

Background: Trans-cellular propagation of aggregation may be important in neurodegeneration, but mechanisms are unknown.
Results: Tau fibrils are secreted into the extracellular space, where they directly trigger aggregation in recipient cells by contacting native protein.
Conclusion: Trans-cellular movement of Tau fibrils seeds subsequent aggregation.
Significance: Therapies that block trans-cellular movement, including antibodies, may have an important role in neurodegenerative diseases.
Keywords: Aggregation, Alzheimer Disease, Fluorescence Resonance Energy Transfer (FRET), Neurodegeneration, Tau, Aggregates, Fibrils, Propagation
Abstract
Aggregation of the microtubule associated protein Tau is associated with several neurodegenerative disorders, including Alzheimer disease and frontotemporal dementia. In Alzheimer disease, Tau pathology spreads progressively throughout the brain, possibly along existing neural networks. However, it is still unclear how the propagation of Tau misfolding occurs. Intriguingly, in animal models, vaccine-based therapies have reduced Tau and synuclein pathology by uncertain mechanisms, given that these proteins are intracellular. We have previously speculated that trans-cellular propagation of misfolding could be mediated by a process similar to prion pathogenesis, in which fibrillar Tau aggregates spread pathology from cell to cell. However, there has been little evidence to demonstrate true trans-cellular propagation of Tau misfolding, in which Tau aggregates from one cell directly contact Tau protein in the recipient cell to trigger further aggregation. Here we have observed that intracellular Tau fibrils are directly released into the medium and then taken up by co-cultured cells. Internalized Tau aggregates induce fibrillization of intracellular Tau in these naive recipient cells via direct protein-protein contact that we demonstrate using FRET. Tau aggregation can be amplified across several generations of cells. An anti-Tau monoclonal antibody blocks Tau aggregate propagation by trapping fibrils in the extracellular space and preventing their uptake. Thus, propagation of Tau protein misfolding among cells can be mediated by release and subsequent uptake of fibrils that directly contact native protein in recipient cells. These results support the model of aggregate propagation by templated conformational change and suggest a mechanism for vaccine-based therapies in neurodegenerative diseases.
Introduction
Aggregation of the microtubule-associated protein Tau in neurons and glia is associated with over 20 neurodegenerative disorders, including Alzheimer disease (AD)2 and frontotemporal dementia (1). Recent evidence from human studies suggests that Tau pathology does not distribute randomly through the brain but instead is linked to existing networks of neuronal connectivity (2, 3). The fibrillar Tau pathology of AD progresses along known anatomical connections, although the mechanisms by which networks degenerate are unknown. Importantly, recent pathological studies suggest that protein aggregates can move from one cell to another in human and mouse brain (4–11). Moreover, fibrillar forms of recombinant, human disease-associated proteins, such as Tau, SOD-1, α-synuclein, and polyglutamines, are readily taken up from the extracellular space to trigger intracellular misfolding (12–16). These phenomena are reminiscent of prion propagation, for which exosomes (17, 18) and tunneling nanotubes (19, 20) have been proposed to mediate trans-cellular spread. It is an open question as to whether Tau aggregates might spread protein misfolding from cell to cell via direct cell-cell contact or through extracellular space. Furthermore, it has not yet been determined whether pathological Tau species can mediate true trans-cellular propagation of aggregation, whereby an aggregate is released from a “donor” cell, enters a second “recipient” cell, and induces further misfolding via direct protein-protein contact, as opposed to more indirect mechanisms. Here we have tested whether Tau fibrils are released directly into the extracellular space and can propagate aggregation by this mechanism.
EXPERIMENTAL PROCEDURES
Antibodies
Rabbit polyclonal antibody directed against Tau (ab64193, epitope located in the repeat domain region) was purchased from Abcam (Cambridge, MA). Mouse monoclonal antibody directed against hemagglutinin (HA) (HA.11 Clone 16B12) was purchased from Covance (Emeryville, CA). Rabbit polyclonal GFP antibody (sc-8334) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse monoclonal affinity-purified antibody directed against Tau (HJ9.3) was produced against full-length recombinant mouse Tau and is directed against the repeat domain of the protein (21).
Plasmids
Sequences encoding the four-repeat domain (RD) of the microtubule-associated protein Tau were used for protein expression. In addition to the wild-type form, various Tau mutants were created: ΔK280 (ΔK), P301L/V337M (LM), and ΔK280/I227P/I308P (PP). These sequences were either subcloned into pcDNA3.1 (Invitrogen) with a C-terminal hemagglutinin (HA) tag or into pEYFP-N1 or pECFP-N1 (Clontech) to create C-terminal fluorescent protein fusions.
Cell Culture and Transfections
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 μg/ml penicillin, and 100 μg/ml streptomycin. Cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C. For transient transfections, cells plated in Opti-MEM medium were transfected using Lipofectamine/Plus reagent and 600 ng of appropriate DNA constructs (Invitrogen) according to the manufacturer's recommendations and harvested 24 or 48 h later for further analyses.
Detergent Fractionation and Western Blot Analyses
HEK293 cells were plated at 400,000 cells/well in a 12-well plate. The following day, cells were transfected with 600 ng of plasmid. After 48 h, cells were harvested with 0.05% trypsin for 3 min at 37 °C, pelleted briefly at 7000 × g, and lysed in 100 μl of 1% Triton in PBS containing protease inhibitors. Soluble cytosolic proteins were then collected by centrifugation at 14,000 × g for 10 min. Insoluble proteins were obtained by resuspending the pellet in radioimmune precipitation/SDS buffer and centrifugation at 20,000 × g for 15 min following benzonase nuclease digestion of nucleic acids. For co-culture experiments, equal numbers of cells transfected with RD(LM)-HA and RD(ΔK)-YFP were co-cultured together for 48 h before harvesting and Western blotting. Equivalent amounts of HEK293 cell protein extract from each fraction were analyzed using 4–20% polyacrylamide gels (Bio-Rad), antibody directed against Tau RD (which recognizes an epitope in the RD region) at a 1:2000 dilution (ab64193, Abcam), or antibody directed against GFP at 1:1000 dilution (sc-8334, Santa Cruz Biotechnology, Inc.). A chemiluminescence-based peroxidase-conjugated secondary antibody reaction was performed and detected by x-ray film. Quantification was performed using ImageJ analysis software.
Co-culture Experiments
Measuring RD-CFP/YFP Co-aggregation by Fluorescence Resonance Energy Transfer (FRET)
HEK293 cells were plated at 300,000 cells/well in a 12-well plate. The following day, cells were transfected with 600 ng of plasmid as described above. Co-transfected cells received a combination of 150 ng of RD-CFP constructs and 450 ng of RD-YFP constructs. 15 h later, cells were harvested with 0.05% trypsin for 3 min at 37 °C, and a fraction of cells was replated in a 96-well plate in quadruplicate or on Ibidi μ-slides (Ibidi GmbH) for imaging by microscopy. Cells were then cultured an additional 48 h before fixation with 4% paraformaldehyde and analysis.
Measuring Induction of RD-YFP Aggregation by RD-HA
HEK293 cells were transfected with either RD(ΔK)-YFP or RD(LM)-HA in 12-well plates. After 15 h, the cells were replated together onto Ibidi μ-slides and co-cultured an additional 48 h. They were then fixed and stained with anti-HA antibody and X-34 for analysis by microscopy.
Propagation Assays in Co-culture
Two populations of HEK293 cells in a 12-well plate were co-transfected with 300 ng of RD(LM)-HA and 300 ng of RD(ΔK)-CFP together or with RD(ΔK)-YFP. After 15 h, equal percentages of the two populations were co-cultured for 48 h in a 96-well plate format. Cells were then fixed with 4% paraformaldehyde, and FRET analysis was performed using a fluorescence plate reader (FPR). For FRET microscopy analysis, two populations of HEK293 cells in a 12-well plate were transfected with 600 ng of RD(LM)-CFP or with RD(LM)-YFP. After 15 h, equal percentages of the two populations were co-cultured for 48 h on Ibidi μ-slides. Cells were then fixed with 4% paraformaldehyde, and FRET acceptor photobleaching was conducted.
Amplification of Tau Aggregation in Serial Culture
HEK293 cells were transfected in a 12-well plate with 600 ng of various forms of non-fluorescent RD-HA and cultured for 24 h. A second group of cells was transfected with CFP or RD(ΔK)-CFP. Equal percentages of the first and second populations were then co-cultured for 48 h. At this point, 50% of this population was plated with a population of cells transfected with RD(ΔK)-YFP in a 96-well plate for 48 h. Cells were then fixed with 4% paraformaldehyde for FRET analyses using the FPR.
Medium Transfer and Conditioned Medium Experiments
HEK293 cells were either transfected in a 12-well plate with 600 ng of RD(LM)-HA or co-transfected with a combination of 150 ng of RD(ΔK)-CFP construct and 450 ng of RD(ΔK)-YFP construct. 15 h later, cells were harvested with 0.05% trypsin for 3 min at 37 °C. An equivalent number of cells expressing RD(ΔK)-YFP/CFP and RD(LM)-HA were co-cultured for 48 h in varying amounts of cell culture medium. Cells were then fixed with 4% paraformaldehyde, and FRET analysis was performed. For the conditioned medium experiments, 15 h after transfection, medium from RD(LM)-HA cells containing transfection complexes was replaced with fresh medium. Cells expressing RD(ΔK)-YFP/CFP were harvested with 0.05% trypsin for 3 min at 37 °C and replated in a 96-well plate. 24 h later, conditioned medium from cells transfected with RD(LM)-HA was collected and added to cells expressing RD(ΔK)-YFP/CFP. 48 h later, cells were fixed with 4% paraformaldehyde, and FRET analysis was performed.
FRET Assays
FRET Measurements by Microscopy with Photobleaching
HEK293 cells transfected for co-transfection and co-culture experiments as described earlier were prepared for FRET acceptor photobleaching microscopy. All images were obtained using a C-Apochromat 40 × 1.2 numerical aperture lens (Carl Zeiss Advanced Imaging Microscopy, 07740 Jena, Germany). Digital images were acquired using a Zeiss LSM510 Meta NLO multiphoton/confocal laser-scanning microscope system on the Zeiss Axiovert 200 M. Channels used for imaging were as follows. The donor CFP was stimulated using a 458-nm argon laser, and fluorescence was collected with a 480–520-nm band pass filter; the acceptor YFP was stimulated using a 514-nm argon laser, and fluorescence was collected with a long pass 560-nm filter. To create an image in which the intensity reflected an estimate of FRET efficiency, the value of the initial CFP image was subtracted from the final CFP image obtained after photobleaching on a pixel-by-pixel basis, and this difference was multiplied by 100 and divided by the final CFP image intensity, 100 × (CFPfinal − CFPinitial)/CFPfinal.
Proper adjustments were made for partial acceptor photobleaching. Image arithmetic and grayscale-to-color image conversion were done using National Institutes of Health ImageJ 1.44 software.
Fluorescence Plate Reader
Spectral FRET measurements (FRET/donor) were obtained using a TecanM1000 fluorescence plate reader according to methods described previously (22). When donor and acceptor are not fused to the same protein, spectral FRET measurements depend on careful control for the relative amount of donor and acceptor proteins expressed within the cell. All values on the plate reader were first background-subtracted against mock-transfected cells. The YFP signal in each well (Smpl485ex/528em) was used to estimate RD-YFP expression levels, and it was likewise assumed that, under experimental conditions, RD-CFP/YFP do not vary independently. This helps eliminate the possibility that changes in apparent FRET are due simply to variations in RD expression levels. Relative contribution of acceptor activation (528 nm) by donor excitation signal (435 nm) to the overall FRET measurement was corrected by determining the “crossover activation” fraction for acceptor, X, where X = RD-YFP signal measured at 435-nm excitation/528-nm emission divided by the signal measured at 485-nm excitation/528-nm emission. This “crossover activation” is essentially constant across different expression levels of RD-YFP encountered in our experiments. The “measured” FRET value in each sample is recorded at 435-nm excitation/528-nm emission, and the “donor” value (CFP) is recorded at 435-nm excitation/485-nm emission. The “actual” FRET/donor value for each well is then reflected as follows.
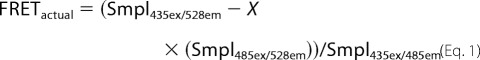 |
This method of measuring protein aggregation by FRET has reliably allowed detection of subtle changes in response to pharmacologic (22) as well as genetic (23) manipulations of androgen receptor and huntingtin protein aggregation that were corroborated by visual and biochemical analyses. Because the relative amount of spectral FRET measured depends on the ratio of acceptor to donor, we use a constant ratio of 3:1 when RD-YFP and RD-CFP are co-expressed within the same cell. This provides close to maximal FRET efficiency while allowing for acceptable signal/noise in the measurement of donor signal.
Atomic Force Microscopy (AFM)
Tau proteins were extracted from transfected HEK293 cells, immunoprecipitated, and incubated on mica chips (Ted Pella, Inc.) for 10 min. Samples were then rinsed twice with 100 μl of double-distilled H2O and left at room temperature to dry. The following day, atomic force microscopy was performed using a MFP-3D atomic force microscope (Asylum Research).
Immunofluorescence and Confocal Microscopy
HEK293 cells transfected for co-culture experiments as described earlier were prepared for immunofluorescence and X-34 staining. After fixation in 4% paraformaldehyde for 15 min at room temperature, cells were washed twice in PBS at room temperature for 5 min and permeabilized in 0.25% Triton X-100 in PBS at room temperature for 10 min. Cells were blocked with a solution containing 1% normal goat serum, 20 mg/ml BSA, 0.25% Triton X-100 in PBS for 3 h at room temperature. Primary mouse monoclonal antibody against HA (Covance, Emeryville, CA) was diluted 1:2000 in blocking solution and applied to cells overnight at 4 °C. Cells were then washed with PBS containing 0.1% Triton X-100 three times for 5 min each and incubated with anti-mouse Alexa 546-conjugated secondary antibody (Invitrogen) diluted at 1:400 in blocking solution. Cells were then washed with PBS containing 0.1% Triton X-100 three times for 5 min each and exposed to 1 μm X-34 prepared in a solution of 40% ethanol, 60% PBS, and 20 mm NaOH for 10 min at room temperature. Cells were then washed three times for 2 min each in 40% EtOH, 60% PBS and rinsed twice in 1× PBS for 5 min each. Images were captured using confocal microscopy (Zeiss confocal microscope). For characterization of the mechanism of HJ9.3 antibody blockade of propagation, HEK293 cells were transfected with RD(ΔK)-YFP or mock-transfected. Following culture of RD(ΔK)-YFP cells or mock-transfected cells in the presence of HJ9.3 for 48 h, cells were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.25% Triton X-100, and then exposed to goat anti-mouse Alexa 546-labeled secondary antibody. Images were captured using confocal microscopy (Zeiss confocal microscope).
Propidium Iodide Cell Death Assay
HEK293 cells were plated at 75,000 cells/well in a 96-well plate. The following day, cells were transfected in quadruplicate with 100 ng of various forms of non-fluorescent RD-HA plasmids or exposed to transfection complexes without DNA. The next day, medium containing transfection complexes was removed and replaced with fresh medium. Non-transfected cells were treated with varying concentrations of staurosporine (1, 2, 4, and 20 μm) for 30 min at 37 °C as a positive control for cell death. Staurosporine solution was then removed, and all cells were exposed to 5 μg/ml propidium iodide for 10 min at 37 °C. The propidium iodide solution was then replaced with phenol-free medium, and fluorescence was read on the plate reader at 535-nm excitation and 617-nm emission.
Immunoprecipitation
Transfected cell populations were co-cultured either alone or in the presence of mouse monoclonal antibody HJ9.3 (1:1000, which is equivalent to 2.5 ng/μl antibody) or pooled mouse IgG antibody for 3, 6, 9, 12, 24, or 48 h. Conditioned medium was collected, and protein G-agarose beads (100 μl of 50% slurry beads from Pierce) were added to the medium and incubated overnight at 4 °C with rotation. 18 h later, 500 μl of binding buffer (Pierce) was added to samples and centrifuged at 2000 × g for 3 min. Supernatant was discarded, and this wash step was repeated three times. Proteins bound to beads were then eluted using a high salt elution buffer (50 μl) with incubation at room temperature for 5 min. Samples were then centrifuged at 2000 × g for 3 min, and supernatant was collected. This elution step was repeated once for a total of 100 μl of eluate. Another sample of conditioned medium not initially exposed to HJ9.3 or IgG was incubated with the HJ9.3 (1:1000) or IgG antibodies overnight at 4 °C with rotation, followed by the same immunoprecipitation protocol as described above. Samples from all conditions were analyzed on 4–20% polyacrylamide gels (Bio-Rad) and detected with rabbit polyclonal antibody directed against Tau RD at 1:2000 dilution in 5% dry milk in TBS/Tween (ab64193, Abcam). A chemiluminescence-based peroxidase-conjugated secondary antibody reaction was performed and detected by x-ray film.
Flow Cytometry
HEK293 cells were plated in a 10-cm plate at ∼80% confluence. Cells were then transfected with 24 μg of RD(LM)-YFP construct or transduced with mCherry lentivirus. The following day, cells were harvested by treatment with 0.05% trypsin for 3 min at 37 °C and then pelleted and resuspended in fresh medium. The two cell populations were co-cultured either alone or in the presence of mouse monoclonal antibody HJ9.3 directed against Tau RD at 1:1000 or 1:10,000 dilutions for 48 h (1:1000 is equivalent to 2.5 ng/μl of antibody). After this time, cells were harvested and resuspended in Hanks' balanced medium containing 1% FBS and 1 mm of EDTA. Cells premixed just prior to cytometry were used as negative controls. Cells were counted using the MoFlo high speed cell sorter (Beckman Coulter), and the percentage of dual positive cells was analyzed for each of the conditions. Each condition had three biological replicates, with 50,000 cells analyzed in each experimental condition.
RESULTS
Tau RD Proteins Form Fibrillar Aggregates in Transfected HEK293 Cells
The Tau gene encodes six protein isoforms, and multiple mutations cause dominantly inherited neurodegenerative disease (1, 24). Depending on splicing, the Tau protein has either three or four repeat regions that constitute the aggregation-prone core of the protein, which we term here the repeat domain (RD). Expression of the Tau RD causes pathology in transgenic mice (25), and there is evidence for truncation of full-length Tau to form fragments that comprise fibrils in patients (26, 27). We used this construct rather than full-length Tau because it reliably forms fibrils in cultured cells. We engineered various mutations known to increase Tau aggregation into a four-repeat domain (RD) protein: ΔK280 (termed ΔK), P301L, and V337M. The P301L and V337M mutants were combined in one protein (termed LM) to create a mutant form of RD with strongly increased aggregation potential, similar to what has been described previously (28). This “non-physiologic” mutant facilitates assays of transfer events and trans-cellular propagation of misfolding that depend on efficient formation of intracellular aggregates and complements similar but less robust aggregation phenotypes of the “physiologic” ΔK mutant. We also engineered two proline substitutions into the ΔK mutant, I227P and I308P (termed PP), which inhibit β-sheet formation and fibrillization (25), although they do not block formation of amorphous aggregates. Each form of mutant Tau was fused either at the carboxyl terminus to cyan or yellow fluorescent protein (CFP or YFP) or to an HA tag. Constructs are diagrammed in Fig. 1A.
FIGURE 1.

Tau RD proteins form fibrillar aggregates in transfected HEK293 cells. A, schematic representation of the different mutant Tau constructs used in this study. Depending on the experimental design, each form of mutant Tau was either fused at the carboxyl terminus to CFP or YFP or to an HA tag. B, AFM performed on SDS-insoluble material from HEK293 cells transiently transfected with the various forms of RD reveals that RD(ΔK)-HA and RD(LM)-HA produced obvious fibrillar species. No fibrils were detected in the aggregation-resistant RD(PP)-HA. (n = 2). Scale bars, 1 μm. C, HEK293 cells transiently transfected with the various forms of RD-YFP and YFP alone were stained with X-34, an amyloid-specific dye. Inclusions formed by RD(WT)-YFP, RD(ΔK)-YFP, and RD(LM)-YFP, visualized by confocal microscopy, also stained positive for X-34. No X-34-positive cells were detected upon expression of YFP alone or RD(PP)-YFP. The arrows indicate inclusions stained with X-34 (n = 3). D, non-transfected cells (NT) and various forms of RD-YFP/CFP were transfected into HEK293 cells, followed by Triton/SDS extraction and Western blotting using an antibody against the RD region. Both monomer and higher order molecular weight species were detected. S, soluble protein; P, pellet-insoluble protein. aa, amino acids. This was repeated three times with identical results.
To evaluate the characteristics of Tau RD intracellular aggregates, we transiently transfected the various forms of RD into HEK293 cells. We used AFM to evaluate SDS-insoluble material. RD(ΔK)-HA and RD(LM)-HA produced evident fibrillar species (Fig. 1B). RD(ΔK)-HA and RD(LM)-HA aggregates within cells also stained positive for X-34, a thioflavin derivative that labels β-sheet fibrils and emits in the blue spectrum (29) (Fig. 1C). Additionally, we used detergent fractionation to test whether the inclusions visible by light microscopy had a biochemical correlate. In SDS-insoluble pellets (1% Triton X-100 in 1× PBS with protease inhibitors for isolation of soluble protein followed by SDS extraction of insoluble pellets), we detected monomer and higher molecular weight species consistent with oligomers (Fig. 1D).
We have previously used FRET to quantitate intracellular huntingtin protein aggregation (22). To test whether this method could be used to track Tau RD aggregation, we fused the various RD mutants (WT, ΔK, PP, and LM) to yellow fluorescent protein (YFP; FRET acceptor) and cyan fluorescent protein (CFP; FRET donor). We co-transfected these constructs into HEK293 cells (denoted as RD-CFP/YFP), and quantified intracellular aggregate formation using FRET acceptor photobleaching confocal microscopy and spectral emission FRET using an FPR. For confocal microscopy, we imaged cells co-expressing RD(LM)-CFP/YFP and measured donor signal before and after partial and complete acceptor photobleaching. The increase in donor signal after photobleaching resulted in a mean FRET efficiency of 18.2 ± 0.058% (n = 6; data are ±S.D.), confirming intermolecular interactions between the FRET-paired RD species (Fig. 2A). To measure RD-CFP/YFP aggregation by spectral FRET with a FPR, we used established methods from our laboratory (30). This was based on co-transfection of RD-YFP and RD-CFP in a 3:1 ratio to maximize donor quenching within the limits of signal detection. We did not observe significant FRET from RD(PP)-CFP/YFP. However, RD(ΔK)-CFP/YFP and RD(LM)-CFP/YFP each produced a strong FRET signal (Fig. 2B), corroborating our findings by microscopy.
FIGURE 2.

Tau RD aggregates in HEK293 cells are detected by FRET. To quantitate intracellular RD protein aggregation by FRET, various RD mutants (WT, ΔK, PP, and LM) fused to YFP and CFP were co-transfected into HEK293 cells. A, HEK293 cells co-transfected with RD(LM)-CFP/YFP were imaged, and intracellular aggregate formation was quantified using FRET acceptor photobleaching microscopy. Donor signal before (Pre) and after (Post) acceptor photobleaching confirmed that RD(LM)-CFP/YFP inclusions produced a mean FRET efficiency of 18.2 ± 0.058% (S.D.) (n = 6). The top and bottom panels depict the acceptor and donor channels, respectively, before and after photobleaching. The top right image is a representative heat map of the calculated FRET efficiency. The scale bar of the histogram depicts the calculated FRET efficiency on a pixel-by-pixel basis. The FRET efficiency of Tau RD aggregate was ∼34% in this cell. B, using an FPR, relative FRET from various constructs was determined. No significant FRET from RD(PP)-CFP/YFP was observed. However, RD(ΔK)-CFP/YFP and RD(LM)-CFP/YFP each produced a strong FRET signal (n = 3). C, HEK293 cells expressing RD(ΔK)-CFP/YFP were exposed to various concentrations of RD(WT)-HA fibrils (monomer equivalents of 0.01, 0.03, 0.1, and 0.3 μm) for 9 h. Extracellular RD(WT)-HA fibrils dose-dependently induced aggregation of RD(ΔK)-CFP/YFP (n = 3). *, p < 0.05; **, p < 0.001; error bars, S.E.
We have previously observed that a variety of cells will take up recombinant Tau fibrils from the extracellular medium (12). This triggers intracellular fibrillization of natively folded, full-length Tau protein fused to YFP (12). To confirm this phenomenon, we used FRET to monitor aggregation of RD(ΔK)-CFP/YFP induced by various amounts of recombinant RD fibrils. HEK293 cells were co-transfected with RD(ΔK)-CFP/YFP and cultured for 15 h. We then added various concentrations of RD-HA fibrils (monomer equivalents of 0.01, 0.03, 0.1, and 0.3 μm) to the medium for 9 h. Fibrils were then removed by changing the medium, and the cells were allowed to recover for 4 h before being fixed and analyzed using FRET. We observed a dose-dependent increase in the FRET signal induced by recombinant fibrils relative to untreated RD(ΔK)-CFP/YFP cells (Fig. 2C). In summary, we observed a correlation between microscopic, molecular, biochemical, and biophysical measures of Tau RD aggregation and fibril formation within cells. Within certain limits, especially with controls for protein expression levels, the plate reader-based FRET assay provides a facile measure of this process.
Trans-cellular Induction of RD Aggregation
We have previously determined that Tau inclusions from one cell will transfer to naive cells in co-culture (12). However, it has not yet been demonstrated that these transferred aggregates can induce further aggregation in the recipient cells; nor has it been shown whether induction of aggregation is based on direct protein-protein interaction. We first tested whether RD(LM)-HA aggregates derived from one donor cell population would form inclusions with RD(ΔK)-YFP in a different recipient population upon co-culture. We transfected one group of cells with aggregation-prone RD(LM)-HA and a separate group with RD(ΔK)-YFP. The next day, we replated the cell populations together and co-cultured them for 48 h. After fixation, we immunostained using an HA antibody and counterstained with X-34. We observed many cells with RD(LM)-HA and RD(ΔK)-YFP colocalized in inclusions (Fig. 3A). Frequently, these inclusions also stained positive for X-34, indicating β-sheet structure. We extended these studies by using the FRET assay to monitor aggregation of RD(ΔK)-CFP/YFP induced by co-culture with cells expressing RD(LM)-HA. In this case, two populations of cells were co-cultured. The donor population expressed RD(LM)-HA, and the recipient population expressed RD(ΔK)-CFP/YFP. As negative controls, we used the β-sheet-resistant form of Tau RD(PP)-HA or mock-transfected cells. After 48 h, we measured FRET from the cell monolayers. We observed a strong increase in FRET induced by co-culture with RD(LM)-HA versus RD(PP)-HA or mock-transfected cells (Fig. 3B). A small increase in FRET signal was observed following co-culture of RD(LM)-HA cells with RD(WT)-CFP/YFP recipient cells (data not shown). These results suggested movement of one aggregation-prone Tau species from one cell to another to trigger co-localization in a β-sheet-rich inclusion. Aggregate release could potentially occur after cell death; however, we observed no evidence for this using propidium iodide staining of the various transfected populations (Fig. 3C).
FIGURE 3.

Tau RD aggregates transfer between cells and induce further aggregation. A, HEK293 cells transfected with RD(ΔK)-YFP were co-cultured for 48 h with an equivalent number of cells expressing RD(LM)-HA. Cells were fixed with 4% paraformaldehyde, and immunofluorescence/X-34 staining was performed. Multiple cells showed colocalization of RD(LM)-HA and RD(ΔK)-YFP within inclusions. These inclusions also stained positive for X-34, indicating β-sheet structure (solid arrows). In addition, some RD(LM)-HA inclusions stained positive for X-34 but did not colocalize with RD(ΔK)-YFP inclusions (open arrow). B, two populations of cells, one expressing RD(ΔK)-CFP/YFP, and the other expressing RD(LM)-HA, were co-cultured for 48 h. RD(PP)-HA or non-transfected cells (NT) were used as controls. FRET was increased by co-culture with RD(LM)-HA-transfected but not with RD(PP)-HA- or mock-transfected cells (n = 3). C, to test for cell death induced by Tau aggregates as a mechanism of Tau release, HEK293 cells were transfected for 48 h with RD-HA (PP, ΔK, or LM) or were mock-transfected. Mock-transfected cells were treated with varying concentrations of staurosporine (1, 2, 4, and 20 μm) for 30 min at 37 °C to induce cell death. Cells were then exposed to 5 μg/ml propidium iodide, and fluorescence was determined via a plate reader. No evidence for cell death in the various transfected populations was observed. **, p < 0.001; error bars, S.E.
Propagation of Misfolding by Direct Protein Contact
Although strongly suggestive, these results could not formally address whether co-aggregation occurred via direct protein contact, with intermolecular association between Tau RD derived from donor contacting the corresponding protein in recipient cells. We used FRET to address this question. First, we co-expressed RD(LM)-CFP within a donor cell population and RD(LM)-YFP in a second recipient population. After 48 h, we measured FRET from the cell monolayers with both confocal microscopy and the FPR. Using confocal microscopy, we measured CFP signal before and after photobleaching of YFP. We recorded a mean FRET efficiency of ∼14.2%, indicating that inclusions contained RD(LM)-CFP and RD(LM)-YFP in direct contact (Fig. 4A). We then compared relative FRET signals via FPR, using different forms of unlabeled RD to induce aggregation of RD-CFP. First, we co-expressed RD(ΔK)-CFP and RD(LM)-HA within a donor cell population and RD(ΔK)-YFP in a second recipient cell population. RD(LM)-HA serves as an enhancer of both RD(ΔK)-CFP aggregation and movement, prompting its subsequent transfer into the RD(ΔK)-YFP recipient cells. This led to a small but reproducible FRET signal increase in the co-cultured cells. This signal disappeared when either the CFP- or YFP-tagged RD constructs contained the PP mutation that blocks β-sheet formation (Fig. 4B), indicating that both members of the pair must have the capacity to form a β-sheet structure. Taken together with the prior experiments, these results suggested that propagation of misfolding by direct contact occurs, i.e. an aggregate from one cell exits to contact and trigger misfolding of natively folded protein in a second cell.
FIGURE 4.

RD aggregates propagate misfolding between cells. HEK293 cells were co-transfected with various RD-CFP and RD-HA constructs. 15 h later, these cells were co-cultured with cells expressing RD(ΔK)-YFP or RD(PP)-YFP for 48 h. A, FRET microscopy was performed to determine whether co-aggregation occurred via direct protein contact. CFP signal was measured before and after photobleaching of YFP. RD(LM)-CFP and RD(LM)-YFP aggregates had a mean FRET efficiency of 14.2 ± 0.053% (S.D.) (n = 11), indicative of RD(LM)-CFP and RD(LM)-YFP in direct contact. The top and bottom panels depict the acceptor and donor channels, respectively, before (Pre) and after (Post) photobleaching. A representative heat map of the calculated FRET efficiency is shown at the top right. The histogram depicts the calculated FRET efficiency on a pixel-by-pixel basis. The FRET efficiency of Tau RD aggregate was ∼25% in this cell. Negative values are derived from unpaired CFP. B, a FRET signal was observed when cells expressing RD(ΔK)-CFP/RD-HA were co-cultured with cells expressing RD(ΔK)-YFP. This signal increased when aggregation of RD(ΔK)-CFP was induced by co-expression of aggregation-prone forms of Tau, either ΔK or LM mutants. No significant signal was noted when either RD-CFP or RD-YFP contained the PP mutation that blocks β-sheet formation (n = 3). C, to test for amplification of misfolding, populations of cells expressing CFP alone or RD(LM)-CFP were pre-exposed for 48 h to cells expressing RD-HA with either PP, ΔK, or LM mutations to promote misfolding to varying degrees. These co-cultured populations were then split and co-cultured for 48 h with cells expressing RD(ΔK)-YFP to determine the degree of aggregation reported by cell-cell transfer and FRET. Prior exposure of RD(LM)-HA cells to the RD(ΔK)-CFP cell population increased FRET signal by 2.6-fold versus prior exposure to RD(PP)-HA. Interposition of cells expressing pure CFP in the second population of cells completely blocked the effect of prior exposure to aggregation-prone RD-HA mutants (n = 3). *, p < 0.05; **, p < 0.001; error bars, S.E.
This data implied that amplification of misfolding might also occur in serial cell co-cultures. We predicted that pre-exposure of a “donor” cell population to aggregation seeds would increase final aggregation detected in a recipient cell population. We tested this by successively culturing three populations of cells. The first population expressed various forms of non-fluorescent RD-HA to form aggregation “seeds.” The second group expressed CFP or RD(ΔK)-CFP, to be either non-permissive (CFP) or permissive (RD(ΔK)-CFP) for aggregate maintenance. These two groups were co-cultured for 48 h to allow amplification of misfolding. Next, 50% of the combined first and second groups were then co-cultured for 48 h with a third group of cells expressing RD(ΔK)-YFP. This third recipient group served as a “reporter” to indicate the degree of RD(ΔK)-CFP intracellular aggregation and propagation. Prior exposure of RD(LM)-HA to the RD(ΔK)-CFP population increased final FRET by 2.6-fold versus cells that had not been pre-exposed to aggregation-prone Tau. As expected, interposition of cells expressing pure CFP in the second population of cells completely blocked the effect of prior exposure to Tau RD seeds (Fig. 4C). Taken together, these data indicate an amplification of Tau aggregation within serially cultured cell populations.
Cell-Cell Propagation Mediated by Release of Aggregates into Extracellular Space
The mechanism by which protein aggregates move between cells is unknown. For example, some have postulated prion protein propagation via tunneling nanotubes (19, 20), whereas others have suggested exosomes (17, 18). Because antibodies against Tau protein have previously been reported to reduce pathology in vivo (31–35), we hypothesized that Tau aggregates might be released directly into the extracellular space.
Because trans-cellular movement based on cell-cell contact should be independent of the volume of extracellular medium, we predicted that trans-cellular movement of Tau might be sensitive to extracellular volume, as has been described for SOD1 (13). We began by testing the effect of co-culture in the setting of various volumes of medium. We observed that increasing the cell culture medium volume reduced the efficiency of trans-cellular movement of aggregates (Fig. 5A). Further, transfer of conditioned medium from cells expressing RD(LM)-HA was sufficient to induce aggregation in cells expressing RD-CFP/YFP (Fig. 5B). These results were consistent with the movement of Tau between cells through the extracellular space but could not determine whether the protein was encapsulated in an endosome.
FIGURE 5.

Propagation of Tau aggregates through the extracellular medium. A, HEK293 cells transfected with RD(LM)-HA were co-cultured for 48 h with an equivalent number of RD(ΔK)-CFP/YFP cells prior to FRET analysis. Increasing the volume of cell culture medium reduced the efficiency of trans-cellular movement of aggregates. B, transfer of conditioned medium from cells expressing RD(LM)-HA to cells expressing RD(ΔK)-CFP/YFP was sufficient to induce aggregation by 60%. C, HJ9.3 antibody added to the medium reduced FRET, consistent with interference with propagation of aggregation. D, nonspecific IgG had no effect on propagation. E, HJ9.3 had no effect on intracellular aggregation of RD(ΔK)-CFP/YFP co-expressed within the same cell. F, HJ9.3 blocked the effect of RD(LM)-HA to induce RD(ΔK)-YFP in co-cultured cells, as determined by detergent fractionation and Western blot. T, total protein; S, soluble protein; P, pellet-insoluble protein. G, quantitative analysis of three independent Western blots revealed a ∼60% decrease in the pellet fraction, relative to the total fraction, after exposure to HJ9.3. H, cells expressing RD(LM)-YFP and mCherry were co-cultured and analyzed by flow cytometry. HJ9.3 decreased the percentage of dual positive cells from 2.07 to 1.31%. Cells mixed just prior to cytometry were a background control. *, p < 0.05; **, p < 0.001; error bars, S.E.
We reasoned that access to encapsulated Tau would be blocked by the lipid membrane, whereas free Tau would be accessible to an antibody. Thus, we tested whether a mouse monoclonal antibody (HJ9.3) that can immunprecipitate Tau (21) would block trans-cellular propagation. We used a modification of the cellular model of Tau RD propagation described above, in which RD(LM)-HA and RD(ΔK)-CFP were co-expressed within one cell population and co-cultured for 48 h with cells that express RD(ΔK)-YFP prior to analysis by FRET. We tested HJ9.3 versus pooled mouse IgG for the 48-h co-culture period. We observed a dose-dependent reduction in trans-cellular propagation with HJ9.3, whereas nonspecific IgG had no effect (Fig. 5, C and D). Importantly, HJ9.3 had no effect on intracellular aggregation of RD(ΔK)-CFP and RD(ΔK)-YFP when the two proteins were co-expressed within the same cell (Fig. 5E), indicating the antibody was not directly inhibiting intracellular aggregation. We further tested the role of free Tau in trans-cellular propagation by evaluating induction of Tau misfolding using biochemistry. We confirmed the induction of aggregation by detergent fractionation and Western blot, which revealed an increase in RD(ΔK)-YFP in the insoluble fraction induced by co-culture with RD(LM)-HA. HJ9.3 blocked the effect of RD(LM)-HA to induce insolubility of RD-YFP in co-cultured cells (Fig. 5, F and G).
The effectiveness of antibody addition suggested that free Tau was directly transferring between cells but left uncertain the mechanism of antibody inhibition. We hypothesized that HJ9.3 was blocking uptake of Tau fibrils into cells. To test this idea, we used flow cytometry to monitor the effect of the antibody on trans-cellular movement of aggregates. We have previously established a cytometry paradigm whereby one population of cells is labeled with mCherry and the second contains Tau-YFP fusions (12). After co-culture, it is possible to monitor trans-cellular movement based on the relative percentage of dual positive (YFP/mCherry) cells. A population of HEK293 cells was transfected with Tau RD(LM)-YFP, and a second population was transduced with lentivirus expressing mCherry. After washing and resuspending the two populations, we then co-cultured the cells for 48 h in the presence or absence of 10-fold dilutions of HJ9.3 in the medium. We harvested cells and measured the relative number of dual positive cells using flow cytometry. Negative controls consisted of the same cell populations mixed prior to sorting. Each data point consisted of biological triplicates. Co-cultured cells had significantly more RD(LM)-YFP/mCherry dual positive cells (2.07%) compared with 0.142% of premixed cells (background). HJ9.3 decreased the percentage of dual positive cells from 2.07 to 1.31% (Fig. 5H). This parallels the effect of this antibody on trans-cellular propagation of aggregation as measured by FRET. The difference in the potency of this antibody in blocking propagation as measured by FRET and flow cytometry is most probably due to the differences between the two techniques used to measure this event.
To further monitor the effect of the HJ9.3 antibody on trans-cellular movement of aggregates, we used direct immunofluorescence in an attempt to define where the HJ9.3-antibody complexes deposited. We cultured RD(ΔK)-YFP cells or non-transfected cells in the presence of HJ9.3 for 48 h. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.25% Triton X-100, and then exposed to goat anti-mouse Alexa 546-labeled secondary antibody. A very small number of HJ9.3-Tau complexes were present inside cells. However, most complexes were found outside of the cells, mainly bound to the cell membrane. This antibody decoration was not present in non-transfected cells, indicating that the signal is specific to the HJ9.3-Tau complexes (Fig. 6). Thus, HJ9.3 blocks Tau aggregate uptake, trapping aggregates outside the cell.
FIGURE 6.

HJ9.3 traps RD(ΔK)-YFP aggregates at the cell surface. HEK293 cells were transfected with RD(ΔK)-YFP or were mock-transfected. HJ9.3 was added to the culture medium for the 48-h period. At the end of the experiment, the cells were fixed, permeabilized, and stained with an anti-mouse secondary antibody (labeled with Alexa 546). Confocal microscopy was used to analyze the localization of HJ9.3-Tau complexes. The top panels show that many complexes are identified when RD(ΔK)-YFP is expressed, but none are identified in its absence (bottom panels). Orthogonal analyses (right panel) demonstrate that most complexes are present at the cell surface, although occasional intracellular complexes were observed. NT, non-transfected; DIC, differential interference contrast.
Tau Fibrils Mediate Cell-Cell Propagation
The activity of HJ9.3 in the propagation assay created an opportunity to define the Tau species responsible. We used HJ9.3 to extract Tau from the cell medium. We added HJ9.3 or control IgG to the medium of cells expressing a variety of RD constructs (WT, PP, ΔK, LM). Antibodies were added either at the beginning or at the end of the 48-h culture period. Medium was harvested for affinity purification of antibody-antigen complexes using protein G-agarose beads. The complexes were washed and then boiled in SDS loading buffer for analysis by Western blot. HJ9.3 specifically captured Tau RD species from the cell medium, whereas IgG had no appreciable effect (Fig. 7A). We observed a ∼10-fold increase in the Tau protein present in the medium when HJ9.3 was present throughout the culture period, as opposed to addition at the end of this period (Fig. 7B). We also noted higher order molecular weight species in the medium of RD(ΔK)-HA- and RD(LM)-HA-transfected cells, consistent with RD aggregates. RD(PP)-HA Tau had the least protein present in the medium, and no higher order species were observed on Western blot. A time course (0, 3, 6, 9, 12, 24, and 48 h) of the previously described experiment showed a time-dependent increase in the levels of Tau in the medium, implying that HJ9.3 incubation was indeed increasing the steady-state level of Tau protein present (Fig. 7C). Taken together, these data indicated that HJ9.3 blocks cell-to-cell propagation by interference with aggregate uptake into cells and is consistent with a steady state flux of Tau aggregates in and out of cells.
FIGURE 7.

Tau fibrils mediate cell-cell propagation. A, conditioned medium was collected from transfected cell populations co-cultured for 0 or 48 h with HJ9.3 or control IgG antibody (1:1000), followed by immunoprecipitation and Western blot. HJ9.3 specifically captured Tau RD species from the cell medium, whereas IgG did not. Higher order aggregated species were present upon expression of RD(ΔK)-YFP or RD(LM)-YFP but not RD(PP)-YFP. B, quantitative analyses of three independent Western blots showed a ∼10-fold increase in the Tau after 48 h of incubation. C, cells were exposed to HJ9.3 for various times. D, purified antibody-antigen complexes from medium exposed for 48 h to HJ9.3 were deposited on AFM chips for imaging. Obvious fibrillar species in the medium of cells expressing RD(ΔK)-HA and RD(LM)-HA were observed, whereas RD(PP)-HA produced only amorphous aggregates. Scale bar, 1 μm. NT, non-transfected.
The precise nature of the Tau species that mediate trans-cellular propagation is not known. Thus, we used HJ9.3 to trap these species for imaging via AFM. We cultured HEK293 cells that were transfected with the various Tau mutants in the presence of HJ9.3. After 48 h, we purified the antibody-antigen complexes with protein G-agarose beads. The complexes were then eluted from the beads in high salt buffer and deposited on AFM chips for imaging. We detected evident fibrillar species in the medium of cells expressing RD(ΔK)-HA and RD(LM)-HA, whereas RD(PP)-HA produced only amorphous aggregates (Fig. 7D), and mock-transfected cells produced no signal (data not shown). These findings are consistent with free Tau fibrils mediating trans-cellular propagation of Tau aggregation by their release into the extracellular space.
DISCUSSION
We and others have previously proposed that prion-like mechanisms involving templated conformational change and trans-cellular propagation of aggregation could explain the relentless progression of tauopathies and other neurodegenerative diseases. This would consist of the release of a protein aggregate from a donor cell, entry into a recipient cell, and direct contact with natively folded protein to amplify the misfolded state. However, mechanistic evidence to support this model of tauopathy has been incomplete, and trans-cellular propagation of Tau misfolding in this manner has not previously been demonstrated. We have now described trans-cellular propagation of Tau aggregation in cultured cells via secreted Tau aggregates and propose a likely mechanism. We first documented spontaneous formation of RD Tau fibrils in transfected cells using X-34 staining and AFM of extracted material. We then observed the co-incidence of Tau derived from two separate cells in intracellular inclusions using confocal microscopy. This was associated with increased detergent insolubility of Tau RD(ΔK)-YFP upon co-culture with cells expressing an aggregation-prone form of the protein, RD(LM)-HA. We also documented this increase in aggregation using FRET between RD(LM)-CFP and RD(LM)-YFP that were co-expressed within the same cells. This was detected by acceptor photobleaching (microscopy), and spectral methods (FPR). We next used FRET between RD(LM)-CFP and RD(LM)-YFP expressed in separate cell populations to document that propagation occurred by direct protein contact. We then extended this method to document amplification of Tau protein misfolding within the cell populations in successive culture conditions. Trans-cellular propagation of Tau aggregation is mediated by fibrils that are released directly into the extracellular space, because transfer is sensitive to extracellular volume, conditioned medium can increase intracellular aggregation, and an anti-Tau antibody (HJ9.3) interfered with cell-cell propagation and trapped extracellular Tau fibrils. Using a variety of techniques, we have thus documented the trans-cellular aggregate propagation via templated conformational change. We propose a simple model to explain these phenomena (Fig. 8).
FIGURE 8.

Trans-cellular propagation of Tau aggregation occurs via transfer of fibrils within the cell medium. We propose a model of Tau propagation whereby a protein aggregate in a donor cell escapes the cell (A), enters a recipient cell (B), and directly contacts natively folded protein (C) to amplify the misfolded state (D). This cell-cell movement is mediated by fibrils that are released directly into the medium. These fibrils can be trapped within the extracellular space by an anti-Tau antibody (HJ9.3) that interferes with cell-cell propagation (E).
Trans-cellular Propagation
Although we previously described spontaneous movement of aggregated Tau between cells (12), it was unknown whether Tau protein aggregates could propagate a misfolded state between cells by direct contact of the proteins, as opposed to indirect effects on protein quality control. Cell culture studies of α-synuclein have also suggested propagation, but the nature of the species (e.g. aggregates versus dimers versus monomer) derived from donor cells and those formed in recipient cells is unclear (6, 36–38). Likewise, SOD1 aggregates can transfer between cells via the medium to induce further aggregation, but the precise nature of the responsible protein conformers and whether direct protein-protein contact occurs are unclear (13). Injection of purified Aβ42 and Tau fibrils into transgenic mouse brain induces aggregation of endogenous Tau (39, 40), with nearby development of Tau fibrils, but it is difficult to rule out seeding by injected protein. Work from our own laboratory (12) and subsequently from others (15) has documented movement of Tau aggregates and induction of aggregation by recombinant protein from the outside to the inside of the cell. However, to our knowledge, no prior study of the Tau protein has demonstrated bona fide propagation: aggregate movement from one cell to another, direct contact with the native protein, conversion of the protein in the recipient cell to a fibrillar state, and amplification of the misfolded species.
In this work, we demonstrated these phenomena in several ways. First, we found that co-culture of an aggregation-prone form of Tau RD(LM)-HA with cells expressing RD(ΔK)-YFP leads to co-localization in β-sheet-positive inclusions. Next, we observed that co-culture of cells expressing RD(LM)-HA with another population expressing both RD(ΔK)-CFP and RD(ΔK)-YFP led to an increase of FRET signal, suggesting that movement of RD(LM)-HA into cells expressing the FRET pair was inducing their aggregation. To demonstrate direct contact and co-aggregation of Tau aggregates moving between cells, we expressed RD(ΔK)-CFP and RD(ΔK)-YFP in separate populations. This led to a FRET signal derived from trans-cellular movement and co-aggregation that disappeared if either one of the constructs contained a double proline mutation to block β-sheet formation. We have also observed induction of full-length Tau-YFP aggregation by transfer of RD-CFP aggregates, but the efficiency is reduced (data not shown). Finally, the efficiency of FRET induced by trans-cellular movement of protein aggregates increased significantly by preliminary co-culture of RD(LM)-HA-expressing cells with those expressing RD(ΔK)-CFP, demonstrating that an aggregated state can be amplified within a population of cells.
Antibody Modulation of Tau Aggregate Propagation
Antibodies against Aβ peptide, which is predominantly extracellular, can prevent Aβ aggregation in the brain and remove existing aggregates (41, 42). Although there are potential side effects, such antibodies hold promise as treatments. However, the success of vaccination in mouse models of tauopathy (31–35) and synucleinopathy (43) has been puzzling in light of the fact that the target proteins are predominantly intracellular. We observed that HJ9.3, a mouse monoclonal antibody against Tau RD, inhibited the trans-cellular propagation of Tau aggregation. However, this antibody had no effect on intracellular aggregation of Tau. Chronic exposure of the cell medium to this antibody strongly increased the steady state Tau levels in the medium. This was corroborated by flow cytometry studies, which indicated that HJ9.3 blocks transfer of aggregates from one cell to another. Finally, we observed HJ9.3-Tau complexes trapped at the cell surface. The effect of this antibody suggested strongly that Tau fibrils are released into the extracellular space and are not propagating misfolding primarily via cell-cell transfer in exosomes or tunneling nanotubes, as has been proposed for prions. Further, aggregates present outside of the cell, if not trapped by HJ9.3, are probably taken up again into cells. Multiple modes of inhibition are conceivable for therapeutic antibodies, including disaggregation of protein fibrils, blockade of conversion within cells, and promotion of intracellular degradation. Our results with HJ9.3 are most consistent with interference with cell uptake as one mechanism that could be used to block tauopathy and suggest new ways to consider development and optimization of therapeutic antibodies for neurodegenerative diseases.
Trans-cellular Propagation via Fibrillar Tau
The effectiveness of HJ9.3 in blocking propagation of Tau aggregation allowed us to use this antibody to trap the responsible species. Immunoaffinity purification of Tau from conditioned medium revealed fibrillar Tau. We observed no Tau fibrils in medium from control cells or from cells expressing the β-sheet-resistant RD(PP)-HA, which produced amorphous aggregates. RD(ΔK)-HA and RD(LM)-HA expression each caused fibril secretion into the extracellular space. It has been unclear how protein aggregation in one cell might influence the aggregation in a neighboring cell, and it was formally possible that cytokines, exosomes, or direct connections between cells might facilitate this process. We cannot completely exclude these possibilities. However, our results are most consistent with free fibrillar species as mediators of propagation through the extracellular space. This work suggests answers to several important questions about the mechanisms by which protein aggregates propagate from one cell to another in culture and thus how they might do so in vivo. In conjunction with the methods described here to monitor trans-cellular propagation, it may be possible to target this process with pharmacological and biological agents for more effective treatment of tauopathies and other neurodegenerative diseases.
Acknowledgments
We thank Josiah Gerdts for help with the schematic design of Figs. 1A and 7. We also thank the Milbrandt laboratory for supplying the mCherry Lentivirus used in the flow cytometry experiments. We thank Mei Li for initiating cloning of the RD Tau constructs.
This work was supported, in whole or in part, by National Institutes of Health (NIH), NINDS, Grant 1R01NS071835-01; NIH Neuroscience Blueprint Interdisciplinary Center Core Grant P30 (NS057105) (to Washington University); and NIH Grant P50AG005681 (to the Knight Alzheimer's Disease Research Center at Washington University). This work was also supported by grants from the Muscular Dystrophy Association, the American Health Assistance Foundation, the Ruth K. Broad Foundation, the Tau Consortium, the Hope Center for Neurological Disorders at Washington University in St. Louis, the Donald H. and Mary Jane Buchanan Foundation Alzheimer's Disease Research Fund, the Barnes-Jewish Hospital Foundation, the Siteman Cancer Center of Washington University in St. Louis, the Bakewell Neuroimaging Core (supported in part by the Bakewell Family Foundation), and the Alafi Neuroimaging Laboratory.
- AD
- Alzheimer disease
- RD
- repeat domain
- FPR
- fluorescence plate reader
- AFM
- atomic force microscopy.
REFERENCES
- 1. Ballatore C., Lee V. M., Trojanowski J. Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 [DOI] [PubMed] [Google Scholar]
- 2. Braak H., Braak E. (1995) Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol. Aging 16, 271–278, discussion 278–284 [DOI] [PubMed] [Google Scholar]
- 3. Seeley W. W., Crawford R. K., Zhou J., Miller B. L., Greicius M. D. (2009) Neurodegenerative diseases target large-scale human brain networks. Neuron 62, 42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kordower J. H., Chu Y., Hauser R. A., Olanow C. W., Freeman T. B. (2008) Transplanted dopaminergic neurons develop PD pathologic changes. A second case report. Mov. Disord. 23, 2303–2306 [DOI] [PubMed] [Google Scholar]
- 5. Kordower J. H., Chu Y., Hauser R. A., Freeman T. B., Olanow C. W. (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506 [DOI] [PubMed] [Google Scholar]
- 6. Desplats P., Lee H. J., Bae E. J., Patrick C., Rockenstein E., Crews L., Spencer B., Masliah E., Lee S. J. (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee H. J., Suk J. E., Patrick C., Bae E. J., Cho J. H., Rho S., Hwang D., Masliah E., Lee S. J. (2010) Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A. K., Beibel M., Staufenbiel M., Jucker M., Goedert M., Tolnay M. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J. Y., Englund E., Holton J. L., Soulet D., Hagell P., Lees A. J., Lashley T., Quinn N. P., Rehncrona S., Björklund A., Widner H., Revesz T., Lindvall O., Brundin P. (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503 [DOI] [PubMed] [Google Scholar]
- 10. de Calignon A., Polydoro M., Suárez-Calvet M., William C., Adamowicz D. H., Kopeikina K. J., Pitstick R., Sahara N., Ashe K. H., Carlson G. A., Spires-Jones T. L., Hyman B. T. (2012) Propagation of Tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu L. (2012) Trans-synaptic spread of Tau pathology in vivo. PLoS One 7, e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frost B., Jacks R. L., Diamond M. I. (2009) Propagation of Tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Münch C., O'Brien J., Bertolotti A. (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3548–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luk K. C., Song C., O'Brien P., Stieber A., Branch J. R., Brunden K. R., Trojanowski J. Q., Lee V. M. (2009) Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. 106, 20051–20056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo J. L., Lee V. M. (2011) Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ren P. H., Lauckner J. E., Kachirskaia I., Heuser J. E., Melki R., Kopito R. R. (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Porto-Carreiro I., Février B., Paquet S., Vilette D., Raposo G. (2005) Prions and exosomes. From PrPc trafficking to PrPsc propagation. Blood Cells Mol. Dis. 35, 143–148 [DOI] [PubMed] [Google Scholar]
- 18. Vella L. J., Sharples R. A., Nisbet R. M., Cappai R., Hill A. F. (2008) The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur. Biophys. J. 37, 323–332 [DOI] [PubMed] [Google Scholar]
- 19. Gousset K., Zurzolo C. (2009) Tunneling nanotubes. A highway for prion spreading? Prion 3, 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gerdes H. H. (2009) Prions tunnel between cells. Nat. Cell Biol. 11, 235–236 [DOI] [PubMed] [Google Scholar]
- 21. Yamada K., Cirrito J. R., Stewart F. R., Jiang H., Finn M. B., Holmes B. B., Binder L. I., Mandelkow E. M., Diamond M. I., Lee V. M., Holtzman D. M. (2011) In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid Tau levels in P301S human Tau transgenic mice. J. Neurosci. 31, 13110–13117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pollitt S. K., Pallos J., Shao J., Desai U. A., Ma A. A., Thompson L. M., Marsh J. L., Diamond M. I. (2003) A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron 40, 685–694 [DOI] [PubMed] [Google Scholar]
- 23. Shao J., Welch W. J., Diamond M. I. (2008) ROCK and PRK-2 mediate the inhibitory effect of Y-27632 on polyglutamine aggregation. FEBS Lett. 582, 1637–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iqbal K., Liu F., Gong C. X., Alonso A. D., Grundke-Iqbal I. (2009) Mechanisms of Tau-induced neurodegeneration. Acta Neuropathol. 118, 53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mocanu M. M., Nissen A., Eckermann K., Khlistunova I., Biernat J., Drexler D., Petrova O., Schönig K., Bujard H., Mandelkow E., Zhou L., Rune G., Mandelkow E. M. (2008) The potential for β-structure in the repeat domain of Tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J. Neurosci. 28, 737–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Basurto-Islas G., Luna-Muñoz J., Guillozet-Bongaarts A. L., Binder L. I., Mena R., García-Sierra F. (2008) Accumulation of aspartic acid 421- and glutamic acid 391-cleaved Tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J. Neuropathol. Exp. Neurol. 67, 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guillozet-Bongaarts A. L., Garcia-Sierra F., Reynolds M. R., Horowitz P. M., Fu Y., Wang T., Cahill M. E., Bigio E. H., Berry R. W., Binder L. I. (2005) Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol. Aging 26, 1015–1022 [DOI] [PubMed] [Google Scholar]
- 28. Vogelsberg-Ragaglia V., Bruce J., Richter-Landsberg C., Zhang B., Hong M., Trojanowski J. Q., Lee V. M. (2000) Distinct FTDP-17 missense mutations in Tau produce Tau aggregates and other pathological phenotypes in transfected CHO cells. Mol. Biol. Cell 11, 4093–4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Styren S. D., Hamilton R. L., Styren G. C., Klunk W. E. (2000) X-34, a fluorescent derivative of Congo red. A novel histochemical stain for Alzheimer's disease pathology. J. Histochem. Cytochem. 48, 1223–1232 [DOI] [PubMed] [Google Scholar]
- 30. Desai U. A., Pallos J., Ma A. A., Stockwell B. R., Thompson L. M., Marsh J. L., Diamond M. I. (2006) Biologically active molecules that reduce polyglutamine aggregation and toxicity. Hum. Mol. Genet. 15, 2114–2124 [DOI] [PubMed] [Google Scholar]
- 31. Boutajangout A., Ingadottir J., Davies P., Sigurdsson E. M. (2011) Passive immunization targeting pathological phospho-Tau protein in a mouse model reduces functional decline and clears Tau aggregates from the brain. J. Neurochem. 118, 658–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Asuni A. A., Boutajangout A., Quartermain D., Sigurdsson E. M. (2007) Immunotherapy targeting pathological Tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 27, 9115–9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boutajangout A., Quartermain D., Sigurdsson E. M. (2010) Immunotherapy targeting pathological Tau prevents cognitive decline in a new tangle mouse model. J. Neurosci. 30, 16559–16566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chai X., Wu S., Murray T. K., Kinley R., Cella C. V., Sims H., Buckner N., Hanmer J., Davies P., O'Neill M. J., Hutton M. L., Citron M. (2011) Passive immunization with anti-Tau antibodies in two transgenic models. Reduction of Tau pathology and delay of disease progression. J. Biol. Chem. 286, 34457–34467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bi M., Ittner A., Ke Y. D., Götz J., Ittner L. M. (2011) Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L Tau transgenic mice. PloS One 6, e26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kondo K., Obitsu S., Teshima R. (2011) α-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol. Pharm. Bull. 34, 1078–1083 [DOI] [PubMed] [Google Scholar]
- 37. Hansen C., Angot E., Bergström A. L., Steiner J. A., Pieri L., Paul G., Outeiro T. F., Melki R., Kallunki P., Fog K., Li J. Y., Brundin P. (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Danzer K. M., Ruf W. P., Putcha P., Joyner D., Hashimoto T., Glabe C., Hyman B. T., McLean P. J. (2011) Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 25, 326–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bolmont T., Clavaguera F., Meyer-Luehmann M., Herzig M. C., Radde R., Staufenbiel M., Lewis J., Hutton M., Tolnay M., Jucker M. (2007) Induction of Tau pathology by intracerebral infusion of amyloid-β-containing brain extract and by amyloid-beta deposition in APP × Tau transgenic mice. Am. J. Pathol. 171, 2012–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Götz J., Chen F., van Dorpe J., Nitsch R. M. (2001) Formation of neurofibrillary tangles in P301l Tau transgenic mice induced by Aβ 42 fibrils. Science 293, 1491–1495 [DOI] [PubMed] [Google Scholar]
- 41. Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P. (1999) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177 [DOI] [PubMed] [Google Scholar]
- 42. Nicoll J. A., Wilkinson D., Holmes C., Steart P., Markham H., Weller R. O. (2003) Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide. A case report. Nat. Med. 9, 448–452 [DOI] [PubMed] [Google Scholar]
- 43. Masliah E., Rockenstein E., Adame A., Alford M., Crews L., Hashimoto M., Seubert P., Lee M., Goldstein J., Chilcote T., Games D., Schenk D. (2005) Effects of α-synuclein immunization in a mouse model of Parkinson's disease. Neuron 46, 857–868 [DOI] [PubMed] [Google Scholar]