

Abstract
The purpose of this study was to examine the effect of short-term high fat feeding on the inflammatory response in polymicrobial sepsis. Male C57BL/6 mice at six-weeks of age were randomized to a high-fat diet (HFD) (60% kcal fat) or control diet (CD) (16% kcal fat) for 3 weeks. After 3 weeks of feeding, sepsis was induced by cecal ligation and puncture (CLP) and animals were monitored for survival. In a separate experiment, after 3 weeks of feeding mice underwent CLP and were sacrificed at various time-points thereafter. Tissue was collected for biochemical studies. Mice fed a HFD gained more weight and had a greater fat mass compared to CD-fed mice. Mice on a HFD had a lower probability of survival and more severe lung injury compared with CD-fed mice following sepsis. Myeloperoxidase activity, an indicator of neutrophil infiltration, was increased in the lung and liver after CLP in HFD-fed mice compared with CD (p<0.05). The plasma cytokines tumor necrosis factor-α (TNFα) and interleukin (IL)-6 were increased in both groups after CLP, however TNFα and IL-6 levels were lower in HFD mice at 3h after CLP compared with CD and consistent with lung, but not liver, mRNA expression. Leptin levels were higher in HFD-fed mice at 18h after sepsis compared to baseline levels (p<0.05). Polymicrobial sepsis increased hepatic nuclear factor-κB (NF-κB) activation in HFD-fed mice after CLP vs. CD-fed mice. Short duration high fat feeding increases mortality and organ injury following polymicrobial sepsis. These effects correspond to changes in NF-κB.
Keywords: infections, high fat diet, inflammation, adipokines
INTRODUCTION
Obesity is an epidemiologic problem with over 1 billion overweight people worldwide (1, 2). In the United States more than 30% of adults and children are overweight or obese (3, 4). Obesity is an important health risk as it complicates many medical conditions including critical illness. Both obese adults and children have longer intensive care unit (ICU) lengths of stay and number of mechanical ventilator days than non-obese patients among survivors of acute lung injury and severe trauma (5–7). These findings were confirmed in a large meta-analysis involving over 62,000 critically ill subjects comparing outcomes in obese and non-obese patients (8). Obesity lowers the odds of survival in pediatric patients after in-hospital cardio-pulmonary arrest (9).
Obesity is associated with low-grade systemic inflammation. Many cells within adipose tissue can contribute to the inflammatory response in obesity and include adipocytes, endothelial cells, leukocytes and monocytes/macrophages (10). Cytokines produced mainly by adipose tissue include adiponectin, leptin, and resistin. Other plasma proteins are also produced from adipose tissue including tumor necrosis factor (TNF)-α, interleukin (IL)-6 and monocyte chemotactic protein-1. Obesity and increased weight are associated with an increase in pro-inflammatory cytokine production which decreases with weight loss (11).
Data suggests that the nuclear factor-κB (NF-κB) pathway is involved in obesity-associated pro-inflammatory responses (12). Mice with loss of function of TLR4 are protected against diet-induced obesity and insulin resistance (12, 13). Mouse models using genetic deletion of leptin (ob/ob) or the leptin receptor (db/db) to study the effects of obesity demonstrate an exaggerated inflammatory response following cecal ligation and puncture (CLP) compared to lean animals (14). However, studies using these transgenic models of obesity may not truly represent clinically relevant obesity as alterations in leptin and the leptin receptor may affect innate and adaptive immunity and not truly reflect inflammation relevant to diet-induced obesity in people (15, 16). Therefore obesity models using diet alone to cause obesity may be more reflective of clinically relevant obesity. Furthermore, most diet-induced obesity models use long periods of high fat feeding to cause obesity. These long-term feeding models may be applicable for obesity in adult patients but may not replicate the inflammatory changes which occur in childhood obesity or in the early stages of adult obesity. Obese children have inflammatory changes like adults but clearly have fewer obesity-years than adults. For example, in a large cross-sectional analysis of children in the United States, associations between inflammatory markers and obesity were evident even in children as young as three years of age (17). Therefore rodent obesity models using long-term high fat feeding may not be an appropriate model to use to investigate the mechanistic changes that occur in children. Therefore, the aim of this study is to determine the effect of obesity, using a model of short-term high fat feeding, on the inflammatory response following polymicrobial sepsis. We hypothesize that short-term high fat feeding increases the inflammatory response and worsen survival following sepsis.
METHODS AND PROCEDURES
Animals
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996) and commenced with the approval of the Institutional Animal Care and Use Committee. Male C57BL/6 mice at six-weeks of age were obtained from Charles River Laboratories International, Inc. (Wilmington, MA). The mice were housed in the animal facility at the Cincinnati Children’s Research Foundation (CCRF). Food and water were provided ad libitum. Animals were randomized to a high-fat diet (HFD) (TestDiet – 58Y1) (60% kcal provided by fat) or a standard-control diet (CD) (Formulab – 5008) (16% kcal provided by fat) for 3 weeks. After 3 weeks of feeding, polymicrobial sepsis was induced by CLP and monitored for survival for 30 h (n=12/group). In a separate experiment, after 3 weeks of feeding on HFD or CD mice underwent CLP and were sacrificed at 0, 1, 3, 6, and 18h thereafter. Plasma samples, lung, and liver were collected for biochemical studies described below.
Mouse model of cecal ligation and puncture (CLP)
CLP was performed as previously described (18). After opening the abdomen, the cecum was exteriorized and ligated by a 6.0 silk ligature at its base without obstructing intestinal continuity. The cecum was punctured twice with a 21-gauge needle and returned to the peritoneal cavity. The abdominal incision was closed with silk running sutures and liquid topical adhesive. After the procedure, animals were fluid resuscitated with sterile saline (0.6 ml) injected subcutaneously.
Imaging
In a separate experiment, mice were randomized to HFD or CD for weeks (n=3/group). After 3 weeks of feeding, animals were transported to the Imaging Research Center at CCRF for imaging. Animals were sedated with isoflurane and fat mass was determined by quantitative magnetic imaging (7T Bruker MRI, Billerica MA). Animals were sacrificed at completion of the study.
Measurement of myeloperoxidase activity
Myeloperoxidase activity was determined as an index of neutrophil accumulation in lung and liver as previously described (18). Tissues were homogenized in a solution containing 0.5% hexa-decyl-trimethyl-ammonium bromide dissolved in 10mM potassium phosphate buffer (pH7) and were centrifuged for 30 min at 20,000 × g at 4°C. An aliquot of the supernatant was allowed to react with a solution of tetra-methyl-benzidine (1.6mM) and 0.1 mM H2O2. The rate of change in absorbance was measured by spectrophotometry at 650 nm. Myeloperoxidase activity was defined as the quantity of enzyme degrading 1 μmol hydrogen peroxide/min at 37°C and was expressed in units per 100 mg of tissue.
Histopathological analysis
Lungs were fixed in 4% paraformaldehyde and embedded in paraffin. Sections were stained with hematoxylin and eosin and evaluated by three independent observers unaware of the experimental protocol. Specifically, lung injury was analyzed by a semi-quantitative score as previously reported (19) based on the following histologic features: a) alveolar congestion; b) hemorrhage; c) infiltration or aggregation of neutrophils in airspace or vessel wall and d) thickness of alveolar wall/hyaline membrane formation. Each feature was graded from 0 to 4 (i.e., no injury, minimal, mild, significant, or severe). The four variables were summed to represent the lung injury score (total score, 0–16).
Bacterial load
Spleen, lung, and liver samples were aseptically excised, weighed and homogenized in sterile saline using sterile tissue homogenizers. Two ml of sterile phosphate buffered saline (PBS) were lavaged into the peritoneum. The peritoneal fluid was placed in a sterile tube. Serial dilutions of tissue homogenates and peritoneal fluid were plated on nutrient agar and incubated overnight at 37°C. Bacterial colony forming units were counted to assess bacterial load.
Subcellular fractionation and nuclear protein extraction
Tissue samples were homogenized in a buffer containing 0.32 M sucrose, 10 mM Tris-HCl, 1 mM ethylene glycol tetraacetic acid (EGTA), 2 mM ethylenediaminetetraacetic acid (EDTA), 5 mM NaN3, 10 mM β-mercaptoethanol, 50 mM NaF, 20 μM leupeptin, 0.15 μM pepstatin A, and 0.2 mM phenylmethylsulphonyl fluoride (PMSF), 1 mM sodium orthovanadate, 0.4 nM microcystin (20). The homogenates were centrifuged (1,000 X g at 4°C, 10 min). The supernatant (cytosol + membrane extract) was collected and stored. The pellets were solubilized in Triton buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl (pH7.4), 1 mM EGTA, 1 mM EDTA, 0.2 mM sodium orthovanadate, 20 μM leupeptin A, and 0.2 mM PMSF). The lysates were centrifuged (15,000 X g, at 4°C, 30 min) and the supernatant (nuclear extract) collected to evaluate the DNA binding of NF-κB. The amount of protein was quantified by Bradford assay.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as described previously (21). An oligonucleotide probe corresponding to NF-κB consensus sequence (5′-AGT TGA GGG GAAC TTT CCC AGG C-3′) were labeled with [γ-32P]ATP using T4 polynucleotide kinase and purified in Bio-Spin chromatography columns (Bio-Rad, Hercules, CA). Ten micrograms of nuclear protein were preincubated with EMSA buffer (12 mmol/L HEPES, pH 7.9, 4 mM Tris-HCl, pH 7.9, 25 mMKCl, 5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, 50ng/ml poly[d(I-C)], 12% glycerol v/v, and 0.2 mM PMSF) on ice for 10 min before addition of the radiobaleled oligonucleotide for an additional 10 min. Protein-nucleic acid complexes were resolved using a nondenaturing polyacrylamide gel consisting of 5% acrylamide (29:1 ratio of acrylamide:bisacrylamide) and run in 0.5X Tris borate-EDTA (45 mM Tris-HCL, 45 mMboric acid, and 1 mMEDTA) for 1 h at constant current (30 mA). Gels were transferred to 3M paper (Whatman, Clifton, NJ), dried under a vacuum at 80°C for 1 h, and exposed to photographic film at −70°C with an intensifying screen. Densitometric analysis was performed using ImageQuant (Molecular Dynamics).
Determination of NF-κB p65 activity
Nuclear protein, 10 μg, was obtained from liver nuclear extracts and added to a 96-well plate to which oligonucleotide containing the NF-κB consensus binding sequence had been immobilized (Active Motif North America, Carlsbad, CA). Antibody directed against the NF-κB p65 subunit was added followed by a secondary horseradish peroxidase conjugate antibody. Developing solution utilizing a colorimetric readout was used. The plate was read by a spectrophotometer at 450 nm with a reference wavelength of 655 nm. A wild-type and mutated oligonucleotides were used for NF-κB binding to monitor the specificity of the assay (data not shown).
Cytokine mRNA isolation and real-time polymerase chain reaction (RT-PCR)
For RNA extractions, the lung and liver were homogenized, and RNA was isolated using the TRIzol method (Invitrogen, Grand Island, NY). RNA was reversely transcribed using the high-capacity cDNA reverse transcription kit according to the manufactures protocol (Applied Biosystems, Grand Island, NY). Relative RT-PCR was performed using TaqMan Gene expression master Mix (Applied Biosystems) with the following primers: GAPDH (Mm99999915_gl), IL-6 (Mm01210732_gl), and TNFα (Mm00443258_ml). The reaction was analyzed using a TaqMan PCR system (Applied Biosystems). Samples were run in duplicate for each time point. The change in expression of IL-6 and TNFα was normalized to GAPDH.
Plasma levels of adipokines and cytokines
Plasma levels of TNFα, IL-6, leptin, and adiponectin were measured by use of the multiplex assay kit (Millipore, Billerica, MA) using the protocol recommended by the manufacturer.
Materials
Oligonucleotide corresponding to NF-κB consensus sequence was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Data Analysis
Data were analyzed using SigmaStat for Windows Version 3.1 (SysStat Software, San Jose, CA). Values in the text and figures are expressed as mean and standard error of the mean (SEM) for parametric data and as median and interquartile range for nonparametric data. Plasma cytokines, tissue gene expression and liver NF-κB activation were analyzed for each time point versus baseline values and Student’s t tests was used for parametric data and Mann-Whitney rank-sum test was used for nonparametric data. Relative gene expression of cytokines in liver and lung was analyzed using the 2−ΔΔCT methodas described by Livak et al. (22). Liver and lung MPO data were analyzed as nonparametric data using Kruskal-Wallis analysis of variance with the Dunn post hoc test. Survival analysis was performed by the log-rank test. A value of p ≤ 0.05 was considered significant.
RESULTS
Mortality increases in HFD-fed mice after polymicrobial sepsis
Mice fed a high fat diet (HFD) for 3 weeks gained significantly more weight compared to mice fed a standard-control diet (CD) (25.2g ± 0.4 vs. 23.4g ± 0.4, p<.01) (Figure 1a). We performed magnetic imaging to confirm adiposity and the location of the adiposity (Figure 1b).
Figure 1.

Mice on a high fat diet gain more weight and more fat than mice on a control chow diet. Male C57BL/6 mice were randomized to a high fat diet (60% kcal fat) or control chow diet (16% kcal fat) for 3 weeks. (a) Body weight of mice on high fat diet or control diets. n=24 mice/group. *p<0.05 vs. normal chow by t-test. (b) Representative image of magnetic imaging with 7T MRI. (c) Fat mass quantified by all areas of high intensity segmented by the computer. n=3 per group.
To determine the effect of high fat feeding on survival from sepsis mice underwent CLP. Mice on a HFD had a lower probability of survival following polymicrobial sepsis compared with mice on a CD (Figure 2). One of the etiologies which could contribute to increased mortality is a higher bacterial load. Therefore, to determine whether the increased mortality was due to an increase in bacterial load we measured the bacterial load in the lung, liver, blood, peritoneal fluid, and spleen at 18h after CLP. We found no difference in bacterial load in any of the organs tested (data not shown).
Figure 2.
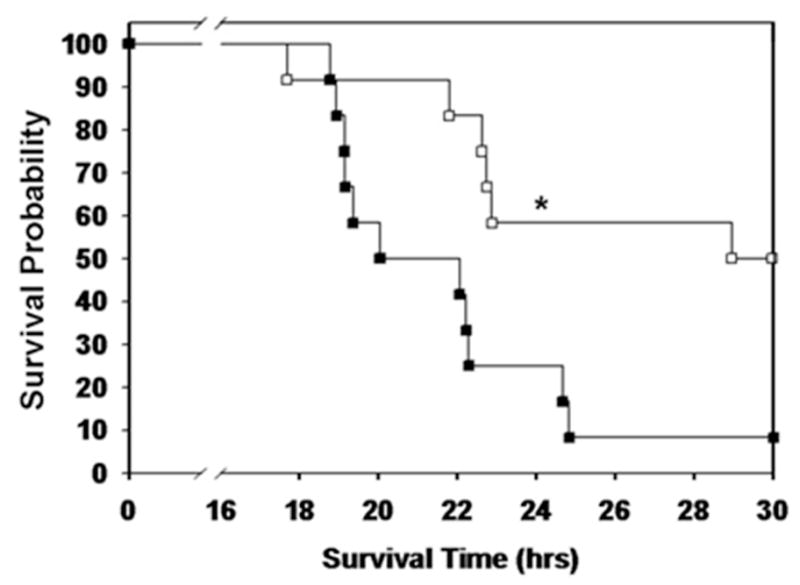
Mice on a high fat diet have higher mortality after sepsis. Mice were randomized to a HFD or CD for 3 weeks. CLP was performed and survival was monitored. Animals were censored at 30 hours. Survival curve demonstrates a lower probability of survival in HFD-fed mice after CLP (p=0.009 by log rank test, n=12/group).
Effects of a HFD on lung injury after induction of polymicrobial sepsis
Histological examination of the lungs of mice fed a HFD at 6h after CLP revealed marked lung injury as characterized by extravasation of red blood cells and accumulation of inflammatory cells into the air spaces compared to CD-fed mice (Figure 3a). This corresponded to a significantly higher lung injury score when compared to CD-fed mice at 6h after CLP (8.6 arbitrary units ± 0.9 vs. 3.5 arbitrary units ± 0.5, p<0.001) (Figure 3b).
Figure 3.

Mice on a high fat diet have worse lung injury. (a) Representative histology of lung sections stained with hematoxylin and eosin is shown at 0 and 6 hours after CLP. At 6h after CLP HFD-fed mice demonstrated interstitial hemorrhage and accumulation of inflammatory cells compared to CD-fed mice. (b) Lung injury was scored form 0 (no damage) to 16 (maximum damage). Box plots represent 25th percentile, median and 75th percentile; error bars define the 10th and 90th percentiles. p<0.001 by t test.
Effects of a HFD on lung and liver neutrophil infiltration after induction of polymicrobial sepsis
Multiple organ failure is a serious complication of sepsis and is usually preceded by accumulation of neutrophils in several vital organs (23). Therefore, we next quantified neutrophil infiltration in the lung and liver by measurement of myeloperoxidase (MPO) activity, an enzyme specific to granulocyte lysosomes. Mice fed a HFD have an increase in liver neutrophil infiltration at baseline prior to CLP (Figure 4a). After induction of sepsis both HFD and CD-fed mice have an increase in neutrophil infiltration. However in CD-fed mice MPO activity peaks in both the liver and the lung at 3 hours after CLP while in HFD-fed mice MPO activity remains elevated (Figure 4a,b).
Figure 4.

Mice on a high fat diet have an increase in liver and lung neutrophil infiltration. 6 week old male C57BL/6 mice were randomized to a HFD (black bars) or CD (white bars) for 3 weeks. Polymicrobial sepsis was induced by CLP. (a) Liver and (b) lung neutrophil infiltration was determined by myeloperoxidase assay. n=3–5 animals/group. *p<0.05 vs. time 0h. #p<0.05 vs. control diet. Data analyzed by t-test and Mann-Whitney Rank Sum test.
Effects of a HFD on the systemic inflammatory response
To determine the effect of high fat feeding on the systemic inflammatory response we measured plasma cytokine levels. As expected, TNFα and IL-6 plasma levels increased in both CD and HFD mice after CLP compared to baseline values (Figure 5a,b). Interestingly, plasma TNFα and IL-6 levels in HFD mice were significantly lower at 3h after CLP. Adiponectin levels were unchanged in CD-fed mice after CLP but decreased in the HFD mice at 6 and 18h after CLP compared to levels obtained at time 0h (Figure 5c). Animals on a HFD had higher leptin levels at baseline compared to CD-fed animals [5,719 (2,995–8.057) vs. 1,520 (1,223–1,820), p<0.05]. Animals on a CD had higher leptin levels at 3 and 6h after CLP compared to baseline leptin levels. HFD-fed also had higher leptin levels at 18h after CLP compared with baseline (time 0h) and CD-fed mice (Figure 5d).
Figure 5.

Effect of high fat feeding on the systemic inflammatory response. Plasma cytokine levels (a) TNFα (b) IL-6 (c) Adiponectin (d) Leptin were measured after CLP. *p<0.05 vs. time 0h. #p<0.05 vs. control diet. Data analyzed by Mann-Whitney Rank Sum test. White bar = CD, Black bar = HFD. n=3–9 animals/group.
Effects of a HFD on cytokine mRNA expression in liver and lung
To determine the effect of high fat feeding on the organ inflammatory response, mRNA expression of TNFα and IL-6 was measured by quantitative real time PCR in the lung and liver after induction of sepsis. Both TNFα and IL-6 mRNA expression increased in CD-fed and HFD-fed mice in the lung and the liver after CLP (Figure 6a–d). However, in the lung TNFα and IL-6 expression were decreased at 6h after CLP in HFD-fed mice compared with CD-fed mice (Figure 6a,b). In contrast, TNFα and IL-6 mRNA expression increased in HFD-fed mice after CLP (18h) compared with CD-fed mice (Figure 6c,d).
Figure 6.

mRNA levels in the lung and liver of pro-inflammatory mediators. The expression of (a) Lung TNFα (b) Lung IL-6 (c) Liver TNFα (d) Liver IL-6 was measured by RT-qPCR after CLP. *p<0.05 vs. time 0h. #p<0.05 vs. control diet. @p<0.05 vs. time 6h. Data analyzed by Mann-Whitney Rank Sum test. White bar = CD, Black bar = HFD. n=5 animals/group.
HFD increases activation of liver NF-κB
To investigate the mechanisms which contribute to the systemic inflammatory response in sepsis we evaluated the nuclear activation of NF-κB p65. Both HFD and CD-fed mice had an increase in liver NF-κB DNA binding at 3h after CLP compared to baseline values as evaluated by transcription factor assay kit (Figure 7a). However in CD-fed mice NF-κB activation decreased thereafter but NF-κB activation remained elevated in HFD-fed mice at 18h after CLP compared to CD-fed mice [0.15 relative units (0.12–0.37) vs. 0.07 relative units (0.06–0.08), p<0.05]. NF-κB DNA binding activation was confirmed by EMSA and correlated with the transcription factor assay findings (Figure 7b).
Figure 7.

HFD increases activation of liver NF-κB. Liver nuclear extracts were obtained from CD and HFD-fed mice at various time points after CLP. (a) Liver NF-κB DNA binding was evaluated by transcription factor assay kit. White bar = CD, Black bar = HFD. *p<0.05 vs. time 0h. #p<0.05 vs. control diet. Data analyzed by t-test. (b) Representative autoradiograph of EMSA for NF-κB.
DISCUSSION
Early mortality during acute sepsis remains a major unsolved problem. It is estimated that 20% of children who die from who die from severe sepsis die within 2 days of admission (24). In this study, we adopted a clinically relevant model of polymicrobial sepsis which mimics the outcomes of acute severe sepsis (18, 25). In this model, we demonstrated that high fat feeding, even for a short duration, increased organ injury and mortality. In our experiments, animals fed a HFD for only 3 weeks had more lung injury and lung and liver neutrophil infiltration following sepsis compared with mice fed a control diet. Additionally, the plasma adipokines, adiponectin and leptin, were altered in high fat-fed mice compared with CD-fed mice following sepsis. Unexpectedly, however, high fat-fed mice exhibited lower plasma levels of IL-6 and TNFα following induction of sepsis, which well correlated with lower TNFα and IL-6 mRNA expression in the lung when compared to control diet fed mice. On the contrary, septic HFD-fed mice exhibited a more severe inflammatory phenotype in the liver, where both TNFα and IL-6 mRNA expression was significantly higher when compared to control diet fed mice. This increased cytokine gene expression corresponded well with a significant increase in hepatic NF-κB activation in high fat-fed mice. Previous studies have suggested that liver and lung may exhibit a different inflammatory response during early and late sepsis and reflect the persistence of a pro-inflammatory response in one organ and the development of a compensatory anti-inflammatory response in another (26). Thus our data suggest that in obese mice a pro-inflammatory response persists longer in the liver than in the lung. Although we did not further explore the possible mechanisms underlying this differential inflammatory response, our data also suggest that obesity may influence the pattern of organ inflammatory response and may contribute to the different vulnerability of organ damage during sepsis.
In the current study, NF-κB DNA binding activity was increased in both CD and HFD-fed mice however the kinetics of NF-κB activation differs in these two groups following sepsis. These findings are consistent with previous results from our laboratory demonstrating that NF-κB DNA binding activity is increased in normal chow-fed animals following polymicrobial sepsis with a peak in the early hours following CLP (21). In this current study while there is an early increase in NF-κB activation in HFD-fed mice there appears to be a second peak of NF-κB activation in the late hours after CLP. Although, we did not detect any baseline differences in NF-κB activation prior to CLP with short term high fat feeding others have demonstrated that NF-κB activity is already increased in the liver in animals on a high fat diet (27).
Obesity alters the systemic inflammatory responses following polymicrobial sepsis. Surprisingly, we found that animals fed a HFD had significantly lower plasma TNFα and IL-6 levels and lower lung TNFα and IL-6 mRNA expression compared with CD-fed animals. These findings are however consistent with previous studies (28). In DIO-mice with experimental periodontitis plasma TNFα levels were lower compared with lean mice (28). Additionally, ex vivo stimulation of peritoneal macrophages with Porphyromonas gingivalis resulted in increased NF-κB gene expression from macrophages but reduced levels of TNFα and IL-6 in macrophage culture from DIO-mice compared with lean mice (28). To characterize these alterations Amar et al. used a chromatin immunoprecipitation assay to demonstrate that the recruitment of NF-κB to the TNFα promoter was substantially reduced in macrophages from DIO mice (28). Further investigation is needed to prove whether this inhibition of NF-κB transactivation may also explain our observations of reduced TNFα and IL-6 gene expression in the lungs of obese mice.
In our current study we demonstrate that mice fed a HFD have a higher mortality following sepsis compared with mice on a CD. Our findings are in agreement with other models of infection which demonstrate that diet-induced obesity results in increased mortality rates following infection. Mice fed a HFD and infected with Staphylococcus aureus or influenza had higher mortality than mice fed a control diet (29, 30). A clinically relevant condition which increases mortality and places patients at risk for multi-system organ failure is the development of leuko-sequestration within organs. In the current study we found that HFD-fed mice have a significant neutrophil infiltration in the lung and liver following polymicrobial sepsis compared with CD-fed mice. Previous studies have demonstrated that following a stimulus like an infection surface markers on leukocytes are activated and allow adherence of white blood cells to the vascular endothelium through an increase in the expression of adhesion molecules (14). Extravasation of leukocytes into tissues results in the release of proteolytic enzymes, cytokines and oxidizing molecules resulting in significant organ damage (31, 32). These alterations involving the immune cells and the vascular endothelium may explain the etiology of tissue injury seen in our study.
This current study demonstrates that mice fed a HFD had significantly lower plasma adiponectin levels following sepsis compared with baseline levels. Adiponectin is an anti-inflammatory adipocyte-derived cytokine which is secreted into human plasma (33). Adiponectin inhibits the NF-κB pathway and decreases NF-κB-dependent pro-inflammatory proteins (34). In a rat model of polymicrobial sepsis, plasma adiponectin levels were significantly decreased after sepsis and inversely correlated with plasma TNFα and endotoxin levels (35). Adiponectin knockout mice had higher inflammatory cytokine production and higher mortality after polymicrobial sepsis compared to wild-type mice (36). Although we found no difference in adiponectin expression in CD-fed mice following sepsis, HFD-fed mice have a decrease in adiponectin. As an anti-inflammatory cytokine decreased levels of adiponectin may play a role in the resultant increase in NF-κB activation.
Leptin, like adiponectin, is secreted mainly by adipocytes and has a role in regulating immune responses (37). Leptin levels were increased in mice after intraperitoneal LPS injection (38). Adults with sepsis had higher plasma leptin levels compared with healthy matched controls (39). In children who died from sepsis, leptin levels were increased compared to survivors (40). In our current study, we found that both CD and HFD-fed mice have an increase in plasma leptin levels but that leptin levels in HFD-fed mice remain significantly increased at 18 hours following sepsis. Changes in these adipose-derived proteins suggest that during sepsis there are alterations in adipose tissue which contributes to the systemic inflammatory response.
Our current study is the first to demonstrate that short duration of high fat feeding increases mortality and organ injury following polymicrobial sepsis. These effects appear to occur through alterations of NF-κB activation and result in an altered inflammatory response. Further studies are needed to elucidate the exact mechanisms which cause these alterations and contribute to the increase in mortality and morbidity in obese patients.
Acknowledgments
Supported, in part, by the National Institutes of Health grants R01 GM067202 (BZ), K08 GM093135 (JK), and P30DK078392.
Footnotes
DISCLOSURE
The authors declared no conflict of interest.
References
- 1.Misra A, Khurana L. Obesity and the metabolic syndrome in developing countries. J Clin Endocrinol Metab. 2008;93:S9–30. doi: 10.1210/jc.2008-1595. [DOI] [PubMed] [Google Scholar]
- 2.James PT, Rigby N, Leach R. The obesity epidemic, metabolic syndrome and future prevention strategies. Eur J Cardiovasc Prev Rehabil. 2004;11:3–8. doi: 10.1097/01.hjr.0000114707.27531.48. [DOI] [PubMed] [Google Scholar]
- 3.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 4.Ogden CL, Carroll MD, Flegal KM. High body mass index for age among US children and adolescents, 2003–2006. JAMA. 2008;299:2401–2405. doi: 10.1001/jama.299.20.2401. [DOI] [PubMed] [Google Scholar]
- 5.Morris AE, Stapleton RD, Rubenfeld GD, Hudson LD, Caldwell E, Steinberg KP. The association between body mass index and clinical outcomes in acute lung injury. Chest. 2007;131:342–348. doi: 10.1378/chest.06-1709. [DOI] [PubMed] [Google Scholar]
- 6.Brown CV, Neville AL, Rhee P, Salim A, Velmahos GC, Demetriades D. The impact of obesity on the outcomes of 1,153 critically injured blunt trauma patients. J Trauma. 2005;59:1048–1051. doi: 10.1097/01.ta.0000189047.65630.c5. discussion 1051. [DOI] [PubMed] [Google Scholar]
- 7.Brown CV, Neville AL, Salim A, Rhee P, Cologne K, Demetriades D. The impact of obesity on severely injured children and adolescents. J Pediatr Surg. 2006;41:88–91. doi: 10.1016/j.jpedsurg.2005.10.012. discussion 88–91. [DOI] [PubMed] [Google Scholar]
- 8.Akinnusi ME, Pineda LA, El Solh AA. Effect of obesity on intensive care morbidity and mortality: a meta-analysis. Crit Care Med. 2008;36:151–158. doi: 10.1097/01.CCM.0000297885.60037.6E. [DOI] [PubMed] [Google Scholar]
- 9.Srinivasan V, Nadkarni VM, Helfaer MA, Carey SM, Berg RA. American Heart Association National Registry of Cardiopulmonary Resuscitation I. Childhood obesity and survival after in-hospital pediatric cardiopulmonary resuscitation. Pediatrics. 2010;125:e481–488. doi: 10.1542/peds.2009-1324. [DOI] [PubMed] [Google Scholar]
- 10.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 11.Ziccardi P, Nappo F, Giugliano G, et al. Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation. 2002;105:804–809. doi: 10.1161/hc0702.104279. [DOI] [PubMed] [Google Scholar]
- 12.Kim F, Pham M, Luttrell I, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. 2007;100:1589–1596. doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- 13.Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) 2008;16:1248–1255. doi: 10.1038/oby.2008.210. [DOI] [PubMed] [Google Scholar]
- 14.Vachharajani V, Vital S, Russell J, Scott LK, Granger DN. Glucocorticoids inhibit the cerebral microvascular dysfunction associated with sepsis in obese mice. Microcirculation. 2006;13:477–487. doi: 10.1080/10739680600777599. [DOI] [PubMed] [Google Scholar]
- 15.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 16.Tian Z, Sun R, Wei H, Gao B. Impaired natural killer (NK) cell activity in leptin receptor deficient mice: leptin as a critical regulator in NK cell development and activation. Biochem Biophys Res Commun. 2002;298:297–302. doi: 10.1016/s0006-291x(02)02462-2. [DOI] [PubMed] [Google Scholar]
- 17.Skinner AC, Steiner MJ, Henderson FW, Perrin EM. Multiple markers of inflammation and weight status: cross-sectional analyses throughout childhood. Pediatrics. 2010;125:e801–809. doi: 10.1542/peds.2009-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zingarelli B, Piraino G, Hake PW, et al. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am J Pathol. 2010;177:1834–1847. doi: 10.2353/ajpath.2010.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu D, Zeng BX, Zhang SH, et al. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, reduces acute lung injury in endotoxemic rats. Crit Care Med. 2005;33:2309–2316. doi: 10.1097/01.ccm.0000183161.81503.7d. [DOI] [PubMed] [Google Scholar]
- 20.Kaplan JM, Cook JA, Hake PW, O’Connor M, Burroughs TJ, Zingarelli B. 15-Deoxy-delta(12,14)-prostaglandin J(2) (15D-PGJ(2)), a peroxisome proliferator activated receptor gamma ligand, reduces tissue leukosequestration and mortality in endotoxic shock. Shock. 2005;24:59–65. doi: 10.1097/01.shk.0000167108.88376.f2. [DOI] [PubMed] [Google Scholar]
- 21.Zingarelli B, Sheehan M, Hake PW, O’Connor M, Denenberg A, Cook JA. Peroxisome proliferator activator receptor-gamma ligands, 15-deoxy-Delta(12,14)-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J Immunol. 2003;171:6827–6837. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Abraham E. Nuclear factor-kappaB and its role in sepsis-associated organ failure. J Infect Dis. 2003;187 (Suppl 2):S364–369. doi: 10.1086/374750. [DOI] [PubMed] [Google Scholar]
- 24.Watson RS, Carcillo JA, Linde-Zwirble WT, Clermont G, Lidicker J, Angus DC. The epidemiology of severe sepsis in children in the United States. Am J Respir Crit Care Med. 2003;167:695–701. doi: 10.1164/rccm.200207-682OC. [DOI] [PubMed] [Google Scholar]
- 25.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu RQ, Xu YX, Song XH, Chen LJ, Meng XJ. Relationship between cytokine mRNA expression and organ damage following cecal ligation and puncture. World J Gastroenterol. 2002;8:131–134. doi: 10.3748/wjg.v8.i1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amar S, Zhou Q, Shaik-Dasthagirisaheb Y, Leeman S. Diet-induced obesity in mice causes changes in immune responses and bone loss manifested by bacterial challenge. Proc Natl Acad Sci U S A. 2007;104:20466–20471. doi: 10.1073/pnas.0710335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strandberg L, Verdrengh M, Enge M, et al. Mice chronically fed high-fat diet have increased mortality and disturbed immune response in sepsis. PLoS ONE. 2009;4:e7605. doi: 10.1371/journal.pone.0007605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith AG, Sheridan PA, Harp JB, Beck MA. Diet-induced obese mice have increased mortality and altered immune responses when infected with influenza virus. J Nutr. 2007;137:1236–1243. doi: 10.1093/jn/137.5.1236. [DOI] [PubMed] [Google Scholar]
- 31.Aldridge AJ. Role of the neutrophil in septic shock and the adult respiratory distress syndrome. Eur J Surg. 2002;168:204–214. doi: 10.1080/11024150260102807. [DOI] [PubMed] [Google Scholar]
- 32.Martins PS, Kallas EG, Neto MC, Dalboni MA, Blecher S, Salomao R. Upregulation of reactive oxygen species generation and phagocytosis, and increased apoptosis in human neutrophils during severe sepsis and septic shock. Shock. 2003;20:208–212. doi: 10.1097/01.shk.0000079425.52617.db. [DOI] [PubMed] [Google Scholar]
- 33.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi N, Argueta JG, Masuhiro Y, et al. Adiponectin inhibits Toll-like receptor family-induced signaling. FEBS Lett. 2005;579:6821–6826. doi: 10.1016/j.febslet.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 35.Tsuchihashi H, Yamamoto H, Maeda K, et al. Circulating concentrations of adiponectin, an endogenous lipopolysaccharide neutralizing protein, decrease in rats with polymicrobial sepsis. J Surg Res. 2006;134:348–353. doi: 10.1016/j.jss.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Teoh H, Quan A, Bang KW, et al. Adiponectin deficiency promotes endothelial activation and profoundly exacerbates sepsis-related mortality. Am J Physiol Endocrinol Metab. 2008;295:E658–664. doi: 10.1152/ajpendo.90384.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loffreda S, Yang SQ, Lin HZ, et al. Leptin regulates proinflammatory immune responses. Faseb J. 1998;12:57–65. [PubMed] [Google Scholar]
- 38.Wang W, Poole B, Mitra A, et al. Role of leptin deficiency in early acute renal failure during endotoxemia in ob/ob mice. J Am Soc Nephrol. 2004;15:645–649. doi: 10.1097/01.asn.0000113551.14276.0b. [DOI] [PubMed] [Google Scholar]
- 39.Bornstein SR, Licinio J, Tauchnitz R, et al. Plasma leptin levels are increased in survivors of acute sepsis: associated loss of diurnal rhythm, in cortisol and leptin secretion. J Clin Endocrinol Metab. 1998;83:280–283. doi: 10.1210/jcem.83.1.4610. [DOI] [PubMed] [Google Scholar]
- 40.Blanco-Quiros A, Casado-Flores J, Arranz E, Garrote JA, Asensio J, Perez A. Influence of leptin levels and body weight in survival of children with sepsis. Acta Paediatr. 2002;91:626–631. doi: 10.1080/080352502760069007. [DOI] [PubMed] [Google Scholar]