

Abstract
Three distinct promoters control the master regulator of MHC class II expression, CIITA, in a cell type specific manner. Promoter I (pI) CIITA, expressed primarily by dendritic cells and macrophages, expresses a unique isoform that contains a caspase recruitment domain. The activity and function of this isoform is not understood but has been thought to enhance the function of CIITA in antigen presenting cells. To determine if isoform I of CIITA has specific functions, CIITA mutant mice were created in which isoform I was replaced with isoform III sequences. Mice in which pI and the CARD encoding exon were deleted were also created. No defect in the formation of CD4 T cells, the ability to respond to a model antigen, or bacterial or viral challenge was observed in mice lacking CIITA isoform I. Although CIITA and MHC-II expression was decreased in splenic DC, the pI knockout animals expressed CIITA from downstream promoters, suggesting that control of pI activity is mediated by unknown s II distal elements that could act at the pIII, the B cell promoter. Thus, no critical function is linked to the CARD domain of CIITA isoform I with respect to basic immune system development, function and challenge.
Keywords: MHC class II, gene expression, CIITA, antigen presentation
INTRODUCTION
The major histocompatibility complex class II (MHC-II) region encodes genes required for the presentation of antigenic peptide to CD4 T cells 1. The process results in a variety of immune responses that range from direct effector responses to those that are regulatory. Constitutive expression of MHC-II is restricted to antigen presenting cells (APC: macrophages, B cells and dendritic cells) and thymic epithelial cells. MHC-II expression can also be induced in most cell types by exposure to interferon-γ (IFN-γ). The expression of MHC-II must be tightly regulated, as aberrant expression can result in immune system dysfunctions, including: autoimmunity, increased susceptibility to cancer, and decreased resistance to infectious organisms 2-4. Loss of MHC-II gene expression results in severe immunodeficiency, as evidenced by patients with bare lymphocyte syndrome (BLS), a disease resulting from mutations within transcription factors that regulate MHC-II expression 5, 6. One BLS complementation group was found to be deficient for the class II transactivator (CIITA), a factor that is essential for MHC-II expression 7.
CIITA functions as a transcriptional coactivator interacting with other MHC-II specific transcription factors bound to regions upstream of each MHC-II gene (reviewed in 8). The recruitment of CIITA to MHC-II promoters orchestrates a set of chromatin modifications and rearrangements that are associated with and are required for MHC-II expression. The gene encoding human CIITA contains four promoters, three of which are conserved in mice (Ciita). Ciita promoters function in a cell type specific manner 9. Ciita promoter I (pI) is utilized by dendritic cells (DC) and macrophages exposed to IFN-γ 10, and appears to be myeloid specific. Ciita pIII is expressed in cells of the lymphoid lineage (B cells, human T cells, and plasmacytoid DCs); whereas pIV is primarily expressed in non-hematopoietic cells upon exposure to IFN-γ 11. Each of the Ciita promoters contains a unique first exon, which splices into a common second exon, resulting in three distinct CIITA isoforms. Isoform I, derived from pI, is particularly intriguing because its unique exon encodes an N-terminal domain of 93 aa that bears homology to a caspase recruitment domain (CARD) 12. Such domains have been shown for other proteins to be important for protein-protein interactions 13, 14. The presence of the CARD domain in addition to other domains led to CIITA being the cardinal member of the family known as nucleotide-binding domain and leucine-rich repeat containing (NLR) proteins 15-17. This family of proteins is related to disease resistance R genes in plants and a number of NLR family members have functions in pathogen sensing, inflammation, cell signaling, and cell death 15, 18, 19. Increasing evidence suggests that many NLRs are cytoplasmic pathogen recognition receptors, activating immune responses to intracellular pathogens 19. Despite being a member of the NLR family, to date, no function outside of transcriptional activation has been ascribed to CIITA. Previous studies that address cell-type specific function of CIITA have focused on promoters III and IV using a knock-out strategy to create mice lacking either pIV or both pIII and pIV 11, 20. Using the Ciita pIV targeted knock-out mouse it was observed that cells of a non-hematopoietic lineage, but not macrophages or microglia, lost the ability to induce Ciita following exposure to IFN-γ, demonstrating a need for pIV in expression of Ciita in non-bone marrow derived cells 11. In addition, positive selection of CD4 T cells was severely impaired due to loss of expression of MHC-II on cortical thymic epithelial cells (cTECs), though MHC-II expression on cells of the thymic medulla was unchanged 11, 21. A deletion of the regions encompassing both pIII and pIV displayed all of the phenotypes observed in the pIV KO, and in addition, resulted in loss of MHC-II expression from B cells and plasmacytoid DCs (pDC), while conventional DCs and macrophages induced with IFN-γ retained MHC-II expression 20. These data point towards the necessity of pI for expression of CIITA and MHC-II in cells of the myeloid lineage.
To address a role for pI and the CARD-containing isoform in regulating CIITA expression and activity, a set of mice were constructed that replaced isoform I of CIITA with the 17 aa exon of isoform III. Effectively, this CIITApI→III knock-in (KI) was designed to create a mouse that would express isoform III CIITA from pI and pIII. Using FLP-mediated recombination, an additional mouse line was created in which pI and its surrounding upstream and downstream DNA were deleted, creating a CIITApI→0 knock-out (KO). Thus, two novel Ciita mouse lines were created. These mice were extensively characterized for their ability to express Ciita and MHC-II gene products and response to pathogen challenge. The results showed that the KI mice expressed MHC-II at levels comparable to wild-type mice. Surprisingly, KO mice still retained Ciita expression in all cell types examined, including splenic DC, which typically use pI nearly exclusively. This was due to redirection of transcript initiation from pI to pIII. Thus, both KI and KO mice lack isoform I CIITA and instead express isoform III in the myeloid and lymphoid compartments where CIITA isoform I is normally expressed. T cell development and activation appeared normal in both KI and KO mice. KO mice were also able to mount normal immune responses to Listeria monocytogenes and lymphocytic choriomeningitis virus (LCMV) infection and rechallenge, suggesting that the isoform I CIITA does not provide an advantage in these settings. Together, these data demonstrate that promoter I of CIITA and its corresponding CARD-containing isoform are not required for proper immune system development or function, and suggest that isoform III is capable of substituting for isoform I.
RESULTS
Generation of Ciita promoter I isoform III knock-in and Ciita promoter I knockout mice
To examine the function of the CARD domain-containing pI isoform in vivo, a targeting vector that replaced the first exon of Ciita’s isoform I with isoform III’s first exon was created using a gene targeting strategy that utilized both Cre-loxP and FLP-FRT mediated recombination. To maximize the utility of this animal, the replaced exon/isoform segment was flanked by FLPe recombination target sites (FRT), which would allow deletion of the entire pI promoter and its first exon in vivo. This includes 298 bp upstream of the transcription start site through 145 bp downstream of pI exon 1 for a total of 798 bp. Thus, the targeting vector contained a 2.5kb fragment upstream of Ciita pI; an HA-tagged pIII exon 1 coding sequence under control of the endogenous isoform I promoter and flanked by FRT sites; a lox-P-flanked neomycin-resistance cassette (neo); and a 2.5kb fragment of Ciita pI intron 1 (Fig. 1A). Homologous recombination was carried out in 129SvEv embryonic stem (ES) cells. Correctly targeted ES cells were injected into C57BL/6 blastocysts and three chimeric males were obtained. These chimeras were crossed to C57BL/6J females, and the agouti F1 heterozygote offspring were examined for the transmission of the targeted allele by PCR. To delete the neo cassette, mice containing the targeted allele were crossed to EIIa-Cre mice (The Jackson Laboratory). Offspring of these crosses were analyzed by PCR for transmission of the targeted allele and for Cre-mediated deletion of the floxed DNA (Fig. 1B). Mice carrying the Cre-deleted locus were then backcrossed to C57BL/6J to generate CIITApI→III (knock-in, KI) mice or crossed to ACT-FLPe mice (The Jackson Laboratory) to delete pI. Offspring of these crosses were analyzed by PCR for FLP-mediated deletion of the DNA between the FRT sites (Fig. 1). Mice carrying the Cre/FLP-deleted locus were then crossed to C57BL/6J to generate CIITApI→0 (knock-out, KO) mice. An example of the PCR assays used to genotype the mice are shown in Fig. 1B.
Figure 1. Ciita targeting construct.
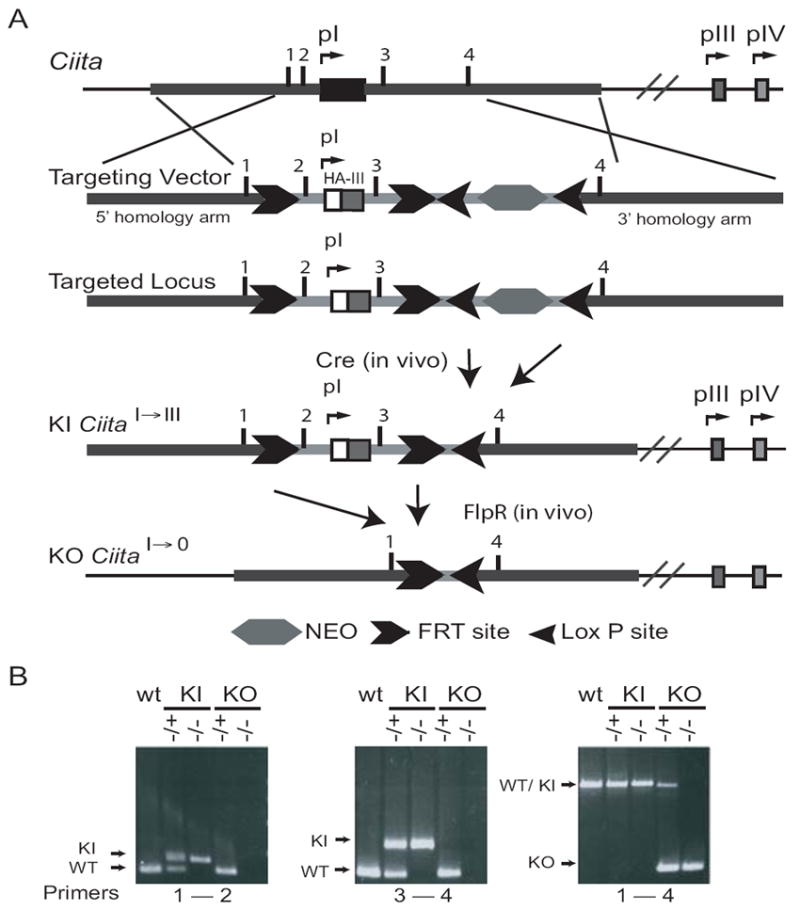
A. Schematic of the targeting construct used to generate CiitapI→III (KI) and CiitapI→0 (KO). Transgene positive mice were breed to a Cre-expressing mouse to delete the neo selective marker. KI mice were further crossed to a Flp-expressing mouse to generate the KO line. B. PCR genotyping results for transgene positive mice, KI, and KO lines.
Speed congenic backcrosses to C57BL/6J females using the male carrying the largest number of C57BL/6J loci were carried out for four generations. Using this speed congenic strategy, only four to five backcrosses are required to obtain a line that is 98-99% C57BL/6J 22. After two backcrosses, at least three markers on each chromosome were of C57BL/6J origin and the CIITA containing chromosome 16 was at least partially C57BL/6J (data not shown). Mice backcrossed to C57BL/6J for four to six generations were used in this study.
CIITA targeted mice are not defective in MHC class II expression
As stated above Ciita is expressed from three promoters in mice. Promoter I has been shown to be specific to cells of the myeloid lineage and specific DC compartments. To determine if the Ciita targeted mice showed defects in expression of MHC-II genes, the levels of I-Ab were examined by flow cytometry on thioglycolate elicited macrophages treated with IFN-γ (Fig. 2A) and splenic DCs (spDC) (Fig. 2B and C). For each strain, wild-type (WT) control mice were compared to either their KI or KO littermates. No difference in MHC-II expression was observed between KO and KI macrophages (CD11b+) compared to littermate controls.
Figure 2. MHC-II surface expression on KI and KO antigen presenting cells is similar to WT.
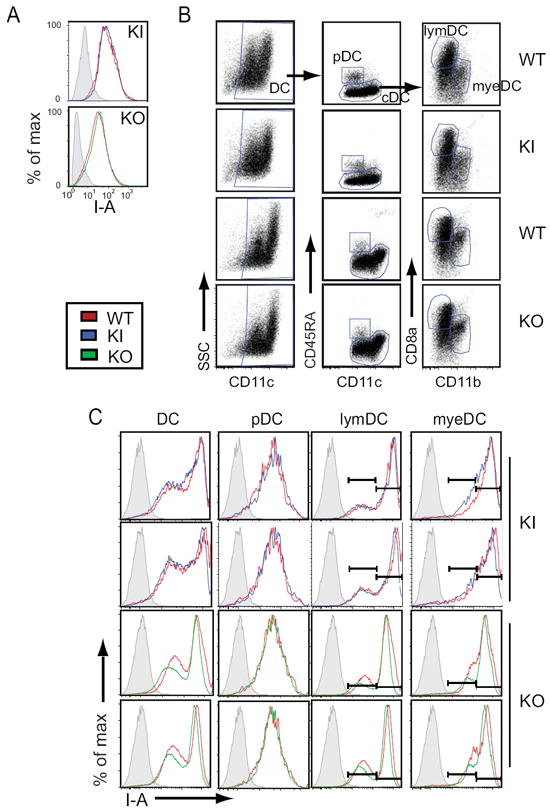
A. Histogram analysis of flow cytometry data for I-Ab expression in KI and KO samples compared to WT littermate controls. Thioglycolate elicited peritoneal macrophages were stimulated in vitro 24 h with 500 U/ml IFN-γ. Live cells were gated on by side scatter properties; macrophages were identified as CD11b+. B. Flt3L elicited, CD11c+ sorted spleen cells were stimulated with 1 μg/ml LPS for 8 h to induce translocation of MHC-II molecules to the surface and analyzed by flow cytometry. Total dendritic cells were identified as CD11c+ and further divided into CD45RA+CD11cint plasmacytoid (pDC) and conventional DCs (cDCs). cDCs were divided into CD11b+ myeloid (myeDC) and CD8a+ lymphoid (lymDC) subpopulations. C. Dendritic cell subsets identified in B were analyzed by flow cytometry for I-Ab expression. The data generated for this figure is representative of four mice for each strain. In panels A and C, WT littermate controls are shown overlaid on KI and KO samples. In panel B, WT littermate controls are shown directly above KI and KO samples.
Splenic dendritic cells were separated into two classes: conventional and plasmacytoid DCs (CD11c+ or CD11cdim, CD45RA+, respectively). Conventional DCs were further divided into myeloid (CD11b+) and lymphoid (CD8a+) compartments 23 (Fig. 2B) and analyzed for MHC-II surface expression (Fig 2C). In addition, fluorescence intensity was calculated in Molecules of Soluble Fluorochrome (MESF) for each subset of dendritic cells and the values were compared between groups of mice (data not shown). In the cases where two MHC-II peaks were observed, each peak was quantitated separately (Fig. 2C). Unlike conventional DCs (cDC), plasmacytoid DCs (pDC) utilize Ciita pIII 20; therefore, it was expected that pI mutations would not affect Ciita expression in this subclass of cells. Indeed, neither KI nor KO strains exhibited changes in MHC-II surface expression from their littermate controls in pDC (Fig. 2C). For the KI animals this was also true of the myeloid and lymphoid DC populations. However, the myeloid and lymphoid populations from KO cells consistently displayed MHC-II at levels that were 70-76% of that seen on WT (Fig. 2C).
MHC-II expression is correlated with Ciita expression in targeted mice
The expression of MHC-II on the surfaces of lymphoid and myeloid DC subsets as well as on macrophages in the KO strain was unexpected as 798 bp surrounding and including pI was deleted and this promoter was reported to control the expression of Ciita in cells derived from the myeloid lineage 20. Isoform specific quantitative real-time RT-PCR (qRT-PCR) using WT mice confirmed that the major Ciita transcript expressed in splenic DC (spDC) and macrophages treated with IFN-γ was isoform I (Fig. 3A). Isoform III was expressed at significant levels in spDC but not macrophages; whereas some isoform IV was detected in macrophages, but only after treatment with IFN-γ (Fig. 3A).
Figure 3. Ciita isoform I is not required for expression of CIITA or MHC-II in cells of the myeloid lineage.
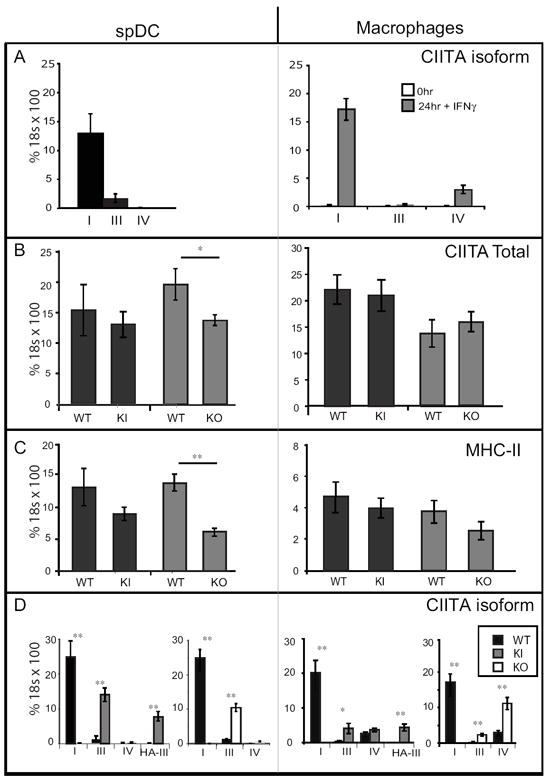
Ciita isoform specific, total Ciita mRNA, or I-Aα mRNAs were quantified in Flt3L-elicited splenic DCs (left panels) or thioglycolate-elicited peritoneal macrophages (-/+IFN-γ) (right panels) were analyzed by qRT-PCR with the primer sets indicated. Flt3L elicited spDC were purified with CD11c+ MACs beads and were >95% CD11c+. Thioglycolate-elicited, peritoneal cavity macrophages were purified by adherence to tissue culture plates and were >95% CD11b+. A. Isoform specific PCR of CIITA in WT splenic dendritic cells (spDC) and macrophages treated for 24h with IFN-γ. B. Total Ciita mRNA levels in KI, KO, and WT littermate controls. C. I-Aα mRNA levels in KI, KO, and WT (littermate controls) spDC or macrophages treated with IFN-γ. D. Isoform specific expression of Ciita. HA-III represents primers specific for the Knock-in allele, HA-tagged isoform III derived from promoter I. Data were averaged from four mice for each genotype, with each representing one biological replicate. Significance was calculated using the students t-test. *p<0.05, **p<0.005.
To investigate the transcriptional basis for cell surface MHC-II gene expression observed on the KI and KO spDC and macrophage cell populations presented in Fig. 2, RNA was also isolated from these samples to perform analyses of Ciita transcripts by qRT-PCR. Cell purity was greater than 95% as measured by CD11c expression on spDC and CD11b expression on macrophages (Fig 2B and data not shown). Except in the case of KO spDC, in which Ciita expression was reduced by 30%, Ciita levels were similar in both wild-type and mutant cells (Fig. 3B). This correlated with the observed decrease in surface MHC-II expression observed only on cDC from the KO line (Fig. 2C). I-Aα mRNA levels were also similar to WT, except in the case of KO spDC, where I-Aα was decreased by 55% (Fig. 3C, left panel).
Isoform specific qRT-PCR was used to determine the origin of the Ciita transcripts that were being detected (Fig. 3D). Again, wild-type cells principally use pI and produce isoform I Ciita transcripts. KI cells were found to express isoform III CIITA, however, this could be a combination of expression from both pI and pIII as the primers cannot distinguish between the two. Using a primer specific for the HA-tag confirmed that KI spDC and macrophages express the KI, HA-tagged-isoform III CIITA allele. Unexpectedly, spDC derived from KO cells expressed high levels of transcripts containing pIII exon 1, producing CIITA isoform III, and low, but detectable levels of isoform IV. Furthermore, in the case of KO derived macrophages, CIITA isoform III, as well as isoform IV, were expressed at significant levels in response to IFN-γ (Fig. 3D, right panel). These results suggest that there may be a change in promoter utilization in the Ciita pI KO mice.
CIITA has been found to be responsible for the regulation of multiple MHC-II associated genes, including H2-D1, H2-DMa, H2-DMb, H2-DOa, H2-DOb, and Ii 24, 25, as well as genes outside of the MHC locus. ChIP-chip experiments on human dendritic and B cells identified the genes KIAA0841, PSMD3, RAB4B, TPP1, and TRIM26 24, and microarray analysis of human B cells 25 and mouse dendritic cells 26 identified KPNA6, RAB4B, and Plxna1 as additional potential targets of Ciita. 24, 25 Additional genes potentially regulated by CIITA are Cste and Col1A1 27,28. To determine if the CARD-containing isoform of CIITA was differentially influential in the expression of these genes, qRT-PCR was carried out on RNA isolated from spDCs and macrophages treated with IFNγ. For the most part, no significant differences in expression for the above genes were observed between KO and wild-type macrophages or dendritic cells (Supplemental Fig. 1A and 1B). There were however two exceptions. In concordance with the level of total CIITA mRNA expressed in KO derived splenic DC, Ii was reduced to a similar extent as I-Ab (Supplemental Fig. 1A and Fig. 3C). The second exception involved H2-DOb expression. In contrast to the other CIITA regulated genes in which IFN-γ treatment of wild-type macrophages either had limited effect or induced their expression, IFN-γ treatment of wild-type macrophages reduced the levels of H2-DOb mRNA by ~7 fold (Supplemental Fig. 1C). Intriguingly, H2-DOb expression only decreased by about half in the IFN-γ treated KO-derived macrophages (Supplemental Fig. 1C). As a point of reference, H2-Ob was expressed at very low levels in both stimulated and unstimulated macrophages, while the expression was much higher in spDC (data not shown). This agrees with previous data that H2-Ob is expressed primarily by B cells, subsets of thymic epithelial cells, and subsets of dendritic cells, but not monocytes or macrophages 29, 30. Therefore, for the most part, expression of non-MHC, as well as MHC genes regulated by CIITA, was not dependent on the CARD domain of CIITA for induction.
spDC use alternate promoters in the absence of pI
Because cells from KO mice expressed transcripts that appeared to derive from promoters III and IV, 5’RACE was conducted to determine the transcriptional start sites of the CIITA transcripts in spDC from KO and KI mice (Fig. 4 and Supplemental Fig. 2). One hundred percent of the transcripts from wild-type spDCs originated at pI. In the KI spDCs, 77, 15.9, and 6.8% initiated their Ciita transcripts from pI, pIII, and pIV, respectively. The majority of transcripts in the KO derived spDCs were initiated in the proximity of pIII (96.3%), with the remaining transcripts being derived from pIV. Thus, spDCs derived from the KO mouse have switched the utilization of their Ciita promoter from pI to pIII.
Figure 4. 5’RACE maps Ciita expression to promoters III and IV in KI and KO spDC.
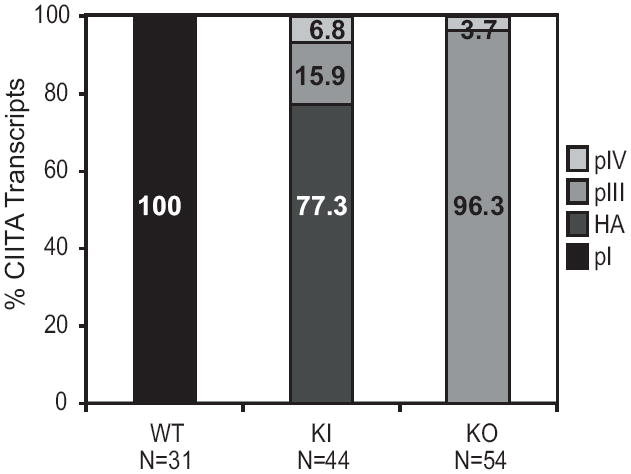
5’RACE was carried out on mRNA from WT, KI, and KO splenic DCs. Wild-type spDC Ciita transcripts initiated from pI, exclusively. Both KI and KO spDC expressed Ciita transcripts from promoters III and IV. The majority of Ciita transcripts in KI cells were the HA-tagged isoform III, and were expressed from promoter I (HA-pI). N refers to the number of cloned sequences analyzed.
CIITA Isoform I is dispensable for proper T cell development
Positive and negative thymic selection of the naïve T cell repertoire is in part controlled by the specific expression of peptide-MHC complexes by specific cell types in the thymus 31. Negative T cell selection is maintained in both Ciita pIV and pIII/pIV knock out mouse models, suggesting that bone marrow derived thymic medullary cells, which are required for negative selection, express CIITA from promoter I and therefore CIITA isoform I 11, 20. Thus, any specific need for CIITA isoform I’s CARD domain might be revealed by a change in the percentage of single positive CD4 T cells when comparing the KI and KO with wild-type thymocytes. It has been shown that impairing negative selection, either by deletion of dendritic cells or knocking down CIITA in mTECs, results in an increased percentage of CD4 T cells in the thymus 32-34. Flow cytometry of CD4 and CD8 single and double positive populations in the thymi of wild-type and Ciita-targeted mice showed that there was no perturbation of the CD4+, CD8+, or CD4+CD8+ T cell compartments when CIITA isoform I was lacking (Fig. 5). These data suggest that CIITA isoform I is not required for the regulation of MHC-II expression to the appropriate levels to ensure proper T cell development in the thymus. Because the KI line has a higher percentage of transcripts derived from pIII and pIV (Fig. 4) and the fact that the HA-tagged allele may add to the stability of the CIITA protein produced 35, the studies described below will use the KO model to assess the role of the pI isoform on a variety of immune responses.
Figure 5. Ciita isoform I is not required for T cell development in the thymus.
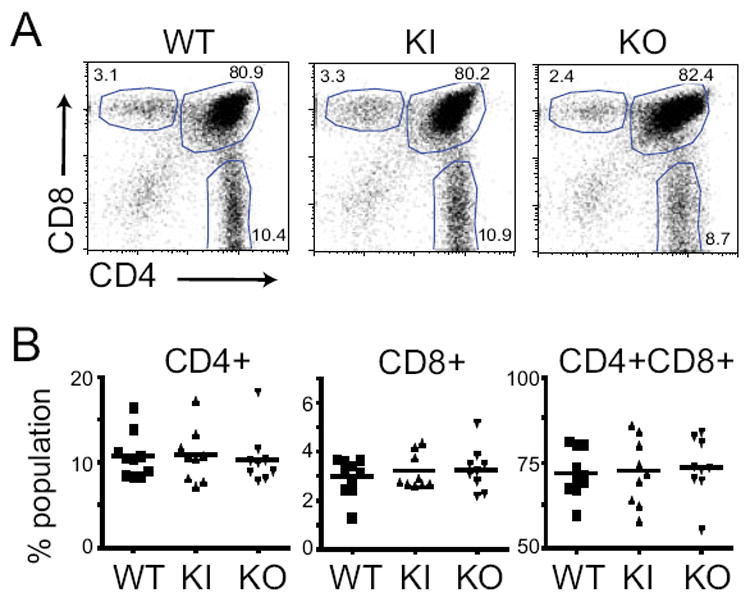
Thymocytes from five-week old mice were analyzed for CD4+, CD8+, and CD4+CD8+ populations using flow cytometry. A. Representative dot plots from individual thymi, gated on live cells and plotted as CD4 vs CD8. Gates used to define CD4+, CD8+, and CD4+CD8+ populations are shown. B. The results from three experiments with 3-4 mice per group are combined (N=10 WT, N=9 KI and KO). None of the samples were found to differ from the wild type by a p value of <0.05. using a student’s t test.
The CARD domain is not required for antigen presentation in vitro
To determine whether CIITA isoform I’s CARD domain is required in antigen presentation, a lymphocyte proliferation assay was used to compare T cell activation between WT and KO mice. Lymph nodes were collected from mice primed with the LCMV, CD4 T cell specific antigen, peptide GP61-80 36. Mice were immunized containing either 1mg/ml GP61-80 in CFA or a ten-fold dilution of peptide to determine whether immunization with suboptimal doses would reveal subtle differences in T cell response. Single cell suspensions were incubated with increasing concentrations of peptide ex vivo and the proliferation of T cells was measured by incorporation of [3H]-thymidine. CD4 T cells derived from the draining lymph nodes of WT and KO mice displayed similar responses to the peptide antigen when immunized with either dose (Fig. 6). Thus, despite loss of isoform I CIITA, there was no defect in activation of T cells in vitro, indicating no defect in antigen presentation in KO APCs.
Figure 6. CiitapI KO T cells proliferate normally in response to antigen.
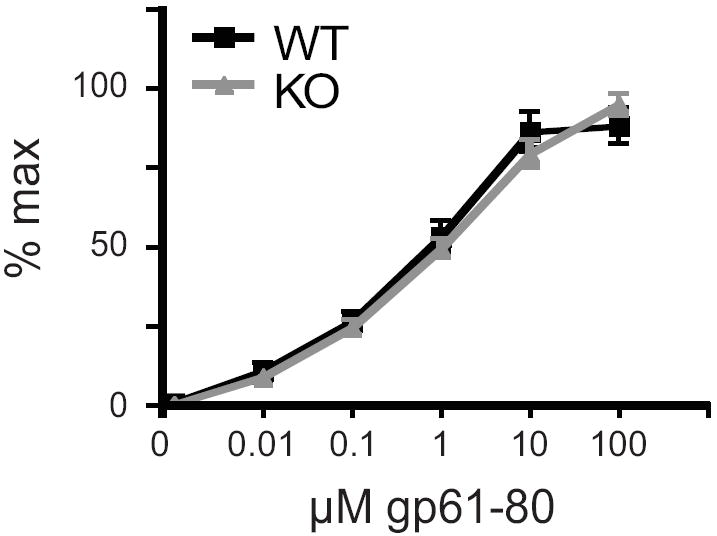
Mice were immunized in the footpad and the base of the tail with the 150μl peptide/CFA emulsion containing A. 1 mg/ml or B. 0.1 mg/ml of the peptide GP61-80. After 12 days, draining lymph nodes were isolated and T cell proliferation was measured by incorporation of 3H-thymidine in response to increasing concentrations of the antigenic peptide. Averaged data from multiple independent experiments are graphed as normalized counts per minute (CPM,) as a function of increasing concentration of peptide. (A. n=9 WT and 8 KO, B. n=6 WT and 7 KO).
CIITA Isoform I does not contribute to the development of experimental autoimmune encephalomyelitis (EAE)
EAE is an inflammatory demyelinating disease that is used as a model of multiple sclerosis. EAE is mediated by CD4 Th1 cells that recognize self-antigens associated with MHC-II molecules. CIITA isoforms 1 and 4 were found in the brain and spinal cord of mice with acute EAE, along with infiltrating DCs 37. Suter et al. found the majority of Ciita was expressed from pI in infiltrating DCs, while astrocytes express isoform IV in response to IFN-γ, and microglia express low levels of both isoforms 1 and 4 in response to IFN-γ. To examine if this model could reveal a need for isoform I, EAE was induced in WT and KO mice, and disease scores were recorded for 4 weeks. Disease severity was compared between the two groups over the entire four weeks, as well as in the last week alone, as this was when the disease scores stabilized. KO mice appeared slightly more sensitive than wild-type mice, with a maximum disease score of 2.5 vs. 2.0 for the WT group, although overall disease severity (combined disease index), disease severity in the fourth week only, and day of onset, were not found to be statistically different between the two groups (Fig. 7).
Figure 7. CiitapI KO mice are not more susceptible to experimentally induced autoimmune disease.
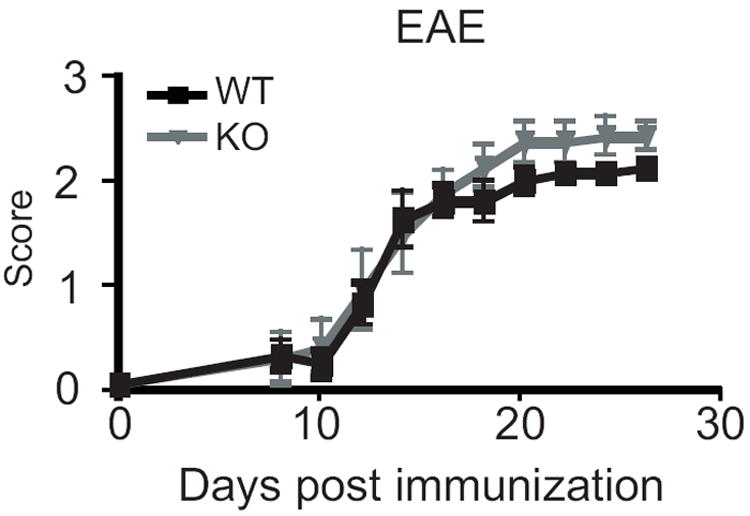
Experimental autoimmune encephalomyelitis was induced in groups of three to five females by injecting an emulsion of MOG and CFA. The experiment was performed three times and results were combined (n=11 WT and 10 KO). No difference was observed in onset or severity of disease. Severity was measured both for the entire four weeks of observation and for week four only. The max score was 2.0 for WT mice and 2.5 for the KO group (p<0.05). Error bars represent standard error. Statistics were done using student’s t-test (onset) or Mann-Whitney (total disease score, max score) using Prism software.
Ciita KO mice are not more susceptible to L. monocytogenes
Listeria monocytogenes is an intracellular pathogen that targets macrophages. Several members of the NOD family have roles in L. monocytogenes recognition including NOD1, NOD2, and NLRP3 15, 19. To determine whether the CARD-containing CIITA plays a role in resistance to L. monocytogenes, mice were infected and bacterial colony forming units (CFU) were counted in the spleen and liver five days after primary infection, or seven days after secondary infection (Fig. 8A). No difference was seen in bacterial burden in spleens or livers of KO mice compared to WT littermate controls after either primary or secondary infection, and all mice cleared the primary infection by day 10 (data not shown). To further dissect the immune response to L. monocytogenes, splenocytes were collected at the peak of the primary and secondary T cell response (ten or six days post infection or rechallenge, respectively), and intracellular cytokine staining for TNF-α, IL-2, and IFN-γ was performed in CD4+ T cells. It has previously been shown that CD4 lymphocytes express a Th1-type cytokine profile after infection with L. monocytogenes 38. There was no difference in cytokine expression in KO CD4+ spleen cells compared to WT control (Fig. 8B, C). These results indicate that KO mice are not deficient in immune response to L. monocytogenes infection and that the other CIITA isoforms can substitute for isoform I.
Figure 8. Ciita isoform I is not required for resistance to the intracellular bacteria Listeria monocytogenes.
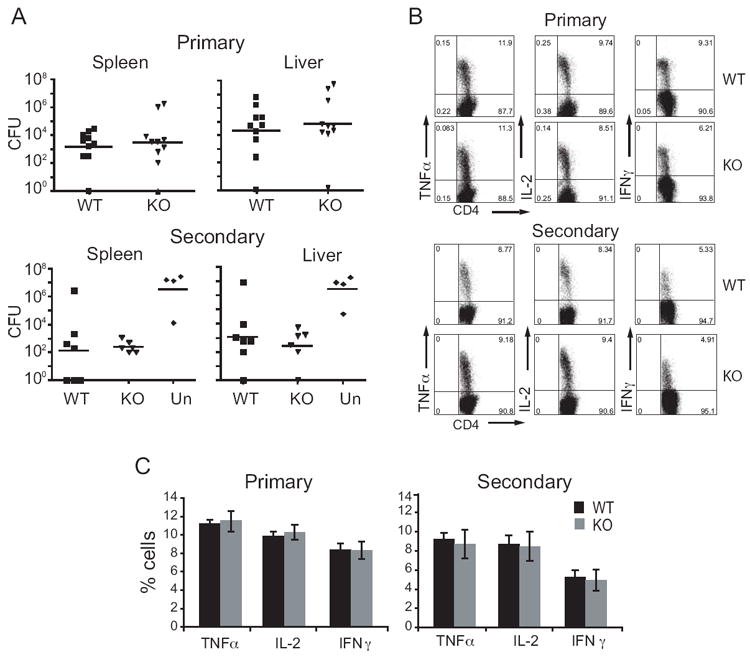
WT and CIITA KO mice were infected with LM, and colony-forming units (CFU) were determined in the spleens and livers. A. For primary infection, mice were infected with 0.5 LD50 LM and CFU were determined at day five post-infection. For secondary infection, mice were immunized with a low dose (0.1 LD50), then re-infected four weeks later with a high dose (10 LD50) of LM, CFU was determined 3 days after re-challenge. Data represent two independent experiments. N=10 WT, 11 KO primary, 7 WT, 6 KO secondary. B. Representative flow cytometry plots of intracellular cytokine expression from CD4 T cells in peritoneum. In primary infection, cells were collected 10 days after infection with 0.8 LD50. For secondary infection, mice were infected with 0.1 LD50, then challenged four weeks later with 6 LD50. Peritoneal exudate cells were collected six days later and CD4 T cell intracellular cytokine levels were measured by flow cytometry. C. Average with standard deviation of the percentage of CD4 T cells (from B) expressing cytokines TNFα, IL-2, and IFN-γ. Each graph is representative of two independent experiments using three to four mice per group. Un, unimmunized.
Ciita pI KO mice mount an effective immune response to lymphocytic choriomeningitis virus (LCMV)
To examine if the CARD domain containing CIITA isoform was required for specific responses to a viral infection, mice were infected with the Armstrong strain of LCMV. This viral strain generates an acute infection that is cleared after eight days 39. Following infection, WT and KO mice were analyzed for a number of parameters depicting a successful immune response to virus challenge and memory. The percentages of virus-specific CD8 or CD4 T cells following initial infection were similar between WT and KO mice (Fig. 9A and E). Virus-specific CD8 T cells from the spleen were analyzed for T cell specific memory markers CD44, CD25, CD62L, CD127, KLrg1, and PD-1 (Fig. 9B). No difference was observed in the expression of these markers as well. When CD4 and CD8 T memory cells were stimulated ex vivo for the production of cytokines, TNF-α and IFN-γ were also found to be similar (Fig. 9C). The LCMV-specific memory B cell pool, as measured by an antigen specific ELISPOT assay revealed higher values for the Ciita pI KO mice, but this value was not found to be statistically significant (Fig. 9D). Together these data indicate an effective adaptive immune response is generated in response to infection with LCMV in the absence of CIITA isoform I.
Figure 9. The adaptive immune response to an acute LCMV infection in KO mice is similar to WT mice.
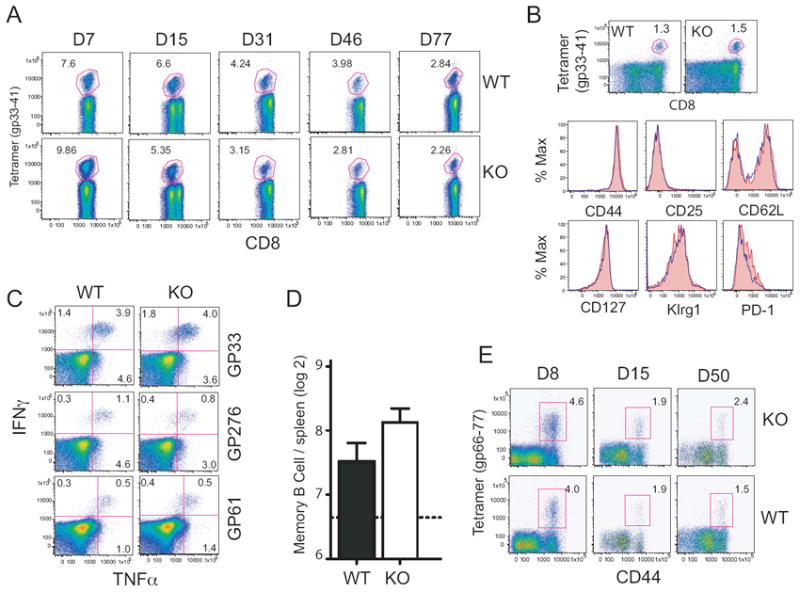
A. Virus-specific CD8 T cells were analyzed using fluorescently labeled tetramer specific for the GP33-41 epitope of LCMV. Mice were serially bled and the percent of LCMV-specific CD8 T cells in PBMCs was determined by flow cytometry analysis at the given time points. B & C. Acutely infected mice were sacrificed at the maintenance stage of T cell memory differentiation, and phenotypic and functional analyses were performed on the LCMV-specific lymphocytes. B. Tetramer specific (GP31-40) CD8 T cells were analyzed by flow cytometry for the T cell markers CD44, CD25, CD28L, CD127, Klrg1, and PD-1. Red filled histograms represent wild-type, blue histograms represent KO CD8 T cells. C. T cell receptor mediated cytokine expression was assessed by culturing splenocytes for 5h in the presence of peptides for the CD8 (GP33 and GP276) and CD4 (GP61) dominant epitopes of LCMV. D. The absolute number of LCMV-specific memory B cells was determined using an LCMV-specific ELISPOT analysis of memory B cells responses. E. Virus-specific CD4 peripheral blood mononuclear T cells were analyzed for CD44 expression and with tetramer specific for the GP66-77 epitope of LCMV.
LCMV and L. monocytogenes elicit primarily a T cell mediated response. In order to test the B cell response in the CIITApI null model, mice were immunized with influenza and antibody titers were measured at various time points post-immunization. No differences were seen between the wild-type and KO mice in antibody responses as measured by hemagglutinin inhibition (HAI) titers or IgG, IgG1, or IgG2a titers in the serum at 8, 14, or 28 days after infection (Supplemental Fig. 3).
DISCUSSION
Expression of MHC-II genes is regulated at the level of transcription by the presence or absence of the master regulator CIITA. The expression of CIITA from pI was of particular interest because this isoform has homology to a CARD domain 12 and its use is restricted to the myeloid and DC compartments 20. Thus, it seemed that this isoform would have unique properties that might be revealed by either substitution or deletion of the isoform. The knock-in/knock-out models created here allowed this hypothesis to be tested. Surprisingly, no gross defect in immune system function or development in either the CIITApI→III KI or CIITApI→0 KO was observed. In the case of the KO model, total levels of Ciita transcript were not reduced in macrophages treated with IFN-γ, and were not reduced by more than 30% compared to WT in spDC. Surface MHC-II expression reflected Ciita mRNA levels. No obvious defect in T cell development was observed either, as the percentage of CD4+ T cells in the thymus was normal. Moreover, T cells from the mutant mice responded normally to in vitro stimulation and showed no significant differences to lower doses of antigen. KO mice were only marginally more susceptible than wild-type in one model for autoimmunity (EAE), but all mice stabilized with the same level of disease. KO mice were equally immune competent (compared to wild-type) when challenged with a viral or bacterial agent (LCMV or L. monocytogenes, respectively), and produced antibodies at levels comparable to wild-type after vaccination to influenza. It remains possible that the decreased levels of I-A and Ii observed in KO spDC have a aubtle effect on immune system function not detected by the experiments presented here. Thus, we conclude that in the models tested, no unique function can be ascribed to the CARD-containing isoform of CIITA.
Interestingly, expression of MHC-II was decreased to a greater extent than Ciita. Nickerson et al. 12 suggested that pI is a more potent transcriptional activator than the other isoforms; however, this was not confirmed by Buttice et. al. 28, who found no difference in transactivation activity between the various isoforms. The reason Nickerson et al. observed greater activity from isoform I may be related to the CARD sequence, which could potentially provide increased stability of the protein 35. The half-lives of isoforms III and IV have been reported to be the same 35, but to date there is no data on the half-life of isoform I. Data presented here argue that equal levels of CIITA either from pI (endogenous) or from the KI lead to similar levels of I-A transcripts, suggesting that CIITA levels are the limiting determinant. Similarly, isoform I is not specifically required for induction of a number of genes identified as CIITA targets, including I-Ab, H2-D1, H2-DOa, H2-DM, Ii, Col1A1, Cste, Kiaa00841, Kpna6, Plxna1, Psmd3, Rab4b, Tpp1, and Trim26, as differences in expression of these genes was not observed in mice expressing only isoform III. With the exception of Plxna1 26, which is specific to DC, all of these genes are also expressed in B cells. KO macrophages treated with IFN-γ had higher levels of H2-DOb mRNA than wild-type macrophages. This result was unexpected for a few reasons. First, H2-DO is strongly expressed in B cells, but it is poorly expressed by macrophages 40,29, 30. Second, the levels of H2-DOb mRNA were reduced in wild-type but not KO IFN-γ treated macrophages. Lastly, the regulation of HLA-DOB/H2-DOb in B cells by CIITA has been controversial, some data suggest that HLA-DOB/H2-DOb is not regulated by CIITA in B cells 41, 42, while other data suggest the opposite 24, 25, 43. These data may indicate that CIITA isoform III, but not isoform I, is important for the proper regulation of H2-Ob. Thus, with the possible exception of H2-DOb, the CARD domain of isoform I does not appear to be required for expression of any of the genes identified as targets of CIITA regulation.
Previous mouse knock-out models showed no evidence for cross-talk between the promoters. In the Ciita pIV KO mouse, expression of Ciita from myeloid and lymphoid cells was not affected 11. Likewise, in the double pIII/pIV KO in which a large region was deleted, expression of Ciita in conventional DCs appeared normal 20. Therefore, it was unexpected that the deletion of pI would have an effect on downstream promoters. The fact that deletion of the 798 bp surrounding and including pI alters expression from other promoters suggests that the mechanisms that control expression from pI also operate on alternate promoters, but are prevented from doing so in cells of the myeloid lineage. Thus, it is most likely that there is an unidentified regulatory region that normally promotes expression of Ciita from promoter I in cells of the myeloid lineage. Expression of Ciita from pIII in the dendritic cells of mice in which pI was deleted suggests that Ciita’s “myeloid specific” regulatory region can direct expression from any (or perhaps the nearest available) promoter. Alternatively, pI and pIII may share transcription factors necessary for expression and that some other mechanism prevents pIII expression in spDC. Previous models that deleted pIV or pIII/pIV failed to identify this possibility, as there was no promoter switching observed in these models and little is known about the regulation of Ciita pI. Recently, Smith et al. 44 characterized the promoter proximal region and identified binding sites for the positive regulatory factors Pu.1 and IRF8, and PRDM1 as a negative regulatory factor. Choi et al. identified two STAT5 binding sites upstream of pI 45. Our data demonstrate that deletion of the proximal promoter region, which includes the regions studied in these reports, is insufficient to silence Ciita expression in cells that primarily use pI.
Transcription factors regulating Ciita pIII and pIV have been well characterized 46-52, and using these as a guide, some suggestions of what may occur at pI and promoter choice can be hypothesized. Several years ago it was found that DNA methylation of the CpG dinucleotides encompassing pIV prevented expression in response to IFN-γ in tumors or cell types refractory to this induction 53, 54. Analyses of the chromatin structure of pIV in human cells showed that other mechanisms may be at play and, by analogy such mechanisms may function with pI as well. In Hela cells, five regions spread across a 110 kb span that contained the CIITA gene were identified that could influence expression of pIV in response to IFN-γ 55. These regions were shown to interact with one another through chromatin conformation capture assays 55. Intriguingly, one of these regions was upstream of pI and was also identified as a DNase hypersensitive site in B cells. This region, termed HSS1, binds PU.1 49, a critical regulator of B cell and myeloid cell fate, as well as STAT1 55, a factor activated when cells are induced by IFN-γ. HSS1 lies three kb upstream of pI and was not deleted from our constructed mouse strains. Thus, the possibility exists that HSS1 could be a pI and pIII control region. However, it was only one of several elements in the long-range control of pIV and one of the other elements, or an as yet undiscovered element could control the use of pI. Thus, whichever element(s) controls expression in myeloid cells, our data suggest that this element targets pI first as opposed to the other Ciita promoters.
Thus, although CIITA in cells of the myeloid lineage express a unique isoform with a CARD homologous domain, this specific domain does not appear to have a unique function with respect to MHC-II expression and the ability of cells using this promoter to mount successful immune responses. Instead, the system appears to be designed to specifically control CIITA and subsequently MHC-II gene expression in a strict tissue specific fashion by having distinct promoters and regulatory mechanisms that restrict expression to each of the promoters.
MATERIALS AND METHODS
Targeting vector and generation of Ciita mutant mice
A Ciita site-specific targeting construct (see Fig. 1A) was generated in PGKneobpA (a gift from Dr. Grant MacGregor, Univ. California, Irvine) by standard PCR and cloning techniques. It contains, in order: a 2.8 kb fragment upstream of Ciita pI (3.1 kb to 0.3 kb upstream of the TSS of pI); a FLP1 recombinase target (FRT) site; upstream sequences -298 through +85 of the pI 5’UTR; an AUG codon and HA-epitope-tag fused to Ciita pIII exon 1 coding sequence; 145 bp from intron 1 of pI; and a second FRT site. A lox-P-flanked neomycin-resistance (neo) cassette and 3.0 kb of Ciita pI intron 1 (0.5kb to 3.5 kb downstream of the TSS) completes the vector and provides the 3’ targeting arm. ClaI and SalI sites were introduced into the construct to facilitate cloning. The linearized targeting vector was transfected into 129 SvEv embryonic stem (ES) cells by electroporation, and G418-resistant clones were selected by inGenious Targeting Labs, Inc. Homologous recombinants were screened by Southern blotting using HindIII, BamHI, or EcoRI digestion in combination with probes either internal or 3’ external to the targeting construct. The presence of the upstream FRT site was determined by PCR, using primers 1 and 2 (Fig. 1 and Supp Table 1). Correctly targeted ES cells were injected into C57BL/6 blastocysts and three chimeric males were obtained. These chimeras were crossed to C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) females and the agouti F1 heterozygote offspring were tested for the transgenic allele by PCR using primers 1 and 2 (Fig. 1). Mice containing the targeted allele were crossed to EIIa-cre mice (Jackson Laboratory). Offspring of these crosses were analyzed by PCR for transmission of the targeted allele and for Cre-mediated deletion of the floxed neo cassette (primer sets 1-2 and 3-4, Fig. 1 ad Supplemental Table 1). Mice carrying the Cre-deleted locus were then backcrossed to C57BL/6J to generate CIITApI→III knock-in (KI) mice or crossed to ACT-FLPe mice (Jackson Laboratory). Offspring of the ACT-FLPe crosses were analyzed by PCR for FLPe-mediated deletion of the DNA between the FRT sites (primer set 1-4, Supp Table 1). Mice carrying the Cre/FLP-deleted locus were then crossed to C57BL/6J to generate CIITApI→0 knockout (KO) mice. Where indicated, C57BL/6J mice from Jackson Laboratory aged six to eight weeks were used. Animals were housed under standard conditions in a conventional mouse facility, where they remained healthy. The Emory University Institutional Animal Care and Utilization Committee approved all animal experiments and protocols.
To generate the targeting construct, we designed primers (D5’d, XhoI and D3’, ClaI) to amplify the upstream flanking region for homologous recombination, introducing XhoI and ClaI sites for cloning into the targeting vector. Another primer pair was designed to introduce a FRT site upstream of promoter I and a SalI site at the start of translation (C5’, ClaI and C3’, SalI). The next fragment inserted into the targeting vector was generated by overlap-PCR using primers that introduced a consensus Kozak sequence and HA tag and amplified the coding region of promoter III exon 1, with the intronic sequence of promoter I exon 1, followed by a FRT site and XhoI for cloning purposes. Primers for this step were: B5’, SalI, B2-5’OL, B1-3’OL, and B2-3’. Finally, A5’, NotI and A3’, SacII were designed to amplify the downstream flanking region, intron 1 of pI, for homologous recombination, introducing NotI and SacII sites for cloning into the targeting vector. All primers are listed in Supplemental Table 1. PCR was performed using genomic DNA isolated from strain 129 mouse tissues. The PCR amplicons were initially cloned into the pCR4-BluntTOPO vector (Invitrogen, Carlsbad, CA) for DNA sequence confirmation and large-scale preparation, then subcloned into PGKneobpA at the XhoI site. The resulting vector was digested with NotI and SacII, and ligated with the downstream flanking region PCR fragment.
Speed congenics
We used a marker assisted selection procedure (MASP), aka “speed congenic” strategy, to construct a C57BL/6 congenic strain containing our targeted locus from a 129/SvEv:C57BL/6J mixed strain background 22, 56. We selected a genome-wide set of markers, spaced approximately 20 cM apart, from the MIT/Whitehead mouse map that were reportedly polymorphic between C57BL/6J and 129/SvEv 22, 56. PCR primers used are available on the Mouse Genome Database at the Mouse Genome Informatics website 57. PCR products were run on polyacrylamide gels and compared to C57BL/6J and 129SvEv control DNA run in parallel. The “best” breeder of each generation was heterozygous by locus-specific PCR for our targeted mutation, and carried the most C57BL/6J alleles by genome-wide SSLP PCR analysis. By the 4th backcross, all markers tested except the one next to the targeted locus were of C57BL/6J origin (data not shown).
Markers used were:
D1Mit211, D1Mit19, D1Mit215, D1Mit139, D1Mit150, D2Mit5, D2Mit61, D2Mit13, D2Mit206, D2Mit304, D2Mit213, D3Mit21, D3Mit40, D3Mit351, D3Mit163, D4Mit89, D4Mit275, D4Mit31, D4Mit204, D4Mit254, D5Mit148, D5Mit20, D5Mit163, D6Mit86, D6Mit33, D6Mit146, D6Mit199, D6Mit15, D7Mit310, D7Mit237, D7Mit31, D7Mit189, D8Mit155, D8Mit191, D8Mit263, D8Mit211, D8Mit93, D9Mit2, D9Mit21, D9Mit306, D9Mit279, D10Mit51, D10Mit40, D10Mit7, D10Mit162, D10Mit180, D11Mit226, D11Mit270, D11Mit5, D11Mit38, D11Mit199, D11Mit184, D12Mit153, D12Mit3, D12Mit231, D13Mit16, D13Mit179, D13Mit193, D13Mit76, D14Mit99, D14Mit60, D14Mit192, D14Mit107, D15Mit13, D15Mit46, D15Mit29, D15Mit42, D16Mit182, D16Mit103, D16Mit64, D16Mit152, D17Mit164, D17Mit51, D17Mit205, D17Mit123, D18Mit19, D18Mit23, D18Mit33, D18Mit25, D19Mit128, D19Mit119, D19Mit91, D19Mit71, DXMit136, DXMit143, DXMit213, DXMit130, DXMit186
5’RACE
To determine the 5’ ends of CIITA transcripts of KO and KI mice, rapid amplification of cDNA ends (RACE) was performed using the FirstChoice RLM-RACE Kit from Applied Biosystems (Carlsbad, CA) following manufacturer’s instructions. Primers used are listed in Supplemental Table 1.
Cell Collection and treatments
Dendritic cells (DC) were collected from the spleen as described in Current Protocols 58. Briefly, mice were injected i.p. with 30 ng Flt3 Ligand-Ig (Flt3-L) for 9 days 59. Flt3-L was generously provided by Dr. R. Mittler (Emory University). Following CO2 asphyxiation, spleens were removed and injected with Dulbeco’s modified Eagle’s media (Cellgro, Manassas, VA) containing 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO) (DMEM-10), 1x non essential amino acids (HyClone Laboratory, Logan VT), 1 M HEPES (HyClone Laboratory), 1 mM sodium pyruvate (HyClone Laboratory), 0.292 mg/ml L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen) and 1mg/mL collagenase D (Roche, Indianapolis, IN), cut into pieces, and incubated at 37°C for 25 min. A single cell suspension was generated by forcing cells through a 40 μm cell strainer (BD Biosciences, San Jose, CA). Red blood cells were lysed with ammonium-chloride potassium-chloride (ACK) lysing buffer. The CD11c+ DC population was purified using CD11c MACS beads (Miltenyi Biotech, Inc., Auburn, CA) according to manufacturer’s protocol. Peritoneal exudate cells were isolated in 10 ml Hanks’ buffered saline solution containing 10 U/ml heparin. For peritoneal macrophages, mice were injected i.p. with 2.5ml 2% solution of thioglycolate four days prior to collection of the peritoneal fluid. Peritoneal exudate cells were plated 0.8-1×106 cells/ml in DMEM-10 containing 0.292 mg/ml L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 50 μM 2-mercaptoethanol (Sigma-Aldrich) and allowed to adhere for 2 h. Non-adherent cells were washed off, and the adherent cells were provided fresh DMEM-10 media and treated with 500 U/mL IFN-γ (Peprotech, Inc., Rocky Hill, NJ) for the indicated time. Total primary peritoneal exudate cells and spleen cells were incubated in RPMI 1640 medium containing 10% FBS, 0.292 mg/ml L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50μM 2-mercaptoethanol, 1 mM sodium pyruvate, and 10mM HEPES buffer. For the collection of B cells and T cells from the spleen or lymph nodes, a single-cell suspension was generated as described above.
Flow cytometry
For cell surface staining, 0.5×106 cells were treated as previously described 38. Briefly, 0.5×106 cells were incubated on ice for 5 min with anti-CD16/32 (FC block clone 2.4G2) prior to addition of antibodies or isotype control for 30 min. Antibodies used for surface staining were purchased from BD Biosciences: anti-CD4 (clone RM4-5 PerCP, GK1.5 PE), anti-CD8 (clone 53-6.7) anti-CD11b (clone M1/70), anti-CD11c (clone HL3), anti-CD45R/B220 (clone RA3-6B2), anti-CD45RA (clone 14.8), anti-I-Ab (clone AF6-120.1), and appropriate isotype controls (A95-1, R35-95, G235-2356, G155-178). For secondary labeling, cells were washed once, followed by 20 min incubation on ice with Pacific Blue labeled streptavidin (Molecular Probes, Eugene, Oregon). Flow cytometry was performed using a BD FACSCalibur or LSRII flow cytometer (BD Biosciences). Fluorescence intensity was calculated in Molecules of Soluble Fluorochrome (MESF) units using Quantum MESF beads (Bangs Laboratories, Inc, Fishers, IN) according to manufacturer’s protocol. All data was analyzed utilizing FlowJo (TreeStar, Inc., Ashland, OR).
For intracellular cytokine staining after infection with Listeria monocytogenes, 3×106 peritoneal exudate cells were cultured for 5 h at 37°C in 1 ml media either without treatment, with 10 ng/ml phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, St. Louis, MO) plus 1 μM ionomycin (Sigma-Aldrich) (positive control), or with 107 cells heat-killed L. monocytogenes. (HKLM). Brefeldin A (BFA) (Sigma-Aldrich) was added (10 μg/ml) after the first 30 min. After LCMV infection, 106 splenocytes were cultured 5-6 h in the absence or presence of the indicated peptide (GP33, GP276, or GP61) plus BFA. For analysis by flow cytometry, extracellular antigens were stained as indicated above, followed by fixation and permeabilization using Fix and Perm cell permeabilization kit (Invitrogen) for intracellular antigens, according to manufacturer’s directions. Antibodies used for intracellular cytokine detection were anti-IFN-γ (clone XMG1.2), anti-IL-2 (clone JES6-5H4), anti-TNF-α (clone MP6-XT22) and appropriate isotype controls (R3-34 and A95-1), staining was done on ice for 30 min. Live cells were gated based on light scatter parameters. The lymphocyte population was gated based on forward and side scatter parameters. All antibodies used were purchased from BD Pharmingen (Franklin Lakes, NJ).
T cell proliferation
To measure MHC-II antigen presentation efficiency, mice were injected in the footpad and base of the tail with an emulsion of Complete Freund’s Adjuvant (CFA) containing 1 mg/ml heat killed Mycobacterium tuberculosis (Difco, Detroit, MI) and the I-Ab specific epitope of LCMV GP61-80 peptide 60. Popliteal and inguinal lymph nodes were collected 12 days later. A single cell suspension was generated, and 5×105 cells were incubated in a 96 well plate with increasing concentrations of peptide for 3 days, with 0.4 μCi/well [3H]thymidine added during the last 24 h. Plates were harvested using a FilterMate harvester (Packard Instrument, Meriden, CT) and 3H-thymidine incorporation was measured on a 1450 LSC Microbeta TriLux counter (PerkinElmer).
Induction of EAE
Experimental autoimmune encephalomyelitis (EAE) was induced in female mice 6-8 weeks of age by subcutaneous injection in the hind flank with 200 μg MOG35-55 peptide emulsified in 200 μl CFA (2.5 mg/ml M. tuberculosis) on days 0 and 7, as previously described 60, 61. On days 0 and 2 mice were given 250 ng pertussis toxin i.p. (List Biological Laboratories, Inc., Campbell, CA). Disease severity was scored according to the following scale: 0, no disease, 1, flaccid tail, 2, hind limb weakness, 3, hind limb paralysis, 4, failure to right (forelimb weakness), 5, moribund.
Peptides
Myelin oligodendrocyte glycoprotein peptide 35-55 (MOG35-55) (MEVGWYRSPFSRVVHLYRNGK) 62, LCMV epitopes GP33-41 (KAVYNFATC), GP276-286 (SGVENPGGYCL), and GP61–80 (GLNGPDIYKGVYQFKSVEFD) 63 peptides were synthesized using standard 9-fluorenylmethyloxycarbonyl chemistry on a Prelude peptide synthesizer (Protein Technologies, Inc).
Listeria monocytogenes infection
A culture of Listeria monocytogenes (strain 10403s) was grown in brain heart infusion (BHI) to OD=0.2, then diluted in phosphate-buffered saline (PBS), the indicated dose was administered in 200 μl. The LD50 for C57BL/6J mice was determined to be 6×106 colony forming units (CFU) (data not shown). Infection dose was verified by plating dilutions of the inoculum and enumerating CFU. Mice were monitored daily for morbidity and mortality. Mice were sacrificed when moribund or weight loss was greater than 25% of initial weight. For enumeration of CFU in organs, spleens and livers were aseptically removed and placed in PBS. Tissue homogenizers were used to generate a single cell suspension, and 1% triton-X100 in PBS was added to final concentration of 0.5% to lyse the cells. Serial dilutions were plated on BHI plates containing 25 μg/ml streptomycin. Colonies were counted the next day, and total CFU/ organ was calculated based on the dilution plated.
LCMV
Acute LCMV infection and cellular analysis were performed as previously described 39. Briefly, mice were infected i.p. with 2×105 pfu LCMV-Armstrong. Mice were serially bled and sacrificed as described in the figure legend. Staining with antibodies and MHC class I and II tetramers complexed with LCMV GP33-41, GP276-286, or GP66-77 for flow cytometric analysis of antigen-specific T cells were performed as previously described 64-66.
Infulenza Immunization
CIITA knockout and wild-type littermate control mice received a single immunization of 5 μg whole inactivated A/California/04/09 influenza virus and they were bled on days 7, 14 and 28. Sera were collected for ELISA and HAI assays. As a negative control group, we included 3 naive wild-type and 2 CIITA knockout mice. Sera were individually collected, and anti-influenza specific antibody levels were determined quantitatively by enzyme-linked immunosorbent assay (ELISA) as described 67, 68. Hemagglutination inhibition (HAI) titers based on the WHO protocol 69 as described previously 67. The HAI titer was read as the reciprocal of the highest dilution of serum that conferred inhibition of hemagglutination. The values were expressed as the geometric mean +/- standard error of the mean.
Quantitative RT-PCR
Total RNA was isolated from cells using RNeasy kit (Qiagen, Valencia, CA). Reverse transcriptase was done using 1 μg of RNA using SuperScrpit II (Invitrogen), RNAse inhibitor (Roche), RQ1 DNase (Promega, Madison, WI), oligo d(T)16, and random hexamers (Applied Biosystems). Real-time PCR using 1/33 of the cDNA product was used to quantify the amount of mRNA. Results were normalized to 18S rRNA or GAPDH mRNA levels and represented using the comparative CT method 70. Data presented represents the average of at least three independent biological replicates, and error bars represent standard error of the mean (SEM). Primers used are listed in Supplemental Table 1.
Supplementary Material
Primer sequences for genotyping, qRT-PCR, 5’RACE, and cloning. Underlined sequences in cloning primers indicates restriction enzyme recognition sequences.
A. Splenic dendritic cells and B. Macrophages treated with IFN-γ 24 h were analyzed by qRT-PCR for the indicated genes identified previously as being regulated by CIITA. Ct values were normalized to the values for actin mRNA and graphed as fold over wild-type. ND: Col1A1 was not detected at significant levels in dendritic cells. C. RNA from control and IFN-γ treated macrophages (24 h) was assayed for the steady state levels of H2-DOb mRNA levels as above. Values are represented as fold over untreated wild-type. Data in this figure were averaged from four independent biological replicates (i.e., four mice), and the statistical significance was calculated using students t-test, ** p<0.01.
Sequences obtained from 5’RACE analysis are shown relative to the predicted transcriptional start sites for each promoter. Colored lines represent transcribed sequence and thicker lines represent translated sequence. The number of transcripts sequenced in each mouse line are shown in the right hand columns.
Mice were immunized and serially bled as described in materials and methods. HAI titer and IgG, IgG1, or IgG2a concentration is plotted for each mouse and each time point.
Acknowledgments
We thank Dr. Joseph Sabatino for help with some of the experiments presented. We also thank the Emory University School of Medicine Core Facility for Flow Cytometry and the NIH tetramer core. This work was supported by the National Institutes of Health grants RO1AI43000 and R56AI34000 (to JMB), PO1 AI080192-01 (to JMB and RA), HHSN266 200700006CMD8 (to IS), and the American Cancer Society postdoctoral fellowship PF-09-134-01-MPC (to BY).
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no competing financial interest in relation to the work describe.
References
- 1.Benacerraf B. Role of MHC gene products in immune regulation. Science. 1981;212(4500):1229–38. doi: 10.1126/science.6165083. [DOI] [PubMed] [Google Scholar]
- 2.Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109(Suppl):S21–33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 3.Reith W, LeibundGut-Landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nature reviews. 2005;5(10):793–806. doi: 10.1038/nri1708. [DOI] [PubMed] [Google Scholar]
- 4.Kanazawa S, Ota S, Sekine C, Tada T, Otsuka T, Okamoto T, et al. Aberrant MHC class II expression in mouse joints leads to arthritis with extraarticular manifestations similar to rheumatoid arthritis. Proc Natl Acad Sci U S A. 2006;103(39):14465–70. doi: 10.1073/pnas.0606450103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krawczyk M, Reith W. Regulation of MHC class II expression, a unique regulatory system identified by the study of a primary immunodeficiency disease. Tissue Antigens. 2006;67(3):183–97. doi: 10.1111/j.1399-0039.2006.00557.x. [DOI] [PubMed] [Google Scholar]
- 6.Reith W, Steimle V, Mach B. Molecular defects in the bare lymphocyte syndrome and regulation of MHC class II genes. Immunol Today. 1995;16(11):539–46. doi: 10.1016/0167-5699(95)80048-4. [DOI] [PubMed] [Google Scholar]
- 7.Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome) Cell. 1993;75(1):135–46. [PubMed] [Google Scholar]
- 8.Choi NM, Majumder P, Boss JM. Regulation of major histocompatibility complex class II genes. Curr Opin Immunol. 2011;23(1):81–7. doi: 10.1016/j.coi.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muhlethaler-Mottet A, Otten LA, Steimle V, Mach B. Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J. 1997;16(10):2851–60. doi: 10.1093/emboj/16.10.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pai RK, Askew D, Boom WH, Harding CV. Regulation of class II MHC expression in APCs: roles of types I, III, and IV class II transactivator. J Immunol. 2002;169(3):1326–33. doi: 10.4049/jimmunol.169.3.1326. [DOI] [PubMed] [Google Scholar]
- 11.Waldburger JM, Suter T, Fontana A, Acha-Orbea H, Reith W. Selective abrogation of major histocompatibility complex class II expression on extrahematopoietic cells in mice lacking promoter IV of the class II transactivator gene. J Exp Med. 2001;194(4):393–406. doi: 10.1084/jem.194.4.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nickerson K, Sisk TJ, Inohara N, Yee CS, Kennell J, Cho MC, et al. Dendritic cell-specific MHC class II transactivator contains a caspase recruitment domain that confers potent transactivation activity. J Biol Chem. 2001;276(22):19089–93. doi: 10.1074/jbc.M101295200. [DOI] [PubMed] [Google Scholar]
- 13.Hofmann K, Bucher P, Tschopp J. The CARD domain: a new apoptotic signalling motif. Trends Biochem Sci. 1997;22(5):155–6. doi: 10.1016/s0968-0004(97)01043-8. [DOI] [PubMed] [Google Scholar]
- 14.Inohara, Chamaillard, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–83. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 15.Ye Z, Ting JP. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr Opin Immunol. 2008;20(1):3–9. doi: 10.1016/j.coi.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28(3):285–7. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harton JA, Linhoff MW, Zhang J, Ting JP. Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol. 2002;169(8):4088–93. doi: 10.4049/jimmunol.169.8.4088. [DOI] [PubMed] [Google Scholar]
- 18.Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nature reviews. 2003;3(5):371–82. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 19.Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the Antimicrobial Response by NLR Proteins. Immunity. 2011;34(5):665–79. doi: 10.1016/j.immuni.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 20.LeibundGut-Landmann S, Waldburger JM, Reis e Sousa C, Acha-Orbea H, Reith W. MHC class II expression is differentially regulated in plasmacytoid and conventional dendritic cells. Nature immunology. 2004;5(9):899–908. doi: 10.1038/ni1109. [DOI] [PubMed] [Google Scholar]
- 21.Waldburger JM, Rossi S, Hollander GA, Rodewald HR, Reith W, Acha-Orbea H. Promoter IV of the class II transactivator gene is essential for positive selection of CD4+ T cells. Blood. 2003;101(9):3550–9. doi: 10.1182/blood-2002-06-1855. [DOI] [PubMed] [Google Scholar]
- 22.Markel P, Shu P, Ebeling C, Carlson GA, Nagle DL, Smutko JS, et al. Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nat Genet. 1997;17(3):280–4. doi: 10.1038/ng1197-280. [DOI] [PubMed] [Google Scholar]
- 23.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nature reviews. 2002;2(3):151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 24.Krawczyk M, Seguin-Estevez Q, Leimgruber E, Sperisen P, Schmid C, Bucher P, et al. Identification of CIITA regulated genetic module dedicated for antigen presentation. PLoS Genet. 2008;4(4):e1000058. doi: 10.1371/journal.pgen.1000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagarajan UM, Bushey A, Boss JM. Modulation of gene expression by the MHC class II transactivator. J Immunol. 2002;169(9):5078–88. doi: 10.4049/jimmunol.169.9.5078. [DOI] [PubMed] [Google Scholar]
- 26.Wong AW, Brickey WJ, Taxman DJ, van Deventer HW, Reed W, Gao JX, et al. CIITA-regulated plexin-A1 affects T-cell-dendritic cell interactions. Nature immunology. 2003;4(9):891–8. doi: 10.1038/ni960. [DOI] [PubMed] [Google Scholar]
- 27.Yee CS, Yao Y, Li P, Klemsz MJ, Blum JS, Chang CH. Cathepsin E: a novel target for regulation by class II transactivator. J Immunol. 2004;172(9):5528–34. doi: 10.4049/jimmunol.172.9.5528. [DOI] [PubMed] [Google Scholar]
- 28.Buttice G, Miller J, Wang L, Smith BD. Interferon-gamma induces major histocompatibility class II transactivator (CIITA), which mediates collagen repression and major histocompatibility class II activation by human aortic smooth muscle cells. Circ Res. 2006;98(4):472–9. doi: 10.1161/01.RES.0000204725.46332.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hornell TM, Burster T, Jahnsen FL, Pashine A, Ochoa MT, Harding JJ, et al. Human dendritic cell expression of HLA-DO is subset specific and regulated by maturation. J Immunol. 2006;176(6):3536–47. doi: 10.4049/jimmunol.176.6.3536. [DOI] [PubMed] [Google Scholar]
- 30.Fallas JL, Yi W, Draghi NA, O’Rourke HM, Denzin LK. Expression patterns of H2-O in mouse B cells and dendritic cells correlate with cell function. J Immunol. 2007;178(3):1488–97. doi: 10.4049/jimmunol.178.3.1488. [DOI] [PubMed] [Google Scholar]
- 31.Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nature reviews. 2009;9(12):833–44. doi: 10.1038/nri2669. [DOI] [PubMed] [Google Scholar]
- 32.Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med. 2009;206(3):549–59. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinterberger M, Aichinger M, da Costa OP, Voehringer D, Hoffmann R, Klein L. Autonomous role of medullary thymic epithelial cells in central CD4(+) T cell tolerance. Nature immunology. 2010;11(6):512–9. doi: 10.1038/ni.1874. [DOI] [PubMed] [Google Scholar]
- 34.van Meerwijk JP, Marguerat S, Lees RK, Germain RN, Fowlkes BJ, MacDonald HR. Quantitative impact of thymic clonal deletion on the T cell repertoire. J Exp Med. 1997;185(3):377–83. doi: 10.1084/jem.185.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnappauf F, Hake SB, Camacho Carvajal MM, Bontron S, Lisowska-Grospierre B, Steimle V. N-terminal destruction signals lead to rapid degradation of the major histocompatibility complex class II transactivator CIITA. Eur J Immunol. 2003;33(8):2337–47. doi: 10.1002/eji.200323490. [DOI] [PubMed] [Google Scholar]
- 36.Oxenius A, Bachmann MF, Ashton-Rickardt PG, Tonegawa S, Zinkernagel RM, Hengartner H. Presentation of endogenous viral proteins in association with major histocompatibility complex class II: on the role of intracellular compartmentalization, invariant chain and the TAP transporter system. Eur J Immunol. 1995;25(12):3402–11. doi: 10.1002/eji.1830251230. [DOI] [PubMed] [Google Scholar]
- 37.Suter T, Malipiero U, Otten L, Ludewig B, Muelethaler-Mottet A, Mach B, et al. Dendritic cells and differential usage of the MHC class II transactivator promoters in the central nervous system in experimental autoimmune encephalitis. Eur J Immunol. 2000;30(3):794–802. doi: 10.1002/1521-4141(200003)30:3<794::AID-IMMU794>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 38.Freeman MM, Ziegler HK. Simultaneous Th1-type cytokine expression is a signature of peritoneal CD4+ lymphocytes responding to infection with Listeria monocytogenes. J Immunol. 2005;175(1):394–403. doi: 10.4049/jimmunol.175.1.394. [DOI] [PubMed] [Google Scholar]
- 39.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77(8):4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karlsson L, Surh CD, Sprent J, Peterson PA. A novel class II MHC molecule with unusual tissue distribution. Nature. 1991;351(6326):485–8. doi: 10.1038/351485a0. [DOI] [PubMed] [Google Scholar]
- 41.Taxman DJ, Cressman DE, Ting JP. Identification of class II transcriptional activator-induced genes by representational difference analysis: discoordinate regulation of the DN alpha/DO beta heterodimer. J Immunol. 2000;165(3):1410–6. doi: 10.4049/jimmunol.165.3.1410. [DOI] [PubMed] [Google Scholar]
- 42.Chang CH, Guerder S, Hong SC, van Ewijk W, Flavell RA. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity. 1996;4(2):167–78. doi: 10.1016/s1074-7613(00)80681-0. [DOI] [PubMed] [Google Scholar]
- 43.Nagarajan UM, Lochamy J, Chen X, Beresford GW, Nilsen R, Jensen PE, et al. Class II transactivator is required for maximal expression of HLA-DOB in B cells. J Immunol. 2002;168(4):1780–6. doi: 10.4049/jimmunol.168.4.1780. [DOI] [PubMed] [Google Scholar]
- 44.Smith MA, Wright G, Wu J, Tailor P, Ozato K, Chen X, et al. Positive regulatory domain I (PRDM1) and IRF8/PU.1 counter-regulate MHC class II transactivator (CIITA) expression during dendritic cell maturation. J Biol Chem. 2011;286(10):7893–904. doi: 10.1074/jbc.M110.165431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi YE, Yu HN, Yoon CH, Bae YS. Tumor-mediated down-regulation of MHC class II in DC development is attributable to the epigenetic control of the CIITA type I promoter. Eur J Immunol. 2009;39(3):858–68. doi: 10.1002/eji.200838674. [DOI] [PubMed] [Google Scholar]
- 46.Ghosh N, Piskurich JF, Wright G, Hassani K, Ting JP, Wright KL. A novel element and a TEF-2-like element activate the major histocompatibility complex class II transactivator in B-lymphocytes. J Biol Chem. 1999;274(45):32342–50. doi: 10.1074/jbc.274.45.32342. [DOI] [PubMed] [Google Scholar]
- 47.van der Stoep N, Quinten E, van den Elsen PJ. Transcriptional regulation of the MHC class II trans-activator (CIITA) promoter III: identification of a novel regulatory region in the 5’-untranslated region and an important role for cAMP-responsive element binding protein 1 and activating transcription factor-1 in CIITA-promoter III transcriptional activation in B lymphocytes. J Immunol. 2002;169(9):5061–71. doi: 10.4049/jimmunol.169.9.5061. [DOI] [PubMed] [Google Scholar]
- 48.van der Stoep N, Quinten E, Rezende MM, van den Elsen PJ. E47, IRF-4, and PU. 1 synergize to induce B-cell-specific activation of the class II transactivator promoter III (CIITA-PIII) Blood. 2004;104(9):2849. doi: 10.1182/blood-2004-03-0790. [DOI] [PubMed] [Google Scholar]
- 49.Yoon H, Boss JM. PU.1 binds to a distal regulatory element that is necessary for B cell-specific expression of CIITA. J Immunol. 2010;184(9):5018–28. doi: 10.4049/jimmunol.1000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris AC, Beresford GW, Mooney MR, Boss JM. Kinetics of a gamma interferon response: expression and assembly of CIITA promoter IV and inhibition by methylation. Mol Cell Biol. 2002;22(13):4781–91. doi: 10.1128/MCB.22.13.4781-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muhlethaler-Mottet A, Di Berardino W, Otten LA, Mach B. Activation of the MHC class II transactivator CIITA by interferon-gamma requires cooperative interaction between Stat1 and USF-1. Immunity. 1998;8(2):157–66. doi: 10.1016/s1074-7613(00)80468-9. [DOI] [PubMed] [Google Scholar]
- 52.Piskurich JF, Linhoff MW, Wang Y, Ting JP. Two distinct gamma interferon-inducible promoters of the major histocompatibility complex class II transactivator gene are differentially regulated by STAT1, interferon regulatory factor 1, and transforming growth factor beta. Mol Cell Biol. 1999;19(1):431–40. doi: 10.1128/mcb.19.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris AC, Spangler WE, Boss JM. Methylation of class II trans-activator promoter IV: a novel mechanism of MHC class II gene control. J Immunol. 2000;164(8):4143–9. doi: 10.4049/jimmunol.164.8.4143. [DOI] [PubMed] [Google Scholar]
- 54.van Eggermond MC, Boom DR, Klous P, Schooten E, Marquez VE, Wierda RJ, et al. Epigenetic regulation of CIITA expression in human T-cells. Biochem Pharmacol. 2011 doi: 10.1016/j.bcp.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 55.Ni Z, Abou El Hassan M, Xu Z, Yu T, Bremner R. The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nature immunology. 2008;9(7):785–93. doi: 10.1038/ni.1619. [DOI] [PubMed] [Google Scholar]
- 56.Estill SJ, Garcia JA. A marker assisted selection protocol (MASP) to generate C57BL/6J or 129S6/SvEvTac speed congenic or consomic strains. Genesis. 2000;28(3-4):164–6. doi: 10.1002/1526-968x(200011/12)28:3/4<164::aid-gene110>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 57.Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT. The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res. 2011;39(Database issue):D842–8. doi: 10.1093/nar/gkq1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inaba K, Swiggard WJ, Steinman RM, Romani N, Schuler G, Brinster C. Isolation of dendritic cells. Curr Protoc Immunol. 2009;Chapter 3:Unit 3 7. doi: 10.1002/0471142735.im0307s86. [DOI] [PubMed] [Google Scholar]
- 59.Zhang B, Zhang Y, Niu L, Vella AT, Mittler RS. Dendritic cells and Stat3 are essential for CD137-induced CD8 T cell activation-induced cell death. J Immunol. 2010;184(9):4770–8. doi: 10.4049/jimmunol.0902713. [DOI] [PubMed] [Google Scholar]
- 60.Ford ML, Evavold BD. Regulation of polyclonal T cell responses by an MHC anchor-substituted variant of myelin oligodendrocyte glycoprotein 35-55. J Immunol. 2003;171(3):1247–54. doi: 10.4049/jimmunol.171.3.1247. [DOI] [PubMed] [Google Scholar]
- 61.Sabatino JJ, Jr, Shires J, Altman JD, Ford ML, Evavold BD. Loss of IFN-gamma enables the expansion of autoreactive CD4+ T cells to induce experimental autoimmune encephalomyelitis by a nonencephalitogenic myelin variant antigen. J Immunol. 2008;180(7):4451–7. doi: 10.4049/jimmunol.180.7.4451. [DOI] [PubMed] [Google Scholar]
- 62.Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25(7):1951–9. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- 63.Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med. 2001;7(8):913–9. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 64.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 65.Fuller MJ, Hildeman DA, Sabbaj S, Gaddis DE, Tebo AE, Shang L, et al. Cutting edge: emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J Immunol. 2005;174(10):5926–30. doi: 10.4049/jimmunol.174.10.5926. [DOI] [PubMed] [Google Scholar]
- 66.Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. 2009;324(5934):1569–72. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skountzou I, Quan FS, Jacob J, Compans RW, Kang SM. Transcutaneous immunization with inactivated influenza virus induces protective immune responses. Vaccine. 2006;24(35-36):6110–9. doi: 10.1016/j.vaccine.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 68.Koutsonanos DG, del Pilar Martin M, Zarnitsyn VG, Sullivan SP, Compans RW, Prausnitz MR, et al. Transdermal influenza immunization with vaccine-coated microneedle arrays. PLoS One. 2009;4(3):e4773. doi: 10.1371/journal.pone.0004773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.WHO/CDS/CSR/NCS. Response DoCDSa. 2002. WHO Manual on Animal Influenza Diagnostics and Surveillance. [Google Scholar]
- 70.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer sequences for genotyping, qRT-PCR, 5’RACE, and cloning. Underlined sequences in cloning primers indicates restriction enzyme recognition sequences.
A. Splenic dendritic cells and B. Macrophages treated with IFN-γ 24 h were analyzed by qRT-PCR for the indicated genes identified previously as being regulated by CIITA. Ct values were normalized to the values for actin mRNA and graphed as fold over wild-type. ND: Col1A1 was not detected at significant levels in dendritic cells. C. RNA from control and IFN-γ treated macrophages (24 h) was assayed for the steady state levels of H2-DOb mRNA levels as above. Values are represented as fold over untreated wild-type. Data in this figure were averaged from four independent biological replicates (i.e., four mice), and the statistical significance was calculated using students t-test, ** p<0.01.
Sequences obtained from 5’RACE analysis are shown relative to the predicted transcriptional start sites for each promoter. Colored lines represent transcribed sequence and thicker lines represent translated sequence. The number of transcripts sequenced in each mouse line are shown in the right hand columns.
Mice were immunized and serially bled as described in materials and methods. HAI titer and IgG, IgG1, or IgG2a concentration is plotted for each mouse and each time point.