription 2 |
| MCAM | Melanoma cell adhesion molecule |
| SIAT1 | ST6 β-galactosamide α-2,6-sialyltransferase 1 |
| FGF18 | Fibroblast growth factor 18 |
| PITX2 | Paired-like homeodomain 2 |
| GNB4 | Guanine nucleotide-binding protein (G protein), β-polypeptide 4 |
| WISP1 | WNT1-inducible signaling pathway protein 1 |
| YWHAZ | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, ζ polypeptide |
| CACNA1C | Calcium channel, voltage-dependent, L-type, α1C subunit |
Retinoic Acid Stimulates KLF15 Expression in Cultured Podocytes
To confirm whether atRA increased KLF15 expression, wild-type and HIV-1-infected murine podocytes were exposed to atRA for 6 h. atRA-treated wild-type and HIV-1-infected murine podocytes revealed a significant increase in KLF15 mRNA expression (Fig. 1A). These findings were replicated in immortalized human podocytes exposed to atRA (Fig. 1B). Time course studies demonstrated that atRA induced KLF15 expression in human podocytes with a peak rise within 1 h after atRA addition, suggesting that KLF15 is an early inducible gene (Fig. 1D). Additionally, these findings were confirmed by Western blot (Fig. 1, C and E). Combined, these findings confirm the microarray gene expression analysis that atRA stimulates KLF15 expression in murine and human podocytes.
FIGURE 1.

RA stimulates KLF15 mRNA expression by real time PCR. KLF15 mRNA expression was measured in atRA (1 μm) treated and untreated wild-type (WT) and HIV-1-infected murine (A) and human (B) podocytes in culture (n = 3, **, p < 0.05 versus control cells without RA treatment). C, KLF15 protein expression was measured in atRA (1 μm)-treated and untreated wild-type (WT) and HIV-1-infected human podocytes in culture. The representative blots of three independent experiments are shown. The densitometry analyses of these blots are shown in the lower panel (n = 3, *, p < 0.05 versus cells without atRA treatment). Human podocytes were treated with atRA (1 μm) for the indicated time intervals, and cells were collected for KLF15 expression by real time PCR (D) and Western (E) (n = 3, *, p < 0.05 versus control cells without atRA treatment). The representative blots of three independent experiments are shown. The densitometry analyses of these blots are shown in the lower panel (n = 3, *, p < 0.05 versus cells without atRA treatment).
Retinoic Acid Increases Binding of KLF15 in Promoter Regions of Podocyte-specific Genes
atRA has been shown to induce the expression of nephrin and podocin in murine podocytes (14). Because we observed that atRA enhanced the expression of KLF15, we examined KLF15 binding to the promoter regions of podocyte-specific genes in response to atRA treatment by ChIP followed by quantitative real time PCR. Binding of KLF15 to the putative KLF15-binding sites in the promoter of nephrin and podocin was significantly increased in atRA-treated murine podocytes compared with the untreated cells (Fig. 2, A and B). In contrast, this finding was not observed in atRA-treated podocytes isolated from KLF15−/− mice (Fig. 2, C and D). These findings suggest that KLF15 likely mediates RA-induced expression of podocyte-specific genes.
FIGURE 2.

Exposure to atRA increased binding of KLF15 in the promoter region of nephrin and podocin. Differentiated wild-type (WT) podocytes were stimulated with or without atRA for 4 h, and nuclear proteins were extracted for ChIP assay as described. Binding of KLF15 to the putative KLF15-binding sites in the promoter of nephrin (A) and podocin (B) was measured in atRA-treated and untreated WT murine podocytes. Primary podocytes were isolated from wild-type and KLF15−/− mice and then stimulated with or without atRA for 4 h, and nuclear proteins were extracted for ChIP assay (n = 3, *, p < 0.05 versus atRA 0 μm). Binding of KLF15 to the putative KLF15-binding sites in the promoter of nephrin (C) and podocin (D) was measured in atRA-treated and untreated primary podocytes from WT mice and KLF15−/− mice.
KLF15 Expression Is Increased in Differentiated Human Podocytes
Because atRA has been previously shown to induce podocyte differentiation (14) and KLF15 is an atRA-induced gene (Fig. 1), KLF15 expression was measured in a known in vitro model of podocyte differentiation. The conditionally immortalized human podocyte cell line harbors a gene encoding a temperature-sensitive T antigen, where T antigen is stable at a permissive temperature of 33 °C and becomes unstable and degraded at a nonpermissive temperature of 37 °C. At 33 °C, this podocyte cell line proliferates and remains in an undifferentiated state, whereas at 37 °C it undergoes differentiation. We observed that KLF15 mRNA and protein expression was significantly increased in the differentiated and nonproliferative state as compared with undifferentiated cells in culture (Fig. 3). These findings indicate that KLF15 expression is increased in a cell culture model of podocyte differentiation.
FIGURE 3.

KLF15 mRNA and protein expression is increased in differentiated human podocytes. Immortalized human podocytes in culture were either incubated and 33 or 37 °C. Cells were collected 1 week after incubation for real time PCR and Western blot. KLF15 mRNA (A) and protein expression (B) were measured in human podocytes in culture at 37 °C compared with 33 °C (n = 3, *, p < 0.01 versus 33 °C; representative blot of three independent experiments is shown). The densitometry analysis is shown in the lower panel (n = 3, *, p < 0.01 versus cells at 33 °C).
Effects of KLF15 on Expression of Differentiated Podocyte Markers
To determine the role of KLF15 in podocyte differentiation, KLF15 was overexpressed in cultured murine podocytes (Fig. 4A), and the expression of differentiated podocyte markers (nephrin, synaptopodin, and podocin) was measured (Fig. 4, B–D). KLF15 overexpression significantly increased the expression of differentiated podocyte markers, suggesting that KLF15 is a key regulator of podocyte differentiation.
FIGURE 4.

Overexpression of KLF15 increases expression of podocyte differentiation markers in both control and HIV-1-infected cells. A, murine podocytes were transiently transfected with control vector or KLF15 construct, and protein was extracted for Western blot. Cells were collected 24 h after transfection for determining mRNA levels of podocyte differentiation markers by real time PCR. Synaptopodin (B), podocin (C), and nephrin (D) were measured in control and HIV-1-infected cells with and without KLF15 overexpression (n = 3, *, p < 0.05 compared with the control cells; n = 3, **, p < 0.05 compared between cells with or without KLF15 overexpression).
KLF15 Null (KLF15−/−) Mice Are More Susceptible to Murine Models of Podocyte-specific Injury
To validate the role of KLF15 in vivo, we initially studied KLF15−/− mice at ages ranging from 6 to 30 weeks. By 30 weeks of age, KLF15−/− mice did not develop significant proteinuria or glomerulosclerosis, suggesting that KLF15 is likely not essential for maintaining podocyte differentiation at base line in the unperturbed state.
To determine the role of KLF15 on podocyte differentiation after injury, two different experimental models of podocyte-specific injury, lipopolysaccharide-induced podocyte injury and adriamycin-induced nephropathy, were examined in KLF15−/− mice. LPS is known to cause significant podocyte effacement and proteinuria (32). We observed that the KLF15−/− mice developed more than 2-fold increase in proteinuria 24 h after LPS injection as compared with the wild-type littermates (497 ± 79 versus 168 ± 74, p < 0.05, n = 6, see Fig. 5A). Coomassie staining revealed that the proteinuria was composed predominantly of albumin (Fig. 5B). No significant change was noted in periodic acid-Schiff-stained kidney sections (data not shown). Electron microscopy evaluation of kidney sections revealed significant podocyte effacement in the LPS-treated KLF15−/− mice as compared with LPS-treated wild-type littermates (Fig. 5, C and D). In addition, the expression of synaptopodin, nephrin, and WT-1 was significantly reduced in LPS-treated KLF15−/− mice as compared with the LPS-treated wild-type littermates as determined by real time PCR analysis of isolated glomeruli and by immunofluorescence staining of kidney sections (Fig. 6, A–C).
FIGURE 5.

LPS-treated KLF15−/− mice had increased albuminuria with podocyte effacement. Urine was collected at 0 h (prior to LPS injection) and subsequently collected at 12-h intervals. All mice were sacrificed and renal cortex fixed for histology at 48 h. A, proteinuria (urine protein/creatinine) was measured in LPS-treated wild-type (WT) and KLF15−/− mice (n = 6, *, p < 0.01 versus LPS-treated WT mice). B, Coomassie stain revealed that the change in proteinuria was mainly albuminuria. The representative gel of three mice in each group is shown. C, podocyte foot process effacement was compared between LPS-treated WT and KLF15−/− mice (×5000). The representative images are shown. D, quantification of foot process effacement is shown (n = 6, *, p < 0.01).
FIGURE 6.

LPS-treated KLF15−/− mice had a reduction in markers of podocyte differentiation. Synaptopodin (A), nephrin (B), and WT-1 (C) mRNA expressions were compared between LPS-treated wild-type (WT) and KLF15−/− mice (n = 6, *, p < 0.01 versus all other groups.) This was confirmed by immunofluorescence as shown in the middle panel. The representative pictures of three mice in each group are shown. The glomerular region was selected, and optical density (OD) was measured and quantified as a relative fold change to wild-type in the lower panel (n = 4, *, p < 0.01).
To further confirm that a loss of KLF15 increases the susceptibility to kidney injury, we treated KLF15−/− and wild-type mice with adriamycin. Adriamycin-induced nephropathy has been previously shown to cause significant glomerular disease and podocyte injury in a BALB/c background (25). A higher dose of adriamycin (20 mg/kg of body weight) was used to induce proteinuria in KLF15−/− mice, which are on a C57BL/6 background. We observed that the KLF15−/− mice developed a significant increase in albuminuria 4 weeks after adriamycin treatment, whereas wild-type C57BL/6 littermates were resistant to adriamycin (1689 ± 392 versus 137 ± 56, p < 0.05; Fig. 7, A and B). Ultrastructural examination of the kidney revealed significant podocyte effacement in the adriamycin-treated KLF15−/− mice as compared with adriamycin-treated wild-type littermates (Fig. 7, C and D). The expression of podocyte-specific genes (synaptopodin, nephrin, and WT-1) in isolated glomeruli of KLF15−/− mice was significantly reduced compared with adriamycin-injected control littermates as determined by real time PCR and immunofluorescence staining (Fig. 8, A–C). Combined, these findings highlight a critical role for KLF15 in regulating podocyte differentiation and response to kidney injury in both in vitro and in vivo models of the disease.
FIGURE 7.

Adriamycin (AD)-treated KLF15−/− mice had increased albuminuria with podocyte effacement. Urine was collected prior to treatment and 4 weeks after treatment. All mice were sacrificed and renal cortex fixed for histology at 4 weeks. A, proteinuria (urine protein/creatinine) was measured in adriamycin-treated wild-type (WT) and KLF15−/− mice (n = 6, *, p < 0.01 versus all other groups). B, Coomassie stain revealed that the change in proteinuria was mainly albuminuria. The representative gel from two mice in each group is shown. C, podocyte foot process effacement was compared between AD-treated WT and KLF15−/− mice (×5000). The representative images are shown. D, quantification of foot process effacement is shown (n = 6, *, p < 0.01).
FIGURE 8.

Adriamycin (AD)-treated KLF15−/− mice had a reduction in markers of podocyte differentiation. Synaptopodin (A), nephrin (B), and Wilms tumor 1 (WT-1) (C) mRNA expressions were compared between adriamycin-treated wild-type (WT) and KLF15−/− mice. (n = 6, *, p < 0.01 versus all other groups). This was confirmed by immunofluorescence as shown in the middle panel. The representative pictures of three mice in each group are shown. The glomerular region was selected, and optical density (OD) was measured and quantified as a relative fold change to wild type in the lower panel (n = 4, *, p < 0.001).
Retinoic Acid Does Not Increase Podocyte Differentiation Markers in Podocytes Lacking KLF15 Expression
Primary podocytes were isolated from wild-type and KLF15−/− mice and confirmed by Western blot (Fig. 9A). As published previously (14), atRA treatment increased the expression of nephrin, podocin, and synaptopodin in podocytes isolated from wild-type mice (Fig. 9, B–D). This finding was not observed in podocytes isolated from KLF15−/− mice (Fig. 9, B–D).
FIGURE 9.

RA does not increase podocyte differentiation markers in podocytes lacking KLF15 expression. A, primary mouse podocytes were initially isolated from wild-type (WT) and KLF15−/− mice, and protein was extracted for Western. These isolated primary podocytes were also stimulated with or without atRA for 8 h, and RNA was extracted for real time PCR. Synaptopodin (B), podocin (C), and nephrin (D) mRNA expressions were measured in atRA-treated and untreated WT and KLF15−/− podocytes (n = 3, *, p < 0.01 versus atRA 0 μm).
Retinoic Acid Is Unable to Attenuate LPS-induced Proteinuria in KLF15−/− Mice
To characterize that atRA-induced podocyte differentiation is dependent on induction of KLF15, wild-type and KLF15−/− mice were treated concurrently with atRA and LPS. Within 24 h, LPS treated wild-type mice had an attenuation in proteinuria with atRA treatment. This finding was not observed in LPS treated KLF15−/− mice (Fig. 10).
FIGURE 10.

RA did not attenuate proteinuria in LPS-treated KLF15−/− mice. First dose of atRA (16 mg/kg) was administered 12 h prior to first dose of LPS with the subsequent two doses administered at 24-h time intervals of the initial dose. Urine was collected at 24 h, and proteinuria was measured in LPS-treated wild-type (WT) and KLF15−/− mice co-treated with and without atRA (n = 4, *, p < 0.0001).
KLF15 Expression Is Reduced in HIV Transgenic Mice
Because the HIV transgenic mouse line (Tg26 mice) is a known model of podocyte dedifferentiation (20) and our studies indicate that KLF15 regulates podocyte differentiation, the expression of KLF15 was determined in this murine model. Glomeruli isolated from Tg26 mice revealed a significant decrease in KLF15 mRNA and protein expression as compared with wild-type mice (Fig. 11, A and B). Immunohistochemistry confirmed the reduced KLF15 expression in Tg26 mice as compared with the wild-type mice (Fig. 11C). Combined, these findings indicate that KLF15 expression is suppressed in Tg26 mice and may contribute to the podocyte dedifferentiation in this HIVAN model.
FIGURE 11.

KLF15 expression is reduced in HIV transgenic (Tg26) mice. Glomeruli were isolated, and RNA was extracted for real time PCR. A, KLF15 mRNA expression was measured in wild-type (WT) and Tg26 mice (n = 4, *, p < 0.05 versus control). B, Western blot analysis was performed in glomerular lysates from WT and Tg26 mice for KLF15 and GAPDH. The representative blot of three independent experiments is shown. The densitometry analysis of these blots is shown in the lower panel (n = 3, *, p < 0.01). C, representative images from the immunostaining of KLF15 in kidney sections from WT and Tg26 mice are shown.
Reduction of KFL15 Expression in Human Glomerular Disease
To ascertain the role of KLF15 in human kidney disease, immunostaining for KLF15 was performed on a renal biopsy specimen from healthy donor nephrectomies, HIVAN, and idiopathic FSGS. We observed that the staining for KLF15 had a nuclear distribution in normal podocytes, parietal cells, and tubular cells from healthy donor nephrectomy specimens. Immunohistochemistry also revealed a significant decrease in KLF15 expression in the podocytes in patients with HIVAN (Fig. 12B) and idiopathic FSGS (Fig. 12C) as compared with healthy donors (Fig. 12A). Quantification of immunostaining in the glomeruli confirmed the changes in KLF15 expression (Fig. 12D). Combined with the rest of our findings, these data suggest that KLF15 expression is reduced in human glomerular disease and may lead to increased susceptibility to podocyte injury in these conditions.
FIGURE 12.

Reduced KLF15 expression in human glomerular disease. A, immunostaining for KLF15 performed on healthy donor nephrectomy specimens shows a nuclear distribution in normal podocytes, parietal cells, and tubular cells. In comparison with biopsy specimen from healthy donor subjects (A), KLF15 expression in the podocytes is shown in biopsy specimens from patients with diagnosed HIVAN (B) and idiopathic FSGS (C). The representative images of three subjects in each group are shown. D, glomerular region was selected, and optical density (OD) was measured and quantified as a relative fold change to healthy donor specimens (n = 3, *, p < 0.0001).
DISCUSSION
A large body of evidence suggests that RA ameliorates kidney injury in several animal models of kidney disease, including HIVAN. However, the transcriptional regulators mediating the effects of RA on podocyte differentiation have not been well characterized. We had previously shown that RA induces podocyte differentiation by activating the cAMP/PKA/CREB pathway (14). Here, we identify that KLF15 is likely one of the mediators for RA-induced podocyte differentiation, and KLF15 deficiency increases the susceptibility of podocytes to injury. This was demonstrated by the following lines of evidence: 1) overexpression of KLF15 induces podocyte differentiation, and RA increases binding of KLF15 to the promoter region of genes encoding podocyte differentiation markers; 2) RA did not induce podocyte differentiation in podocytes lacking KLF15 expression; 3) an exaggerated proteinuric response and a significant reduction in expression of podocyte differentiation markers occur in KLF15−/− mice subjected to two experimental models of podocyte injury (LPS and adriamycin); 4) RA was unable to attenuate LPS-induced proteinuria in KLF15−/− mice; and 5) reduced KLF15 expression occurs in the murine HIVAN model and in renal biopsy specimens from human subjects with podocyte disease.
Transcriptional regulators of podocyte differentiation have remained largely unknown. Notch signaling, Wilms tumor 1 (WT1), and Forkhead box protein C2 (FOXC2) are key regulators in the Xenopus podocyte gene network that specify the expression of podocyte terminal differentiation markers (33). WT1 is known to mediate podocyte differentiation and kidney development in mice as well as in humans (34). Although RA stimulates the expression of several podocyte differentiation markers (14) and CREB is a key transcription factor that mediates the effects of RA on podocyte differentiation (35), most markers/genes of podocyte terminal differentiation lack CREB-binding elements and RA-response elements in their promoters. This was the impetus for our approach to identify additional transcriptional regulators of podocyte differentiation that are RA-induced and CREB-mediated. We found that KLF15 is able to bind directly to the promoter of nephrin and podocin and stimulate expression of these two key podocyte differentiation markers. Consistent with this, a comparative promoter analysis by Cohen et al. (19) revealed that KLF motif is one of the four most common bindings sites in the promoter region of many podocyte-specific genes. We recognize that other factors may also mediate RA-induced podocyte differentiation. For example, our data suggest that GDNF is also CREB-targeted and highly regulated by RA. Previous studies have already shown that GDNF is implicated in podocyte differentiation (31). Although the KLF15−/− mice developed minimal podocyte injury at base line, the murine models of podocyte injury used in our study revealed that the KLF15 deficiency increased the susceptibility of KLF15−/− mice to LPS- or adriamycin-induced albuminuria and foot process effacement. These data indicate that KLF15 is dispensable for normal podocyte development and maintenance of the glomerular filtration barrier in health, but KLF15 is required for protection against podocyte injury. We speculate a classic “two-hit” model for podocyte injury, the loss of KLF15 expression and the insult from either LPS or adriamycin. Additionally, as with other members of the KLF family (36), the lack of significant podocyte injury in the KLF15−/− mice may be attributed to the compensation by the other factors in the KLF family. Further studies will be required to fully characterize the role of KLF15 in this two-hit model.
We also confirmed that KLF15 expression is reduced in Tg26 mice and in human glomerular diseases, such as HIVAN and idiopathic FSGS, where podocyte injury is prominent. Podocyte dedifferentiation is a hallmark of collapsing FSGS in HIVAN. KLF15 expression was markedly reduced in Tg26 mice as well as in human HIVAN kidneys suggesting that a loss of KLF15 expression may contribute to podocyte dedifferentiation in HIVAN kidneys. Therefore, knock-out of KLF15 in Tg26 mice may not yield a significant change in an already severely diseased phenotype. Consequently, future studies are needed to determine whether overexpression of KLF15 protects against kidney injury in Tg26 mice. However, the KLF15 gene was recently found to be associated with the HIVAN2 gene.4 Although this finding alone does not establish causality between the lack of KLF15 and human kidney disease, our animal and in vitro studies suggest that KLF15 may have a protective role in human kidney disease.
Regulation of KLF15 in human kidney disease is not known. Promoter analysis of KLF15 revealed transcription factor-binding sites for NF-κB. Because HIV-1 infection of kidney cells is known to transactivate NF-κB (37) in Tg26 mice (38) and patients with HIVAN (39), future studies are required to examine whether NF-κB mediates regulation of KLF15 in kidney disease such as HIVAN.
Here, we report that KLF15 is a novel transcriptional regulator of podocyte differentiation. Our studies indicate that the loss of KLF15 leads to increased susceptibility to podocyte injury. In addition, KLF15 expression is suppressed in human kidney disease. This study provides new insight into podocyte biology and pathology, as well as a potential new target for therapy of kidney diseases with podocyte injury.
This work was supported, in whole or in part, by National Institutes of Health Grants 1R01DK078897-01 (to J. H.), R01HL084154 (to M. K. J.), P01DK056492 (to P. K.), 5K08DK082760 (to P. Y. C.), and T32 DK07757-12 (to S. K. M.) from NIDDK.
Ali G. Gharavi, personal communication.
- FSGS
- focal segmental glomerulosclerosis
- RA
- retinoic acid
- KLF
- Krüppel-like factor
- HIVAN
- HIV-associated nephropathy
- atRA
- all-trans-retinoic acid
- RARE
- retinoic acid-response element
- CREB
- cAMP-response element-binding protein
- WT-1
- Wilms tumor 1
- F
- forward
- R
- reverse
- GDNF
- glial cell-derived neurotrophic factor
- GBM
- glomerular basement membrane.
REFERENCES
- 1. Wolf G., Chen S., Ziyadeh F. N. (2005) From the periphery of the glomerular capillary wall toward the center of disease. Podocyte injury comes of age in diabetic nephropathy. Diabetes 54, 1626–1634 [DOI] [PubMed] [Google Scholar]
- 2. Kriz W., Gretz N., Lemley K. V. (1998) Progression of glomerular diseases. Is the podocyte the culprit? Kidney Int. 54, 687–697 [DOI] [PubMed] [Google Scholar]
- 3. Barisoni L., Kriz W., Mundel P., D'Agati V. (1999) The dysregulated podocyte phenotype. A novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 10, 51–61 [DOI] [PubMed] [Google Scholar]
- 4. Barisoni L., Bruggeman L. A., Mundel P., D'Agati V. D., Klotman P. E. (2000) HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int. 58, 173–181 [DOI] [PubMed] [Google Scholar]
- 5. Bruggeman L. A., Dikman S., Meng C., Quaggin S. E., Coffman T. M., Klotman P. E. (1997) Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J. Clin. Invest. 100, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwartz E. J., Cara A., Snoeck H., Ross M. D., Sunamoto M., Reiser J., Mundel P., Klotman P. E. (2001) Human immunodeficiency virus-1 induces loss of contact inhibition in podocytes. J. Am. Soc. Nephrol. 12, 1677–1684 [DOI] [PubMed] [Google Scholar]
- 7. Husain M., Gusella G. L., Klotman M. E., Gelman I. H., Ross M. D., Schwartz E. J., Cara A., Klotman P. E. (2002) HIV-1 Nef induces proliferation and anchorage-independent growth in podocytes. J. Am. Soc. Nephrol 13, 1806–1815 [DOI] [PubMed] [Google Scholar]
- 8. He J. C., Husain M., Sunamoto M., D'Agati V. D., Klotman M. E., Iyengar R., Klotman P. E. (2004) Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1/2 pathways. J. Clin. Invest. 114, 643–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evans T. R., Kaye S. B. (1999) Retinoids. Present role and future potential. Br. J. Cancer 80, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu Q., Lucio-Cazana J., Kitamura M., Ruan X., Fine L. G., Norman J. T. (2004) Retinoids in nephrology. Promises and pitfalls. Kidney Int. 66, 2119–2131 [DOI] [PubMed] [Google Scholar]
- 11. Lehrke I., Schaier M., Schade K., Morath C., Waldherr R., Ritz E., Wagner J. (2002) Retinoid receptor-specific agonists alleviate experimental glomerulonephritis. Am. J. Physiol. Renal Physiol. 282, F741–F751 [DOI] [PubMed] [Google Scholar]
- 12. Suzuki A., Ito T., Imai E., Yamato M., Iwatani H., Kawachi H., Hori M. (2003) Retinoids regulate the repairing process of the podocytes in puromycin amino nucleoside-induced nephrotic rats. J. Am. Soc. Nephrol. 14, 981–991 [DOI] [PubMed] [Google Scholar]
- 13. Pérez de Lema G., Lucio-Cazaña F. J., Molina A., Luckow B., Schmid H., de Wit C., Moreno-Manzano V., Banas B., Mampaso F., Schlöndorff D. (2004) Retinoic acid treatment protects MRL/lpr lupus mice from the development of glomerular disease. Kidney Int. 66, 1018–1028 [DOI] [PubMed] [Google Scholar]
- 14. He J. C., Lu T. C., Fleet M., Sunamoto M., Husain M., Fang W., Neves S., Chen Y., Shankland S., Iyengar R., Klotman P. E. (2007) Retinoic acid inhibits HIV-1-induced podocyte proliferation through the cAMP pathway. J. Am. Soc. Nephrol. 18, 93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vaughan M. R., Pippin J. W., Griffin S. V., Krofft R., Fleet M., Haseley L., Shankland S. J. (2005) ATRA induces podocyte differentiation and alters nephrin and podocin expression in vitro and in vivo. Kidney Int. 68, 133–144 [DOI] [PubMed] [Google Scholar]
- 16. Mori T., Sakaue H., Iguchi H., Gomi H., Okada Y., Takashima Y., Nakamura K., Nakamura T., Yamauchi T., Kubota N., Kadowaki T., Matsuki Y., Ogawa W., Hiramatsu R., Kasuga M. (2005) Role of Krüppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J. Biol. Chem. 280, 12867–12875 [DOI] [PubMed] [Google Scholar]
- 17. Feinberg M. W., Lin Z., Fisch S., Jain M. K. (2004) An emerging role for Krüppel-like factors in vascular biology. Trends Cardiovasc. Med. 14, 241–246 [DOI] [PubMed] [Google Scholar]
- 18. Pearson R., Fleetwood J., Eaton S., Crossley M., Bao S. (2008) Krüppel-like transcription factors. A functional family. Int. J. Biochem. Cell Biol. 40, 1996–2001 [DOI] [PubMed] [Google Scholar]
- 19. Cohen C. D., Klingenhoff A., Boucherot A., Nitsche A., Henger A., Brunner B., Schmid H., Merkle M., Saleem M. A., Koller K. P., Werner T., Gröne H. J., Nelson P. J., Kretzler M. (2006) Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 5682–5687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng X., Lu T. C., Chuang P. Y., Fang W., Ratnam K., Xiong H., Ouyang X., Shen Y., Levy D. E., Hyink D., Klotman M., D'Agati V., Iyengar R., Klotman P. E., He J. C. (2009) Reduction of Stat3 activity attenuates HIV-induced kidney injury. J. Am. Soc. Nephrol. 20, 2138–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee T. I., Johnstone S. E., Young R. A. (2006) Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 1, 729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dickie P., Felser J., Eckhaus M., Bryant J., Silver J., Marinos N., Notkins A. L. (1991) HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology 185, 109–119 [DOI] [PubMed] [Google Scholar]
- 23. Fisch S., Gray S., Heymans S., Haldar S. M., Wang B., Pfister O., Cui L., Kumar A., Lin Z., Sen-Banerjee S., Das H., Petersen C. A., Mende U., Burleigh B. A., Zhu Y., Pinto Y. M., Pinto Y., Liao R., Jain M. K. (2007) Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 104, 7074–7079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reiser J., Mundel P. (2004) Danger signaling by glomerular podocytes defines a novel function of inducible B7-1 in the pathogenesis of nephrotic syndrome. J. Am. Soc. Nephrol. 15, 2246–2248 [DOI] [PubMed] [Google Scholar]
- 25. Lee V. W., Harris D. C. (2011) Adriamycin nephropathy. A model of focal segmental glomerulosclerosis. Nephrology 16, 30–38 [DOI] [PubMed] [Google Scholar]
- 26. Koop K., Eikmans M., Baelde H. J., Kawachi H., De Heer E., Paul L. C., Bruijn J. A. (2003) Expression of podocyte-associated molecules in acquired human kidney diseases. J. Am. Soc. Nephrol. 14, 2063–2071 [DOI] [PubMed] [Google Scholar]
- 27. Takemoto M., Asker N., Gerhardt H., Lundkvist A., Johansson B. R., Saito Y., Betsholtz C. (2002) A new method for large scale isolation of kidney glomeruli from mice. Am. J. Pathol. 161, 799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katsuya K., Yaoita E., Yoshida Y., Yamamoto Y., Yamamoto T. (2006) An improved method for primary culture of rat podocytes. Kidney Int. 69, 2101–2106 [DOI] [PubMed] [Google Scholar]
- 29. Gassmann M., Grenacher B., Rohde B., Vogel J. (2009) Quantifying Western blots. Pitfalls of densitometry. Electrophoresis 30, 1845–1855 [DOI] [PubMed] [Google Scholar]
- 30. Potapova T. A., Sivakumar S., Flynn J. N., Li R., Gorbsky G. J. (2011) Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol. Biol. Cell 22, 1191–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsui C. C., Shankland S. J., Pierchala B. A. (2006) Glial cell line-derived neurotrophic factor and its receptor ret is a novel ligand-receptor complex critical for survival response during podocyte injury. J. Am. Soc. Nephrol. 17, 1543–1552 [DOI] [PubMed] [Google Scholar]
- 32. Sun Y., He L., Takemoto M., Patrakka J., Pikkarainen T., Genové G., Norlin J., Truvé K., Tryggvason K., Betsholtz C. (2009) Glomerular transcriptome changes associated with lipopolysaccharide-induced proteinuria. Am. J. Nephrol. 29, 558–570 [DOI] [PubMed] [Google Scholar]
- 33. White J. T., Zhang B., Cerqueira D. M., Tran U., Wessely O. (2010) Notch signaling, wt1, and foxc2 are key regulators of the podocyte gene regulatory network in Xenopus. Development 137, 1863–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palmer R. E., Kotsianti A., Cadman B., Boyd T., Gerald W., Haber D. A. (2001) WT1 regulates the expression of the major glomerular podocyte membrane protein Podocalyxin. Curr. Biol. 11, 1805–1809 [DOI] [PubMed] [Google Scholar]
- 35. Lu T. C., Wang Z., Feng X., Chuang P., Fang W., Chen Y., Neves S., Maayan A., Xiong H., Liu Y., Iyengar R., Klotman P. E., He J. C. (2008) Retinoic acid utilizes CREB and USF1 in a transcriptional feed-forward loop to stimulate MKP1 expression in human immunodeficiency virus-infected podocytes. Mol. Cell. Biol. 28, 5785–5794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eaton S. A., Funnell A. P., Sue N., Nicholas H., Pearson R. C., Crossley M. (2008) A network of Krüppel-like factors (Klfs). Klf8 is repressed by Klf3 and activated by Klf1 in vivo. J. Biol. Chem. 283, 26937–26947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ross M. J., Martinka S., D'Agati V. D., Bruggeman L. A. (2005) NF-κB regulates Fas-mediated apoptosis in HIV-associated nephropathy. J. Am. Soc. Nephrol. 16, 2403–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bruggeman L. A., Adler S. H., Klotman P. E. (2001) Nuclear factor-κB binding to the HIV-1 LTR in kidney: implications for HIV-associated nephropathy. Kidney Int. 59, 2174–2181 [DOI] [PubMed] [Google Scholar]
- 39. Ross M. J., Fan C., Ross M. D., Chu T. H., Shi Y., Kaufman L., Zhang W., Klotman M. E., Klotman P. E. (2006) HIV-1 infection initiates an inflammatory cascade in human renal tubular epithelial cells. J. Acquir. Immune Defic. Syndr. 42, 1–11 [DOI] [PubMed] [Google Scholar]