

Background: Phosphorylation of amyloid β-precursor protein (APP) at Thr668 alters the conformation of its cytoplasmic domain.
Results: Phosphorylation of APP C-terminal fragments (pCTFs) at Thr668 decreases membrane lipid binding.
Conclusion: Phosphorylation at Thr668 regulates the localization of pCTFs away from γ-secretase-containing, lipid raft-like membrane microdomains.
Significance: Preservation of the phosphorylation of APP CTFs at Thr668 may be a useful treatment to lower amyloid β-protein generation.
Keywords: Alzheimers Disease, Amyloid Precursor Protein, Lipid Raft, Protein Phosphorylation, Secretases
Abstract
Amyloid β-precursor protein (APP) is primarily cleaved by α- or β-secretase to generate membrane-bound, C-terminal fragments (CTFs). In turn, CTFs are potentially subject to a second, intramembrane cleavage by γ-secretase, which is active in a lipid raft-like membrane microdomain. Mature APP (N- and O-glycosylated APP), the actual substrate of these secretases, is phosphorylated at the cytoplasmic residue Thr668 and this phosphorylation changes the overall conformation of the cytoplasmic domain of APP. We found that phosphorylated and nonphosphorylated CTFs exist equally in mouse brain and are kinetically equivalent as substrates for γ-secretase, in vitro. However, in vivo, the level of the phosphorylated APP intracellular domain peptide (pAICD) generated by γ-cleavage of CTFs was very low when compared with the level of nonphosphorylated AICD (nAICD). Phosphorylated CTFs (pCTFs), rather than nonphosphorylated CTFs (nCTFs), were preferentially located outside of detergent-resistant, lipid raft-like membrane microdomains. The APP cytoplasmic domain peptide (APP(648–695)) with Thr(P)668 did not associate with liposomes composed of membrane lipids from mouse brain to which the nonphosphorylated peptide preferentially bound. In addition, APP lacking the C-terminal 8 amino acids (APP-ΔC8), which are essential for membrane association, decreased Aβ generation in N2a cells. These observations suggest that the pCTFs and CTFΔC8 are relatively movable within the membrane, whereas the nCTFs are susceptible to being anchored into the membrane, an interaction made available as a consequence of not being phosphorylated. By this mechanism, nCTFs can be preferentially captured and cleaved by γ-secretase. Preservation of the phosphorylated state of APP-CTFs may be a potential treatment to lower the generation of Aβ in Alzheimer disease.
Introduction
Alzheimer disease (AD)3 is the most common of the senile neurodegenerative disorders. Amyloid β-protein (Aβ) is a causative peptide of AD and is believed to show toxicity to neurons by forming oligomers (1). Aβ is generated from the amyloid β-precursor protein (APP), which is a type I membrane protein composed of three major spliced isoforms, APP770, APP751, and APP695, with APP695 being expressed exclusively in neurons (2, 3). APP is primarily cleaved by either non-amyloidogenic α-secretase or amyloidogenic β-secretase at the juxtamembrane region, with a secondary cleavage by γ-secretase in the transmembrane domain. APP cleavage by the combination of β- and γ-secretases generates Aβ, whereas that of the combination of α- and γ-secretases generates an amyloidolytic (non-amyloidogenic) product (4). Thus, suppression of APP amyloidogenic cleavages and its associated Aβ generation or facilitation of the non-amyloidogenic cleavage of APP are thought to potentially be useful therapies for AD (5).
Cleavage of APP by β-secretase generates a large N-terminal sAPPβ fragment and a short, membrane-associated C-terminal fragment, CTFβ, whereas the cleavage by α-secretase occurs within the Aβ domain and generates sAPPα and CTFα. Both amyloidogenic CTFβ and non-amyloidogenic CTFα are subsequently cleaved by the γ-secretase complex composed of nicastrin (NCT), presenilin enhancer 2 (pen-2), anterior pharynx defective 1 (APH-1), and presenilin 1 or 2 (PS1 or PS2) (6). Following endoproteolysis to generate its N- and C-terminal fragments, presenilin forms the catalytic site of the γ-secretase complex. Both PS1 and PS2 are linked, along with APP, to causative genes for familial AD (FAD) (7). Over 150 FAD mutations in PS1 and PS2 that increase the generation of pathogenic types of Aβ species are known (8). Sporadic AD also shows a similar pathogenic process but is associated with later onset and has no mutation(s) in the causative APP and PS genes. This points to other sources in the sporadic AD brain, which alter the APP processing. It is known that some of the APP-interacting proteins regulate the APP metabolism, including Aβ generation. Dysfunction of and/or aberrant regulation by these APP-interacting proteins may alter the APP metabolism without mutations in causative genes (9, 10) and suggest(s) a possible cause for sporadic AD (11, 12).
Post-translational modifications of APP are also involved in the regulation of APP function and metabolism. APP is subject to N-glycosylation (immature APP or imAPP) in the endoplasmic reticulum and the imAPP is further modified with O-glycosylation in the Golgi complex. The mature APP (mAPP), with both N- and O-glycosylation, localizes to the late protein secretory pathway in which mAPP is cleaved by secretases (4, 11). The mAPP is further modified by phosphorylation, also occurring in the late secretory pathway. Phosphorylation of mAPP at Thr668 (numbering for APP695 isoform) in the cytoplasmic region is predominantly observed in neurons in the brain (13). Both neurite outgrowth of differentiating PC12 cells and interaction of APP with a neural adaptor FE65 protein are affected by the phosphorylation of APP at Thr668 (14–16). These observations suggest that phosphorylation of APP at Thr668 plays an important role in the expression and/or regulation of APP function (11).
In contrast to this, the role of the phosphorylation in APP metabolism is controversial. Mutant mice possessing Ala substitution for Thr668 of APP did not show significant differences in APP metabolism, including Aβ generation, when compared with the wild-type mouse brain (17). This analysis would suggest that the phosphorylation state of APP at Thr668 does not play an obvious role in the direct governing of the APP metabolism in the brain in vivo. However, contrary to this observation, several reports have indicated that phosphorylation of Thr668, or amino acid substitutions mimicking phosphorylated Thr, could regulate the cleavage by γ-secretase in cultured cells (18–23). Therefore, the ability of phosphorylation at Thr668 to induce alterations in the cleavage of APP at β- and/or γ-secretase sites is still controversial in AD brains.
The phosphorylation of APP at Thr668 can likely induce a change in the overall structure of its cytoplasmic domain because Thr668 is located in the 667VTPEER672 motif, which forms a type I β-turn and amino-terminal helix-capping box structure to stabilize its carboxyl-terminal helix structure (24, 25). The usual procedure to explore the function of phosphorylation of a protein is to mimic the phosphorylation state by amino acid substitutions of Asp or Glu for the appropriate Thr and Ser residues (26). However, this strategy might not be suitable in the case of APP phosphorylation as the substitution of Asp for Thr668 did not alter the carboxyl-terminal helix state as remarkably as did phosphorylation of Thr668 (supplemental Fig. S1). In contrast, substitution with Ala668 for Thr668 of APP is found to effectively mimic the nonphosphorylated state in the helix structure of the APP cytoplasmic domain (supplemental Fig. S1) and in localization and metabolism of APP in the brain (17). Therefore, to reveal the role of APP phosphorylation at Thr668, we carefully examined the phosphorylation state of both APP and APP metabolic fragments in the brain, in vivo. Furthermore, because the phosphorylation state of the CTFs is higher than that of mAPP, we focused on the features of the phosphorylated forms of the CTFs as regards to their susceptibility to γ-secretase cleavage and lipid binding, in vitro. Finally, we analyzed the age-dependent changes of the phosphorylation state of CTFβ in cynomolgus monkey brain in which amyloid plaque formation has progressed in an age-dependent manner (27).
EXPERIMENTAL PROCEDURES
Plasmid Construction
The cDNA encoding the cytoplasmic domain of APP (APPcyt) was inserted into pGEX4T-1 (GE Healthcare) at the EcoRI/BamHI sites to produce a GST fusion protein (28, 29). The cDNA encoding APPcyt ΔC8 (APP(649–687), a cytoplasmic domain of APP695 lacking C-terminal 8 amino acids) was produced by PCR and cloned into pGEX4T-1. pcDNA3-FLAG-APP695 was described (14). To prepare pcDNA3-FLAG-APPΔC8, the cDNA in pcDNA3-FLAG-APP695 was amplified by PCR using specific primers. The PCR products were digested with HindIII/XbaI and cloned into the pcDNA3 (Invitrogen).
Animals
All of the animal studies were conducted in compliance with the guidelines of the Animal Studies Committee of Hokkaido University, Shiga University of Medical Science, and the National Institute of Biomedical Innovation. The C57BL/6 line of mice housed in an SPF environment were used throughout these studies. Brain samples of cynomolgus monkey (Macaca fascicularis) were obtained from the Shiga University of Medical Science and the National Institute of Biomedical Innovation. Monkeys were housed in individual cages and maintained according to guidelines for experimental animal welfare.
Preparation and Detection of Membrane Proteins by Immunoblotting
Membrane fractions were prepared from brains of wild-type mice (C57BL/6, 4 months old) as described (16). To dephosphorylate proteins, membrane lysates (30 μg of protein) were treated with λ-protein phosphatase (400 unit; Sigma) for 4 h at 30 °C. The immunoblotting procedure was described previously (17). In brief, for detection of APP CTFs, membrane lysate proteins were separated by electrophoresis on a 17.5% (w/v) polyacrylamide Tris-Tricine gel, transferred onto a nitrocellulose membrane, and incubated with anti-APP C-terminal A8717 (Sigma) or G369 (30) (gift from Dr. S. Gandy), anti-Thr(P)668 of APP (anti-pAPP, Cell Signaling), or anti-actin (Chemicon) antibodies. Immunoreactants were further reacted with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG antibody (GE Healthcare) and detected with ECL Plus (GE Healthcare). Proteins were separated by a standard SDS electrophoresis with 8 (w/v) or 15% (w/v) polyacrylamide Tris glycine gel and detected by immunoblotting with the indicated antibodies; PS1 N-terminal Ab14 (31) (gift from Dr. S. Gandy), PS1 C-terminal (Chemicon), pen-2 (Zymed Laboratories Inc.), and GST (Upstate Biotechnology). The protein band intensities detected by ECL were quantified with an imaging analyzer, VersaDoc (Bio-Rad).
Cynomolgus monkey brain lysates were prepared from frozen stocks of temporal cortices. The tissue blocks were homogenized in a 10-fold volume of a lysis buffer (50 mm Tris-HCl, pH 8.0, containing 0.5% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40, and 150 mm NaCl) containing a protease inhibitor mixture (Sigma) and 1 μm microcystin-LR (Wako Pure Chemical) and then lysed by sonication in ice. The resulting supernatant was used for immunoblot analysis with mouse monoclonal anti-Tau HT7 (Thermo Scientific) and anti-phosphorylated Tau AT8 (Ser(P)202 and Thr(P)205, Innogenetics) antibodies, along with immunoblotting with anti-APP C-terminal A8717 (Sigma), anti-phosphorylated APP (Thr(P)668, Cell Signaling), and anti-actin (Chemicon) antibodies.
Estimation of APP CTFs Level
APP CTFs detected by immunoblotting with anti-APP C-terminal and anti-pAPP antibodies were quantified with VersaDoc (Bio-Rad). The values of phosphorylated C99 (pC99) CTFβ were set to 1.0 and the level of the other CTF species are defined as a ratio to that of pC99. In practice, we estimated the ratio of CTFs using the following calculations: pC99:nC99:pC89:(pC83 + nC89):nC83 = 1:a:b:c:d, these ratios being determined by immunoblotting with anti-C-terminal antibody, and pC99:pC89:pC83 = 1:b:e, these ratios being determined by immunoblotting with anti-pAPP antibody. Therefore, the relative ratios of all six CTFs were expressed as: pC99:nC99:pC89:nC89:pC83:nC83 = 1:a:b:(c-e):e:d. Data were obtained from three independent experiments. The levels of phosphorylated (pCTFs) and nonphosphorylated CTFs (nCTFs) were combined and indicated as total levels of CTFs.
In Vitro γ-Secretase Assay
Wild-type C57BL/6 mouse (4 months old) brains were suspended in 5 volumes of buffer H (20 mm Hepes-NaOH, pH 7.4, 150 mm NaCl, 5 mm EDTA, 10% (v/v) glycerol) including a protease inhibitor mixture (Sigma) and 1 μm microcystin LR. The suspension was then homogenized with a Dounce homogenizer on ice. After centrifugation at 1,000 × g for 10 min, the postnuclear supernatant was further centrifuged at 100,000 × g for 60 min. The precipitated membrane fraction (P100) was suspended in the same buffer, including a protease-inhibitor mixture and 1 μm microcystin LR, and used as the mouse brain membrane preparation. Aliquots (25 μg of protein) of the mouse brain membrane preparation were incubated at 37 °C for the indicated times, as described (32), in buffer H containing 5 mm EGTA, 5 mm phenanthroline, protease inhibitor mixture (Sigma), and 2× PhosSTOP (Roche Diagnostics) to prevent protein degradation and dephosphorylation. Samples were then separated by electrophoresis in Tris-Tricine gels (17.5% (w/v) polyacrylamide). The separated proteins were transferred onto a nitrocellulose membrane, boiled in PBS for 5 min, and probed with the indicated antibodies. Reactive proteins were detected using an ECL plus detection system (GE Healthcare).
Isolation of CHAPSO-resistant Membrane Fraction
Detergent-resistant membrane (DRM) fractions were prepared according to the established DRM isolation procedure (9). Briefly, the brain membrane preparation from each mouse (3 months old) was homogenized in TNE buffer (10 mm Tris-HCl, pH 7.5, 0.15 m NaCl, 5 mm EDTA) containing 1% (v/v) CHAPSO, a protease inhibitor mixture, and 1 μm microcystin-LR. The sucrose concentration of the extract was adjusted to 42.5% (w/v) by the addition of 85% sucrose in TNE buffer; the extract was then placed at the bottom of an ultracentrifuge tube and overlaid with 5 (4 ml) and 35% (4 ml) discontinuous sucrose gradients in TNE buffer. The gradients were centrifuged at 39,000 × g for 20 h with an SW41 rotor (Beckman Coulter). Fractions (1 ml) were collected from the top (fraction 1) of the ultracentrifuge tubes and proteins in the respective fractions were analyzed by immunoblotting.
Neuronal Culture and Treatment of Neurons with Methyl-β-cyclodextrin (MβCD)
The primary culture of cortical neurons was as described (9). Briefly, the cortex of mice at embryonic day 15.5 was dissected, and neurons were spread in a buffer containing papain and cultured at 1.5 × 105 cells in Neurobasal medium containing B27, Glutamax, and antibiotics (Invitrogen) on a poly-d-lysine-treated dish. Neurons at in vitro day 7 were incubated with Neurobasal medium containing MβCD for 2 h. After incubation, neuronal membrane fractions (P100) were isolated by ultracentrifugation. The membrane preparation was used for the in vitro γ-secretase assay and samples were analyzed by immunoblotting as described above. The labeling of membrane cholesterol in neurons (in vitro day 7) was investigated by microscopy analysis using the cholesterol cell-based detection assay kit including Filipin III (Cayman Chemical).
Liposome Binding Assay of Peptide
Lipids were extracted with chloroform/ethanol mixtures from brain membrane preparations of C57BL/6 mouse and evaporated using a SpeedVac vacuum concentrator to prepare a lipid film. The lipid film was dissolved in phosphate-buffered saline (PBS) and agitated to prepare liposomes (33). The brain-lipid liposomes were incubated with the indicated peptide in PBS for 1 h at room temperature, and peptide bound to liposomes was recovered by ultracentrifugation at 100,000 × g for 10 min as described (34).
Preparation of Recombinant Proteins in Vitro and Synthetic Peptides
Production and purification of recombinant GST fusion proteins were as described (28, 29). Briefly, GST fusion proteins were generated in Escherichia coli BL21 transformed with pGEX-4T-1 cDNA constructs and affinity purified with glutathione-Sepharose 4B (GE Healthcare). The cytoplasmic peptides of APP(648–695), with or without phosphate at the Thr668 residue, Aβ(1–40) and Aβ(1–42) peptides (for sELISA standard) were synthesized using solid phase N-tert-butyloxycarbonyl chemistry. The peptides were purified by reverse-phase high pressure liquid chromatography to greater than 90% purity, and their expected molecular weights were confirmed by mass spectroscopy.
Quantification of Aβ40 and Aβ42
N2a cells (total ∼1.0 × 106) were transiently transfected with 1.0 μg of pcDNA3-FLAG-APP695 or pcDNA3-FLAG-APP695ΔC8 and then cultured in DMEM containing 10% (v/v) fetal bovine serum for 24 h. Aβ40 and Aβ42 secreted into the medium were quantified by a sandwich ELISA (35).
RESULTS
Phosphorylation State of APP Carboxyl-terminal Fragments at Thr668 in Mouse Brain
For investigating the cleavage of CTFs (nCTFs plus pCTFs) in mouse brain, we first estimated the levels of the respective CTF species in the wild-type mouse brain membrane fractions. As shown in Fig. 1A, six CTF species composed of CTFβ (pC99 and nC99), CTFβ′ (pC89 and nC89), and CTFα (pC83 and nC83) were detected as five protein bands by immunoblotting with anti-APP C-terminal antibody (upper panel) as nC89 and pC83 are indistinguishable on the blotted membrane (see review in Refs. 11 and for CTFβ′, see Ref. 36). Identification of the CTF species has already been described (11). However, to better understand the following studies, we show the CTF species detected by the immunoblot as the first figure in Fig. 1A. All three phosphorylated proteins are discernible with anti-APP Thr668 phosphorylation state-specific (pAPP) antibody (lower panel). The slowest migrating protein band represents pC99, which was detected with both anti-APP C-terminal and anti-pAPP antibodies. This allowed us to estimate the ratio of the other CTF proteins relative to the levels of pC99 as described under “Experimental Procedures.” In Fig. 1B, we quantified each CTF species as a relative ratio to pC99. Phosphorylated CTFβ species, pC99 and pC89, were significantly higher in amounts as compared with their nonphosphorylated forms, nC99 and nC89; whereas phosphorylated CTFα, pC83, trended to a lower amount when compared with the level of nonphosphorylated CTFα, nC83. The total level of phosphorylated CTFs is equivalent to that of the nonphosphorylated CTFs in their ratios (Fig. 1C), even though phosphorylated CTFβ and CTFβ′ are predominant compared with their nonphosphorylated forms. These observations indicate that pCTFs and nCTFs are equally present in the brain as potential substrates for γ-secretase.
FIGURE 1.

Level of CTF species in brain membrane fraction. A, detection of CTF species in brain membrane preparation. Membrane fractions (30 μg of protein) prepared from 3 wild-type mouse brains were treated with (+) or without (−) λ-protein phosphatase (λPPase) and analyzed by immunoblotting with anti-APP C-terminal (upper panel) and anti-Thr668 phosphorylation state-specific (lower panel) antibodies. pC99 and nC99, phosphorylated and nonphosphorylated CTFβ; pC89 and nC89, phosphorylated and nonphosphorylated CTFβ′; pC83 and nC83, phosphorylated and nonphosphorylated CTFα. Identification of APP CTFs in mouse brain was previously published (Fig. 1A of Ref. 9). B and C, levels of CTF species in brain membrane preparations. Respective CTFs shown in panel A were quantified with the VersaDoc imaging system. The amount of pC99 was set to 1.0 and other species were calculated (see text) and shown in panel B as the relative ratio to pC99. Total amounts of pCTFs and nCTFs are indicated in panel C. Results are presented with mean ± S.D. Asterisk represents the statistical significance (p < 0.05) with Student's t test (n = 3).
Role of APP Phosphorylation in CTFs Cleavage by γ-Secretase
Because almost identical amounts of nCTFs and pCTFs were found in mouse brain, APP intracellular cytoplasmic domain fragments, nonphosphorylated AICD (nAICD), and phosphorylated AICD (pAICD) are expected to be generated at the same level if γ-secretase cleaves nCTFs and pCTFs on an equal basis. To examine this, whether the phosphorylation of CTFs at Thr668 influences the γ-cleavage of CTFs, we performed an in vitro γ-secretase assay with membrane preparations from mouse brain. The membrane preparations were incubated for 0 to 4 h and the generation of AICD, which is the product of the γ-cleavage of CTFs, was followed by immunoblotting (Fig. 2A). Membrane fractions of mouse brain, which includes almost equal amounts of pCTFs and nCTFs (Fig. 1), generated large amounts of nAICD and small amounts of pAICD. We confirmed that the production of AICD ((3,5-difluorophenylacetyl)-l-alanyl-l-2-phenylglycine t-butyl ester) was due to γ-secretase by using the γ-secretase inhibitor DAPT (37), which inhibited the production of both nAICD and pAICD (data not shown). The molecular identification of nAICD and pAICD has already been published (16) and confirmed here (supplemental Fig. S2A). Simple interpretation of these results is that nCTFs may be more susceptible to γ-cleavage, or that pCTFs may be more resistant to γ-cleavage. Therefore, we next compared the production rate of nAICD from nCTFs with that of pAICD from pCTFs using the in vitro γ-secretase assay. Incubation of membrane preparations showed a time-dependent, nearly linear increase in the generation of nAICD and pAICD during the 0–2-h time period and the reaction essentially reached a plateau in the 2–4-h period (Fig. 2, B and C). We confirmed that dephosphorylation or degradation of pAICD did not occur in this assay by assaying the synthetic APP cytoplasmic domain peptide, APP(648–695) with phosphate at the Thr668 residue (pC47), for pAICD production (supplemental Fig. S2B). Importantly, the ratio of pAICD to AICD generation was constant throughout the incubation time (1–4 h) with the relative ratio (amount of pAICD/amount of nAICD) measuring 0.35 ± 0.10 (n = 3) at the 2-h point (Fig. 2D). Taken together, the results indicate that phosphorylated and nonphosphorylated CTFs are kinetically equivalent as a substrate for γ-secretase, but the results also show that the generation of pAICD was significantly lower when compared with that of nAICD. These observations suggest that, in mouse brain, pCTFs are located at a distance from active γ-secretase in the membrane, whereas nCTFs are positioned nearer the active enzyme. This is significantly different from an interpretation that phosphorylation of CTFs directly interferes in the cleavage by γ-secretase (20, 23, 38).
FIGURE 2.

In vitro kinetic analysis of phosphorylated and nonphosphorylated CTF cleavage by γ-secretase. A, in vitro γ-secretase assay with membrane preparations from wild-type mouse brain. Membrane preparations of the wild-type mouse brains were incubated at 37 °C for the indicated period of time (h). The samples were subjected to immunoblotting with anti-APP C-terminal to detect pAICD and nAICD, along with CTFs. Actin was also detected with anti-actin antibody. B and C, kinetic analysis of AICD generated by incubation of membrane preparations. The nAICD and pAICD in panel A were quantified with VersaDoc imaging analyzer and the levels of nAICD (panel B) and pAICD (panel C) at the respective incubation times were indicated as a relative ratio to the level at 4 h (1.0). D, the production ratio of pAICD to nAICD (pAICD/nAICD) at the indicated times are shown. Results are presented with mean ± S.D. (n = 3).
Nonphosphorylated CTFs Are Predominant in Lipid Raft-like Membrane Microdomains
To clarify our hypothesis, we isolated γ-secretase-enriched lipid raft-like membrane microdomains, which were prepared as DRMs using CHAPSO. Application of CHAPSO is suitable for the isolation of DRMs, including active γ-secretase complexes, rather than procedures using other detergents (39, 40). As shown in supplemental Fig. S3A, the buoyant DRM fraction (fraction 5) was identified by the recovery of flotillin-1 (DRM marker), whereas the non-DRM heavy fractions (fractions 9–10) contained calnexin (non-DRM marker) and the majority of the APP CTFs (∼80% measured). Components of the active γ-secretase complex, both PS1 N- and C-terminal fragments and pen2, were predominantly recovered in fraction 5 along with a small amount of the APP CTFs (∼20% measured). Phosphorylation levels of APP CTFs in the membrane preparation and DRM fraction did not change significantly for 24 h, which is a significant amount of time beyond that needed for membrane preparation and DRM fractionation (supplemental Fig. S3B). Thus, the phosphorylation levels of APP CTFs are stable during membrane preparation and DRM fractionation.
We measured the phosphorylation level of APP CTFs in the DRM and non-DRM fractions. CTFs were detected by immunoblotting with anti-APP and anti-pAPP antibodies (Fig. 3A), and the respective CTFs and pCTFs were quantified as a relative ratio in which pC99 in the DRM was set to 1.0 (Fig. 3B). The phosphorylated species, pC99, pC89, and pC83, showed significantly higher levels in the non-DRM fractions compared with those in the DRM fractions. Additionally, the phosphorylation level of total APP CTFs in the DRMs was significantly lower than that in the non-DRMs (Fig. 3C). The phosphorylated CTFs seem to be preferentially localized outside of the DRM/lipid raft-like membrane microdomain and, thus, prevented from active cleavage by γ-secretase.
FIGURE 3.
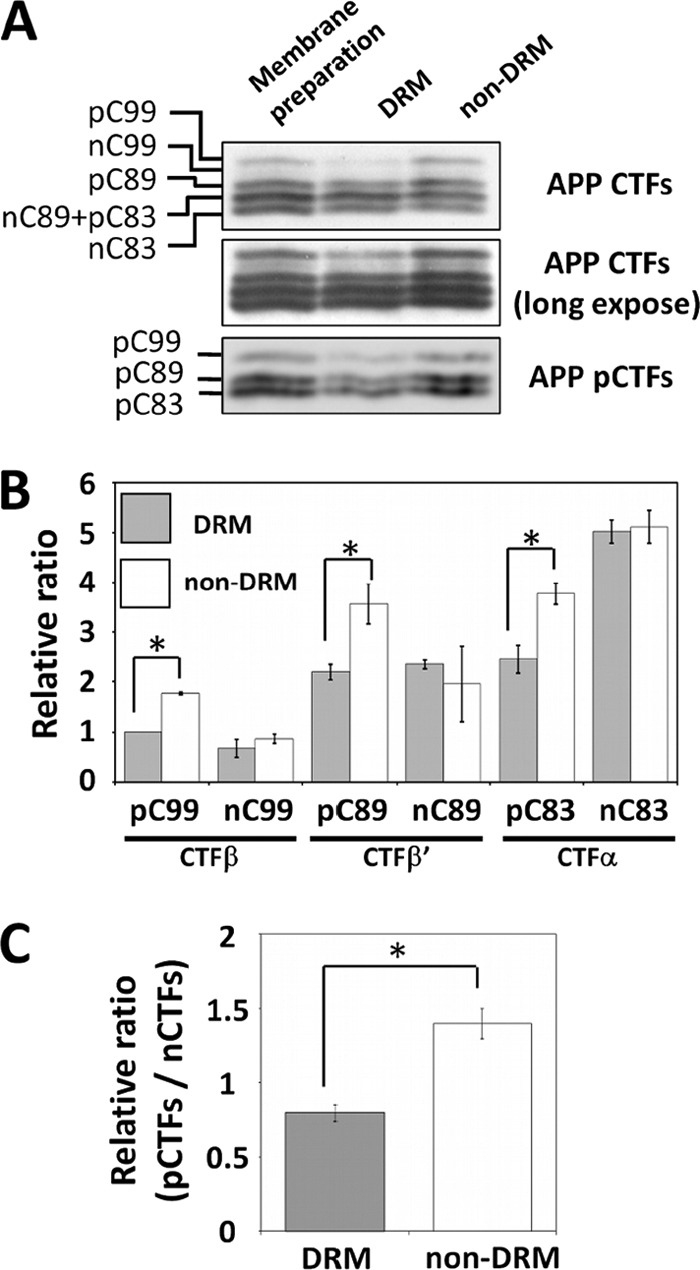
Quantification of pCTFs and nCTFs in DRM and non-DRM fractions. A, identification of APP CTFs in DRM and non-DRM fractions. Proteins in CHAPSO DRM and non-DRM fractions were analyzed by immunoblotting with anti-APP C-terminal (upper and middle) and anti-Thr668 phosphorylation state-specific (lower) antibodies. The membrane preparation is a sample prior to fractionation. A representative result is indicated. B, CTFs levels in DRM and non-DRM fractions. Respective CTFs shown in panel A were quantified by setting the amount of pC99 in the DRM to 1.0 and measuring the amounts of other CTF species as a ratio to pC99. Results are presented with mean ± S.D. The asterisk represents the statistical significance (p < 0.05) with Student's t test (n = 3). C, ratio of phosphorylated to nonphosphorylated CTFs in DRM and non-DRM fractions. Ratios of pCTFs/nCTFs in DRM and non-DRM fractions were compared. The ratio of membrane preparations prior to fractionation was set to 1.0. The error bars indicate ± S.D. (n = 3). Statistical analysis was performed using Tukey's test. *, p < 0.001.
The level of CTFs in the non-DRM fraction of the membrane preparation from mice possessing an Ala substitution for Thr668 of APP, which has been found to mimic the wild-type levels of nCTFs (17), was compared with the level of CTFs in the non-DRM fraction of wild-type mice. Because CTFs of wild-type mice include pCTFs, we treated the sample with λ-protein phosphatase and analyzed by immunoblotting (supplemental Fig. S4). The CTFs recovered in the DRM fraction increased and those in the non-DRM fraction decreased in mutant mice when compared with the levels in wild-type mice, supporting our results that nCTFs (and CTFs with Ala668 as a mimic to nCTFs) tend to stay in the DRM rather than the non-DRM fraction. However, as reported previously (17), the brain Aβ levels were not significantly altered in either mutant or wild-type mouse brain. These observations are consistent with the in vitro γ-secretase assay results that point toward a localization of the pCTFs away from the γ-secretase location.
Depletion of Cholesterol from Neurons Accelerates γ-Cleavage of Phosphorylated CTFs
As described above, the localization of pCTFs is largely in non-DRM fractions (using CHAPSO), thus the phosphorylation of APP CTFs at Thr668 may regulate membrane microlocalization of the CTFs. To test this idea, we sought to disrupt the lipid raft-like membrane microdomain structure and then remeasure the γ-cleavage of the pCTFs. It is known that MβCD can deplete cholesterol from membranes and disrupt the lipid raft-like membrane microdomain structure (41). When mouse primary cultured cortical neurons were treated with MβCD (1 mm), cholesterol staining of the neurons with filipin almost disappeared, indicating remarkable depletion of cholesterol from the neural membrane (Fig. 4A). We estimated that almost 75% of the cellular cholesterol was removed from the neurons by treatment with 1 mm MβCD (data not shown). A membrane fraction was prepared from cortical neurons with and without MβCD treatment and an in vitro assay for production of AICD from CTFs through cleavage by γ-secretase was performed (Fig. 4B). Significantly, the levels of pAICD increased in a dose-dependent manner with the concentration of MβCD, whereas those of nAICD were largely unaffected (Fig. 4B). The results were quantified and the production of AICDs was standardized using actin. The results indicate that the production of pAICD was significantly increased in the disrupted membranes prepared from neurons treated with MβCD (Fig. 4C). The ratio of pAICD to nAICD generation also increased as a function of the MβCD concentration (Fig. 4C). Because in the control, the content of the pCTFs in primary cultured neurons was not significantly altered by incubation with MβCD (Fig. 4D), the results using membranes treated with MβCD indicate that more pCTFs were cleaved by γ-secretase to generate pAICDs. One possibility is that the depletion of cholesterol disrupted the lipid raft-like DRM structure such that both the nCTFs and pCTFs were more equally diffused throughout the membrane. Alternatively, both CTF types may be equivalently available for reaction with the active γ-secretase complex, which, itself, has diffused throughout the membrane. In fact, PS1 NTF, a component of the active γ-secretase complex, decreased in the buoyant DRM fraction and showed dispersion into the non-DRM fraction when cells were treated with MβCD (supplemental Fig. S5, A and B). Similarly, APP CTFs localization in the DRM fraction decreased and dispersed into the non-DRM fraction by the treatment with MβCD (supplemental Fig. S5, A and C). We next examined a mechanism whereby nCTFs tend to localize preferentially to the DRM domain and pCTFs tend to localize away from the DRM.
FIGURE 4.

Influence of methyl-β-cyclodextrin on the γ-cleavage of CTFs. A, detection of cholesterol in neurons. Primary cultured mouse cortex neurons (in vitro day 7) were treated with (MβCD) or without (control) methyl-β-cyclodextrin (1 mm) at 37 °C for 1 h. After incubation, neurons were fixed and stained with filipin. DIC indicates differential interference contrast microscopy. Scale bar, 50 μm. B, generation of AICD from membranes of neurons treated with or without MβCD. Membrane fractions were prepared from primary cultured neurons (in vitro day 7) treated with MβCD (0, 1, and 10 mm) and were incubated at 37 °C for the indicated time (0 or 2 h). The membrane lysates were subjected to immunoblotting with anti-APP C-terminal antibody to detect AICDs (upper) along with anti-actin antibody (lower). C, relative ratios of AICD production from membrane preparations of neurons treated with MβCD. The nAICD (left) and pAICD (middle) generated from membranes of neurons treated with MβCD (0, 1, and 10 mm) for 2 h were standardized with actin, and the relative ratios against control neurons (0 mm, set to 1.0) are presented with mean ± S.D. (n = 3). The production ratios of pAICD to nAICD (pAICD/nAICD) at the indicated MβCD concentrations are shown in the right panel. The error bars indicate ± S.D. (n = 3). Statistical significance is indicated with asterisks (*, p < 0.05) with the Tukey's test. D, changes in the phosphorylation levels of APP CTFs by treatment of neurons with MβCD. Primary cultured neurons were incubated in the presence of MβCD at the indicated concentrations for 1 h, and APP CTFs, APP pCTFs, and actin were detected by immunoblotting.
Phosphorylation of APP at Thr668 Suppresses Lipid Association of Cytoplasmic Terminus of APP
Recent structural studies of the cytoplasmic domain of APP revealed that APP and APP CTFs can interact with the cytoplasmic-membrane region through binding of the APP C-terminal tail (composed of 7 to 9 amino acids) with membrane lipids (42). Because phosphorylation of APP at Thr668 induces a significant change in its cytoplasmic domain conformation (15, 24, 25) (supplemental Fig. S1), we examined whether phosphorylation of the APP cytoplasmic domain at Thr668 influences the association of the APP cytoplasmic tail with membrane lipids.
We extracted endogenous lipids from membrane fractions of mouse brain to prepare liposomes to serve as a model for neural membranes (34, 43–45). Synthetic cytoplasmic peptides of APP sequence 648–695, one with phosphate at Thr668 (pC47) and one without phosphate at Thr668 (nC47) were incubated with the liposomes prepared from mouse brain lipids and the liposome-bound peptides were recovered and analyzed by immunoblotting. Even though the protein components of the brain, such as protein kinases and protein phosphatases, are thought to be excluded by lipid extraction, we still confirmed that pC47 was not dephosphorylated and nC47 was not phosphorylated during this assay (supplemental Fig. S6). Significantly, nonphosphorylated APP cytoplasmic peptide (nC47) strongly bound to the liposomes, whereas phosphorylated peptide (pC47) showed no detectable association (Fig. 5A). This trend is also confirmed by examining the AICDs, which lack the transmembrane domain due to ϵ-cleavage by γ-secretase (32, 46). Most of the nAICD was found in the brain-membrane fraction (∼75%) rather than in the soluble cytoplasmic fraction (∼25%), whereas comparatively more pAICD was found in the cytoplasmic fraction (∼45%) (Fig. 5B). Therefore, the nonphosphorylated forms of APP, APP CTFs, and AICD tend to bind with membrane lipids, mediated by their C termini, and phosphorylation of APP, APP CTFs, and AICD at Thr668 functions to prevent direct membrane association by apparently changing the conformation of their cytoplasmic regions. One possible concern in this study was that the increased negative charge of the C47 peptide by phosphorylation might influence its interaction with the liposome regardless of any conformational change induced by the phosphorylation of Thr668. However, we consider that this concern is addressed by results showing that the GST-C47 peptide with Glu668 could bind liposome just as well as nC47 (supplemental Fig. S7). The charge of GST-C47 with Glu668 (−1.50 at pH 7) is almost the same as that of pC47 (−1.59 at pH 7), whereas the charges of the peptides that could bind to liposome were calculated as +0.41 (pH 7) for the nC47 synthetic peptide and −0.50 (pH 7) for GST-nC47 with Thr668 (dnastar). The analysis indicates that the overall net charge of the APP cytoplasmic region does not influence the association with liposome.
FIGURE 5.

Liposome-binding ability of the APP cytoplasmic domain peptide with phosphorylated Thr668 residue compared with the nonphosphorylated peptide. A, the binding ability of the phosphorylated APP cytoplasmic domain peptide with liposomes composed of lipids from mouse brain membranes. Synthetic APP cytoplasmic peptide APP(648–695) with (pC47) or without (C47) phosphate at the Thr668 residue was incubated with liposomes composed of lipids extracted from wild-type mouse brain membrane preparations. Liposome-bound peptides were recovered by centrifugation and analyzed by immunoblotting with anti-APP C-terminal antibody, along with the applied peptide (Input). B, distribution of AICD endogenously generated in mouse brain. Mouse brain homogenate was prepared in a buffer including 1 μm microcystin LR and fractionated into crude membrane (M) and cytoplasmic (C) fractions. Proteins (200 μg) of the respective fractions were immunoprecipitated with anti-APP C-terminal antibody and analyzed by immunoblotting with the same antibody to examine the levels of membrane-bound and cytosolic pAICD and nAICD.
We confirmed that the direct interaction between the APP cytoplasmic region and liposomes is mediated by the APP cytoplasmic tail (41). The GST-APP cytoplasmic domain peptide (GST-APPcyt/C47) and the peptide lacking the C-terminal 8 amino acids (GST-ΔC8) were prepared and measured for their binding to liposomes (Fig. 6, A–C). As expected, GST-ΔC8 and GST alone showed only a background level of binding to the liposomes, whereas GST-APPcyt/C47 showed significant binding, as did the synthetic nC47 peptide (shown in Fig. 5). The experiment confirmed that the APP cytoplasmic tail can bind to liposomes composed of lipids derived from mouse brain membranes, and supports the idea that phosphorylation of the APP cytoplasmic domain at Thr668 alters its overall cytoplasmic conformation which, in turn, weakens the interaction of the cytoplasmic tail with membrane lipids. In other words, GST-ΔC8 can mimic pAICD, at least for the binding to membrane lipids. Again, the charge of GST-ΔC8 (−0.50 at pH 7) was identical to that of GST-APPcyt/C47 (−0.50 at pH 7) (dnastar).
FIGURE 6.

Liposome association of the APP cytoplasmic peptide through its C-terminal region and Aβ generation from the APP lacking the lipid-interaction region. A, schematic representation of the GST-APPcyt fusion protein. GST-APPcyt, GST-APP(649–695) fusion protein; GST-ΔC8, GST-APP(649–687) fusion protein that lacks the C-terminal 8 amino acids; GST, GST protein alone. Numbering for APP695 isoform. B and C, binding of GST fusion proteins to brain-lipid liposome. GST fusion proteins were incubated with liposomes, and the liposome-bound GST fusion proteins were recovered as described previously and detected by immunoblotting with anti-GST antibody (B). GST-ΔC8 shows a slower mobility than GST-APPcyt on the gel of SDS-PAGE. Quantified liposome-bound proteins were normalized with the amount of GST-APPcyt (set to 1.0) and shown as a relative ratio (C). Results are presented with mean ± S.D. Asterisk represents statistical significance (p < 0.05) with the Tukey's test (n = 4). D, Aβ generation from APP with or without the C-terminal lipid-binding region. Aβ40 and Aβ42 secreted from N2a cells (3.5 × 105) transiently expressing FLAG-APP695 or FLAG-APPΔC8 were quantified with sandwich ELISA, normalized to the amount of mAPP, and shown as relative ratios (Aβ amount generated from FLAG-APP was set to 1.0). Results are presented with mean ± S.D. Asterisk represents the statistical significance (p < 0.05) with Student's t test (n = 3).
APP-ΔC8 is expected to have an increase in its membrane fluidity as it lacks the interaction between the C-terminal 8 residues and the membrane lipids (42). Therefore, APP-ΔC8 may disperse throughout the membrane instead of staying in a restricted area such as the DRM, which contains the active γ-secretase. In this case, we expected that APP-ΔC8 can, in part, mimic APP phosphorylated at Thr668. If this consideration is reasonable, APP-ΔC8 should generate a lesser amount of Aβ than intact APP, which can associate with membrane lipid. We examined this assumption: Neuro 2a cells were transiently expressed with FLAG-APP and FLAG-APP-ΔC8 and the Aβ generation into the medium was assayed (Fig. 6D). In cultured cell lines, the phosphorylated level of mAPP is extremely low, even though imAPP is subject to phosphorylation transiently in the mitotic phase of the cell cycle (30, 47). Therefore, we can largely exclude the effect of APP phosphorylation in the cultured cells. Production of both Aβ40 and Aβ42 from APP-ΔC8 was significantly decreased when compared with that from intact APP. Although we cannot rule out the possibility of other functions of the APP cytoplasmic tail, it is suggested that, in neurons, the presence of phosphorylation at Thr688 may decrease neurotoxic Aβ generation in much the same way as APP-ΔC8; that is, a decrease in lipid association results in an increase in membrane fluidity and, therefore, an increase in dispersal throughout the membrane.
We explored the change of a phosphorylation level of the amyloidogenic CTFβ/C99 in an age-dependent manner. Because the phosphorylation state of proteins in the brain is thought to change significantly relative to the time of postmortem analysis, we used freshly frozen monkey brain tissue. Freshly frozen tissue blocks of temporal cortex of 4- to 36-year-old cynomolgus monkeys (n = 47) were analyzed for nC99 and pC99 levels, along with the total Tau and pTau (Ser(P)202/Thr(P)205) levels, by immunoblotting the ratios of pC99/nC99 and pTau/total Tau were determined (Fig. 7). As can also be seen in mice, the ratio of pC99/nC99 is almost 1.0 in young monkeys (<10 years old), however, the ratio tended to decrease gradually in older monkeys. Decrease of the phosphorylation level of CTFβ/C99 correlated weakly but significantly with aging (R2 = 0.1573, p = 0.0125). In contrast to APP CTFβ, the phosphorylation level of pTau/total Tau (pTau/total Tau) tended to increase versus aging but not significantly (R2 = 0.0389, p = 0.2229). Because cynomolgus monkeys have been reported to show amyloid plaques in individuals over 20 years of age (27, 48), changes in the phosphorylation level of CTFβ and Tau were analyzed both in younger (<20 years old) and older (>20 years old) individuals, respectively (supplemental Fig. S8). In younger populations, no correlations between the pCTFβ/nCTFβ ratio and age or between the pTau/total Tau ratio and age were observed (panels A and C in supplemental Fig. S8). However, a strong and significant negative correlation between the pCTFβ/nCTFβ ratio and age (R2 = 0.4677, p < 0.0001) was observed and a weak positive correlation between the pTau/total Tau ratio and age (R2 = 0.1389, p = 0.0698) was found in older populations (panels B and D). These analyses suggest there is a meaningful decrease of the APP CTFβ phosphorylation level in aged monkey brain. The monkey study supports our hypothesis that the preservation of the phosphorylation level of APP CTFs correlates with the suppression of γ-cleavage.
FIGURE 7.

Age-dependent decrease of phosphorylated CTFβ/C99 levels in the brain of cynomolgus monkeys. A, immunoblot analysis of APP C99 species and Tau proteins in monkey brain lysate. Representative blots of pC99, C99, Tau, and pTau proteins with brain lysates (10 μg of protein) of young (4 years old), middle (20 years old), and aged (30 years old) monkeys are shown. Proteins were detected by immunoblot with antibodies: anti-APP C-terminal antibody for CTFβ, HT-7 for total Tau proteins, and AT-8 for pTau proteins. B, correlations of the ratio of pC99 to nC99 and the ratio of pTau to total Tau with age. The band densities of proteins were quantified with the VersaDoc imaging system, and the pC99/nC99 (upper panel) and pTau/total Tau (lower panel) ratios were plotted versus age. Analysis was performed with Spearman correlation (n = 47). R2 and the significance with p values (p < 0.05) are indicated. Detailed analysis divided into younger (<20 years old) and older (>20 years old) populations are shown in supplemental Fig. S8.
DISCUSSION
The roles of APP phosphorylation at Thr668 in APP metabolism are controversial. The presence of this phosphorylation has been reported by others to contribute to amyloidogenic metabolism (19, 20). Furthermore, substitution of Glu668 or Asp668 for Thr668 to mimic phosphorylation may contribute to the controversial conclusions in the previous studies using cultured cells (23). As is presented in supplemental Fig. S1, comparison of CD spectra among the APP cytoplasmic domain peptides (648–695) show that phosphorylated Thr668 (APPcyt-Thr(P)668) is distinctly different in the secondary structure from T668A, T668D, and nThr668. T668D showed an intermediate α-helicity when compared with Thr(P)668 and T668A or nThr668, the latter two having almost identical CD spectra. Thus, the purported mimic for threonine phosphate, T668D, does not show a similar secondary structure to that of the phosphate peptide, Thr(P)668, regardless that both T668(E/D) and Thr(P)668 shows almost identical negative charge levels.
Although we have generated mutant knock-in mice expressing endogenous APP with Ala668 and Asp668 instead of phosphorylatable Thr668 to analyze the role of APP phosphorylation at Thr668 in vivo (17), the CD results described above indicate that the APP-Asp668 knock-in mice would not be a good mimic for the phosphorylated state of APPcyt-Thr(P)668. In contrast, APP-Ala668 knock-in mice appear by both CD and previous work to mimic well the nonphosphorylated state of APP (17).
We previously reported that the APP-Ala668 knock-in mice did not alter the production of Aβ when compared with the wild-type mice at 24 months (17). Based on the present results, these mutant mice would have been expected to increase the amount of brain Aβ. Because APP processing is regulated by many steps, including intracellular protein sorting (4), it may prove difficult to sort out the effect of APP CTF phosphorylation on γ-cleavage in brain, in vivo. Instead, to reveal the function of APP phosphorylation at Thr668, we used wild-type mouse brain in vivo to examine, in detail, the phosphorylation state of the APP metabolic fragments and then analyzed the role of these fragments, as regards to phosphorylation, using in vitro measurements.
First, we found that almost equal amounts of pCTFs and nCTF existed in mouse brain, whereas lower amounts of pAICD compared with nAICD were found using a membrane fraction γ-secretase assay. Second, we showed that both pAICD and nAICD were kinetically equivalent substrates for the γ-secretase, again using the in vitro AICD generation assay. Both the in vivo and in vitro analyses suggested that pCTFs are sequestered in a separate membrane region away from the membrane region where γ-secretase is active (DRM/lipid raft-like membrane microdomain) (39, 40), and the present results further suggested that the pCTFs locate outside of the DRMs/lipid raft-like membrane microdomain due to a change in the conformation of their cytoplasmic tail to which the membrane lipids bind. In other words, the pCTF can quickly disperse from the DRMs/lipid raft-like membrane microdomain with their increased mobility in the membrane (supplemental Fig. S9). Our interpretation, taken together with previous biophysical/biochemical studies (15, 24, 25, 42), indicates that γ-secretase cleavage of CTFs was decreased by an indirect rather than a direct action due to phosphorylation (23). Although we cannot rule out the possibility that phosphorylation at several sites in CTFs may directly prevent cleavage by the γ-secretase complex, such multiple phosphorylations of the APP cytoplasmic domain have not been observed, at least in healthy mouse brain (11, 17).
Recent structural analysis of APP CTFs proposed that the C termini of the CTFs associate with the cytoplasmic membrane surface (42). We also confirmed this observation with a nonphosphorylated synthetic APP cytoplasmic peptide that could bind to liposomes composed of lipids from mouse brain membranes. Significantly, the same peptide with Thr(P)668 showed little binding to the liposomes even though the peptide possesses an intact C terminus that is, except for the phosphorylation, sequentially the same as the nonphosphorylated peptide. Previous studies on the effect of APP phosphorylation at Thr668 clearly demonstrated that the phosphorylation induces an overall conformational change in the cytoplasmic domain (15, 24, 25). Therefore, it is very reasonable that the conformational alteration of the APP cytoplasmic domain by Thr668 phosphorylation affects the binding ability of the C terminus to membrane lipids. Indeed, the pAICD, which was endogenously generated in the brain, showed a trend to localize in the cytoplasmic fraction, whereas nAICD was largely found in the membrane fraction. This observation also supports our conclusion that phosphorylation of the APP cytoplasmic domain at Thr668 prevents interaction with membrane lipids.
Taken together, the previous work (42) and our current observations suggest that the C-terminal region composed of 8 amino acids, APP(688–695), is at least a significant portion of the lipid-interaction domain of APP. We showed that APP lacking this region results in a decreased generation of both Aβ40 and Αβ42. In a similar manner, the increase of the fluidity of APP-pCTFs within the membrane resulting from phosphorylation at Thr668 contributes to localizing away from γ-secretase and results in a decrease in Aβ generation. As a conclusion, the phosphorylation of CTFs at Thr668 reduces rather than increases γ-cleavage of these CTFs, despite suggestions to the contrary (20, 23, 38). In fact, the phosphorylation levels of amyloidogenic CTFβ in monkey brains significantly decreased in an age-dependent manner. One of the possible causes of sporadic AD may be the decrease of the CTF phosphorylation state as a function of aging. Therefore, preservation of the phosphorylation state of APP and its metabolic products, APP CTFs, may be a useful treatment to lower Aβ generation.
Supplementary Material
Acknowledgments
We thank Drs. Koich Kato (Okazaki Institution for Integrate Bioscience, Division of Biomolecular Science) and Yoshiki Yamaguchi (Riken Advanced Science Institute, System Glycobiology Research Group) for structural analyses of the APP cytoplasmic domain. We sincerely thank Margaret M. Elliott for careful reading of the manuscript.
This work was supported in part by Grant-in-aid for Scientific Research 2339001 (to T. S.) and 23790069 (to Y. S.) from MEXT Japan.

This article contains supplemental Figs. S1–S9.
- AD
- Alzheimer disease
- Aβ
- amyloid β-protein
- AICD
- APP intracellular domain peptide
- APP
- amyloid β-precursor protein
- CTF
- C-terminal fragment of APP truncated at primary α- or β-cleavage site
- DRM
- detergent-resistant membrane
- PS
- presenilin
- mAPP
- mature APP
- imAPP
- immature APP
- MβCD
- methyl-β-cyclodextrin
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- pCTF
- phosphorylated C-terminal fragment
- nCTF
- nonphosphorylated C-terminal fragment
- pAPP
- APP phosphorylated at Thr668.
REFERENCES
- 1. Haass C., Selkoe D. J. (2007) Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 2. Kang J., Lemaire H. G., Unterbeck A., Salbaum J. M., Masters C. L., Grzeschik K. H., Multhaup G., Beyreuther K., Müller-Hill B. (1987) The precursor of Alzheimer disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736 [DOI] [PubMed] [Google Scholar]
- 3. Rohan de Silva HA, Jen A, Wickenden C, Jen LS, Wilkinson SL, Patel AJ. (1997) Cell-specific expression of β-amyloid precursor protein isoform mRNAs and proteins in neurons and astrocytes. Mol. Brain Res. 47, 147–156 [DOI] [PubMed] [Google Scholar]
- 4. Thinakaran G., Koo E. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aydin D., Weyer S. W., Müller U. C. (2012) Exp. Brain Res. 217, 423–434 [DOI] [PubMed] [Google Scholar]
- 6. Steiner H., Fluhrer R., Haass C. (2008) Intramembrane proteolysis by γ-secretase. J. Biol. Chem. 283, 29627–29631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Strooper B., Annaert W. (2010) Novel research horizons for presenilins and γ-secretases in cell biology and disease. Annu. Rev. Cell Dev. Biol. 26, 235–260 [DOI] [PubMed] [Google Scholar]
- 8. De Strooper B. (2007) Loss-of-function presenilin mutations in Alzheimer disease. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saito Y., Sano Y., Vassar R., Gandy S., Nakaya T., Yamamoto T., Suzuki T. (2008) X11 proteins regulate the translocation of amyloid β-protein precursor (APP) into detergent-resistant membrane and suppress the amyloidogenic cleavage of APP by β-site-cleaving enzyme in brain. J. Biol. Chem. 283, 35763–35771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kondo M., Shiono M., Itoh G., Takei N., Matsushima T., Maeda M., Taru H., Hata S., Yamamoto T., Saito Y., Suzuki T. (2010) Increased amyloidogenic processing of transgenic human APP in X11-like deficient mouse brain. Mol. Neurodegener. 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki T., Nakaya T. (2008) Regulation of amyloid β-protein precursor by phosphorylation and protein interactions. J. Biol. Chem. 283, 29633–29637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Taru H., Suzuki T., (2009) Regulation of the physiological function and metabolism of AβPP by AβPP binding proteins. J. Alzheimers Dis. 18, 253–265 [DOI] [PubMed] [Google Scholar]
- 13. Iijima K., Ando K., Takeda S., Satoh Y., Seki T., Itohara S., Greengard P., Kirino Y., Nairn A. C., Suzuki T. (2000) Neuron-specific phosphorylation of Alzheimer β-amyloid precursor protein by cyclin-dependent kinase 5. J. Neurochem. 75, 1085–1091 [DOI] [PubMed] [Google Scholar]
- 14. Ando K., Oishi M., Takeda S., Iijima K., Isohara T., Nairn A. C., Kirino Y., Greengard P., Suzuki T. (1999) Role of phosphorylation of Alzheimer amyloid precursor protein during neuronal differentiation. J. Neurosci. 19, 4421–4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ando K., Iijima K. I., Elliott J. I., Kirino Y., Suzuki T. (2001) Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of β-amyloid. J. Biol. Chem. 276, 40353–40361 [DOI] [PubMed] [Google Scholar]
- 16. Nakaya T., Suzuki T. (2006) Role of APP phosphorylation in Fe65-dependent gene transactivation mediated by AICD. Genes Cells 11, 633–645 [DOI] [PubMed] [Google Scholar]
- 17. Sano Y., Nakaya T., Pedrini S., Takeda S., Iijima-Ando K., Iijima K., Mathews P. M., Itohara S., Gandy S., Suzuki T. (2006) Physiological mouse brain Aβ levels are not related to the phosphorylation state of threonine 668 of Alzheimer APP. PLoS ONE 1, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu F., Su Y., Li B., Zhou Y., Ryder J., Gonzalez-DeWhitt P., May P. C., Ni B. (2003) Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35Cdk5 and p25Cdk5. FEBS Lett. 547, 193–196 [DOI] [PubMed] [Google Scholar]
- 19. Lee M. S., Kao S. C., Lemere C. A., Xia W., Tseng H. C., Zhou Y., Neve R., Ahlijanian M. K., Tsai L. H. (2003) APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 163, 83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pastorino L., Sun A., Lu P. J., Zhou X. Z., Balastik M., Finn G., Wulf G., Lim J., Li S. H., Li X., Xia W., Nicholson L. K., Lu K. P. (2006) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 440, 528–534 [DOI] [PubMed] [Google Scholar]
- 21. Pierrot N., Santos S. F., Feyt C., Morel M., Brion J. P., Octave J. N. (2006) Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-β accumulation. J. Biol. Chem. 281, 39907–39914 [DOI] [PubMed] [Google Scholar]
- 22. Rockenstein E., Torrance M., Adame A., Mante M., Bar-on P., Rose J. B., Crews L., Masliah E. (2007) Neuroprotective effects of regulators of the glycogen synthase kinase-3β signaling pathway in a transgenic model of Alzheimer disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 27, 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feyt C., Pierrot N., Tasiaux B., Van Hees J., Kienlen-Campard P., Courtoy P. J., Octave J. N. (2007) Phosphorylation of APP695 at Thr668 decreases γ-cleavage and extracellular Aβ. Biochem. Biophys. Res. Commun. 357, 1004–1010 [DOI] [PubMed] [Google Scholar]
- 24. Ramelot T. A., Gentile L. N., Nicholson L. K. (2000) Transient structure of the amyloid precursor protein cytoplasmic tail indicates preordering of structure for binding to cytosolic factors. Biochemistry 39, 2714–2725 [DOI] [PubMed] [Google Scholar]
- 25. Ramelot T. A., Nicholson L. K. (2001) Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR. J. Mol. Biol. 307, 871–884 [DOI] [PubMed] [Google Scholar]
- 26. Tomita S., Stein V., Stocker T. J., Nicoll R. A., Bredt D. S. (2005) Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron 45, 269–277 [DOI] [PubMed] [Google Scholar]
- 27. Kimura N., Tanemura K., Nakamura S., Takashima A., Ono F., Sakakibara I., Ishii Y., Kyuwa S., Yoshikawa Y. (2003) Age-related changes of Alzheimer disease-associated proteins in cynomolgus monkey brains. Biochem. Biophys. Res. Commun. 310, 303–311 [DOI] [PubMed] [Google Scholar]
- 28. Tomita S., Ozaki T., Taru H., Oguchi S., Takeda S., Yagi Y., Sakiyama S., Kirino Y., Suzuki T. (1999) Interaction of a neuron-specific protein containing PDZ domains with Alzheimer amyloid precursor protein. J. Biol. Chem. 274, 2243–2254 [DOI] [PubMed] [Google Scholar]
- 29. Taru H., Iijima K., Hase M., Kirino Y., Yagi Y., Suzuki T. (2002) Interaction of Alzheimer β-amyloid precursor family proteins with scaffold proteins of the JNK signaling cascade. J. Biol. Chem. 277, 20070–20078 [DOI] [PubMed] [Google Scholar]
- 30. Oishi M., Nairn A. C., Czernik A. J., Lim G. S., Isohara T., Gandy S. E., Greengard P., Suzuki T. (1997) The cytoplasmic domain of Alzheimer amyloid precursor protein is phosphorylated at Thr654, Ser655, and Thr668 in adult rat brain and cultured cells. Mol. Med. 3, 111–123 [PMC free article] [PubMed] [Google Scholar]
- 31. Thinakaran G., Borchelt D. R., Lee M. K., Slunt H. H., Spitzer L., Kim G., Ratovitsky T., Davenport F., Nordstedt C., Seeger M., Hardy J., Levey A. I., Gandy S. E., Jenkins N. A., Copeland N. G., Price D. L., Sisodia S. S. (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190 [DOI] [PubMed] [Google Scholar]
- 32. Gu Y., Misonou H., Sato T., Dohmae N., Takio K., Ihara Y. (2001) Distinct intramembrane cleavage of the β-amyloid precursor protein family resembling γ-secretase-like cleavage of Notch. J. Biol. Chem. 276, 35235–35238 [DOI] [PubMed] [Google Scholar]
- 33. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 34. Sumioka A., Yan D., Tomita S. (2010) TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron 66, 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tomita S., Kirino Y., Suzuki T. (1998) A basic amino acid in the cytoplasmic domain of Alzheimer β-amyloid precursor protein (APP) is essential for cleavage of APP at the α-site. J. Biol. Chem. 273, 19304–19310 [DOI] [PubMed] [Google Scholar]
- 36. Liu K., Doms R. W., Lee V. M. (2002) Glu11 site cleavage and N-terminal truncated Aβ production upon BACE overexpression. Biochemistry 41, 3128–3136 [DOI] [PubMed] [Google Scholar]
- 37. Kornilova A. Y., Das C., Wolfe M. S. (2003) Differential effects of inhibitors on the γ-secretase complex. Mechanistic implications. J. Biol. Chem. 278, 16470–16473 [DOI] [PubMed] [Google Scholar]
- 38. Maudsley S., Mattson M. P. (2006) Protein twists and turns in Alzheimer disease. Nat. Med. 12, 392–393 [DOI] [PubMed] [Google Scholar]
- 39. Wahrle S., Das P., Nyborg A. C., McLendon C., Shoji M., Kawarabayashi T., Younkin L. H., Younkin S. G., Golde T. E. (2002) Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23 [DOI] [PubMed] [Google Scholar]
- 40. Vetrivel K. S., Cheng H., Lin W., Sakurai T., Li T., Nukina N., Wong P. C., Xu H., Thinakaran G. (2004) Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rajendran L., Masilamani M., Solomon S., Tikkanen R., Stuermer C. A., Plattner H., Illges H. (2003) Asymmetric localization of flotillins/reggies in preassembled platforms confers inherent polarity to hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 100, 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beel A. J., Mobley C. K., Kim H. J., Tian F., Hadziselimovic A., Jap B., Prestegard J. H., Sanders C. R. (2008) Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP). Does APP function as a cholesterol sensor? Biochemistry 47, 9428–9446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cremona O., Di Paolo G., Wenk M. R., Lüthi A., Kim W. T., Takei K., Daniell L., Nemoto Y., Shears S. B., Flavell R. A., McCormick D. A., De Camilli P. (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188 [DOI] [PubMed] [Google Scholar]
- 44. Peter B. J., Kent H. M., Mills I. G., Vallis Y., Butler P. J., Evans P. R., McMahon H. T. (2004) BAR domains as sensors of membrane curvature. The amphiphysin BAR structure. Science 303, 495–499 [DOI] [PubMed] [Google Scholar]
- 45. Yan J., Wen W., Xu W., Long J. F., Adams M. E., Froehner S. C., Zhang M. (2005) Structure of the split PH domain and distinct lipid-binding properties of the PH-PDZ supramodule of α-syntrophin. EMBO J. 24, 3985–3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qi-Takahara Y., Morishima-Kawashima M., Tanimura Y., Dolios G., Hirotani N., Horikoshi Y., Kametani F., Maeda M., Saido T. C., Wang R., Ihara Y. (2005) Longer forms of amyloid β protein. Implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci. 25, 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suzuki T., Oishi M., Marshak D. R., Czernik A. J., Nairn A. C., Greengard P. (1994) Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. EMBO J. 13, 1114–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oikawa N., Kimura N., Yanagisawa K. (2010) Alzheimer-type tau pathology in advanced aged nonhuman primate brains harboring substantial amyloid deposition. Brain Res. 1315, 137–149 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.