

Abstract
Over-expression of MYC transforms cells in culture, elicits malignant tumors in experimental animals and is found in many human tumors. We now report the paradoxical finding that this powerful oncogene can also act as a suppressor of cell motility, invasiveness and metastasis. Overexpression of MYC stimulated proliferation of breast cancer cells both in culture and in vivo as expected, but inhibited motility and invasiveness in culture, and lung and liver metastases in xenografted tumors. We show further that MYC represses transcription of both subunits of αvβ3 integrin, and that exogenous expression of β3 integrin in human breast cancer cells that do not express this integrin rescues invasiveness and migration when MYC is downregulated. These data uncover an unexpected function of MYC, provide an explanation for the hitherto puzzling literature on the relationship between MYC and metastasis and reveal a variable that should influence the development of therapeutics that target MYC.
The proto-oncogene MYC encodes an exceptionally pleiotropic transcription factor (MYC) that participates in the control of a wide variety of genes1-3. Included among these genes are functions vital to regulation of the cell cycle, cell growth, apoptosis, cell adhesion, and genomic stability3-5. Over-expression of MYC transforms cells in culture6, elicits tumors in experimental animals7, and is found in as many as 50% of all human cancers and 25% of human breast cancers8-13. Paradoxically, such overexpression is on occasion dissociated from the propensity to invade and metastasize10,13-18. These observations raise the possibility that MYC may inhibit cellular properties such as motility and invasion that are essential to metastasis. Consistent with this possibility, overexpression of MYC in mouse skin causes a severe impairment in wound healing and in keratinocyte migration, whereas deletion of MYC causes increased migration of keratinocytes19,20.
Cellular invasion and migration are governed by extracellular and intracellular signals, and depend on the interaction of the cell with ligand molecules in the extracellular matrix (ECM)21-23. The principal ECM receptors are the integrins, and altered expression of integrins is associated with tumor growth and metastasis24,25. Overexpression of MYC was found to inhibit the spreading and adhesion of primary keratinocytes, which exhibited decreased expression of α6, β1 and β4 integrins in response to MYC26. When MYC was overexpressed in cells from neuroblastomas and sarcomas, the levels of α3 and β1 integrins were down-modulated27,28, whereas genetic ablation of MYC in hematopoietic stem cells stimulated the expression of α2, α5, and β1 integrins, and other ECM proteins29. In addition, MYC binds to the E-box sequence of the promoters of the α3 and α7 integrin genes and silences their transcription in mouse myoblast and human sarcoma cells27,30.
We report here that elevated expression of MYC reduced the motility and invasiveness of breast cancer cells in vitro, their capacity for local invasion, and their ability to seed distant metastases. Concomitantly, MYC overexpression inhibited the formation of stress fibers and focal adhesions. These effects of MYC could be attributed to the decreased expression of αv and β3 integrins, mediated by repression of transcription from the corresponding genes. Our results provide an explanation for the dissociation between overexpression of MYC and metastasis, point to αv and β3 integrins as crucial elements in the metastasis of malignant cells, and uncover a variable that may be important in the development of therapeutics that target MYC.
RESULTS
MYC inhibits invasion and metastasis
We explored the role of MYC in metastasis by manipulating expression of the gene in four established cell lines of human breast cancer, two of which express little MYC (MDA-MB-231 and BD549), and two of which express MYC at high levels (MCF-7 and T47D) (Fig. 1a), and in human retinal pigment epithelium (RPE) cells, a cell line that has been used previously for investigating the effects of MYC on induction of proliferation and apoptosis.
Figure 1.
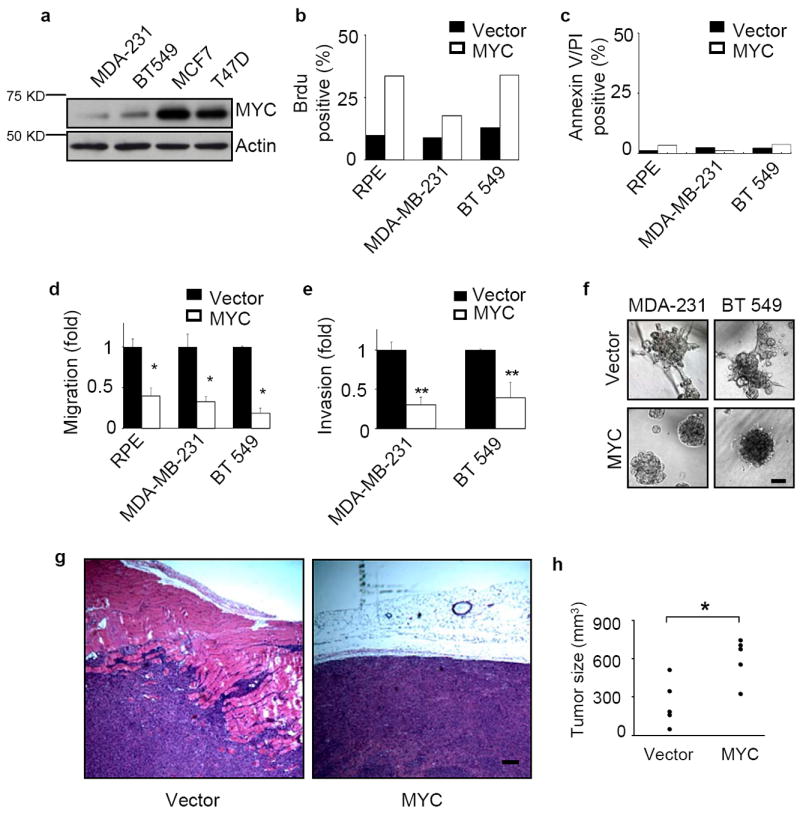
Elevated MYC expression impedes the invasiveness of human breast cancer cells. (a) MYC expression levels in four breast cancer cell lines. The endogenous MYC expression levels of MDA-MB-231, BT549, MCF7, and T47D cells were measured by western blots. (b-c) Ectopic overexpression of MYC increases proliferation but does not affect apoptosis. Cells were labeled with Brdu on tissue culture plastic for 30 minutes at 37°C (b, n=2) or incubated with annexin V and propidium iodide (PI) for 5 minutes at room temperature (c, n=2). Both positive and total nuclei were counted and the results are expressed as mean±SD, * p<0.05. (d) MYC overexpression inhibits cell migration. Migration of MDA-MB-231, BT549 and RPE cells was measured in Boyden-chamber assays (n=3 for each experiment). The cells were transfected with either vector control or a MYC construct. (e) The invasiveness of breast cancer cells is inhibited by MYC overexpression. Cell invasion through Matrigel-coated transwells was measured for MDA-MB-231 (n=3) and BT549 (n=3) cells stably transduced with vector control or exogenous MYC. (f) High level of MYC expression abrogates the invasive phenotype of breast cancer cells grown in 3D Matrigel. Results are shown for day 6. Scale bars: 50 μm. (g-h) MYC overexpression enhances tumor growth but reduces tumor invasion into nearby tissues. MDA-MB-231 cells stably expressing vector or exogenous MYC were inoculated subcutaneously into nude mice. The tumors were collected 4 weeks after injection, sectioned and stained with hematoxilin and eosin (H&E, g) and assessed for size (h, n=5 for each group, p<0.01). Scale bar: 200 μm. Results are expressed as mean± SD. * p<0.05; ** p<0.01.
Exogenous overexpression of human MYC in MDA-MB-231, BT549, and RPE cells significantly increased proliferation assessed by BrdU labeling (Fig. 1b) without enhancing basal levels of cell death (Fig. 1c). However, overexpression of MYC significantly inhibited migration of these cells (Fig. 1d) and reduced invasiveness of MDA-MB-231 and BT549 cells (Fig. 1e). Overexpression of MYC in MDA-MB-231 and BT549 cells also inhibited the invasive growth pattern of these cells when cultured in three-dimensional, laminin-rich gels (lrECM) such as Matrigel (Fig. 1f). When injected subcutaneously, MYC-expressing MDA-MB-231 cells produced tumors that showed substantially reduced local invasion (Fig. 1g), even though the expression of MYC led to significantly larger tumors (Fig. 1h). These results showed that overexpression of MYC simultaneously promotes cell proliferation while inhibiting invasion, both in culture and in vivo.
Invasion and metastasis are both characteristics of cancer progression. We found that MDA-MB-231 cells overexpressing MYC showed significantly reduced incidence of lung metastases following injection into the tail vein (Fig. 2a,b), although occasional MYC-expressing cells that did metastasize resulted in larger tumors (Fig. 2c). To assess tumor growth and metastasis from the orthotopic site, MDA-MB-231-vector and MDA-MB-231-MYC cells were transduced with luciferase-expressing virus (Fig. 2d) and injected into the inguinal mammary gland. We found that the MYC-expressing tumors, as expected, grew more rapidly at the orthotopic site (Fig. 2e-g). However, isolation and quantification of luminescence of lungs (Fig. 2h,i) and livers (Fig. 2m,n) revealed decreased metastatic burden at 6 weeks following injection, though the difference was not statistically significant. Metastatic burden is a combination of both number of metastases and the size of the metastatic tumors; we found that expression of MYC significantly reduced the number of metastases but led to increased size of individual metastases in both lung (Fig. 2j-l) and liver (Fig. 2o-q). We conclude that overexpression of MYC can stimulate cell proliferation producing larger tumors that have reduced invasiveness and metastatic potential. These findings are consistent with recent indications that dissemination of tumor cells may occur early during tumor progression and is not necessarily linked to primary tumor size31.
Figure 2.
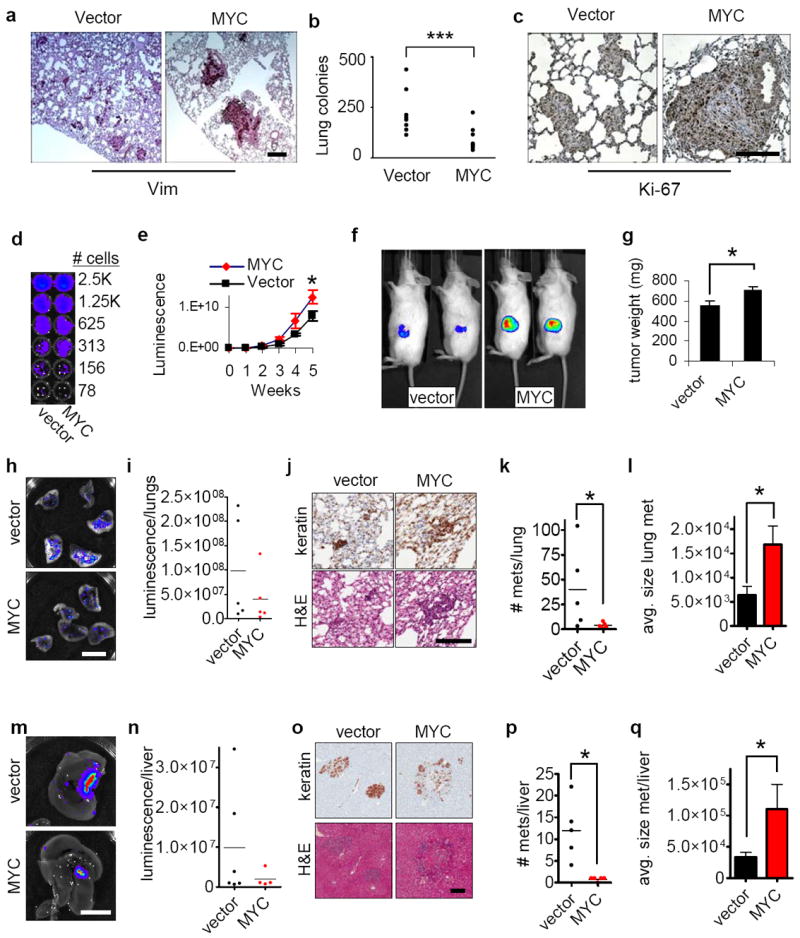
Elevated MYC expression inhibits metastasis of human breast cancer cells. (a-c) Over-expression of MYC significantly decreases lung metastases of breast cancer cells. MDA-MB-231 cells stably expressing vector or ectopic MYC were injected into nude mice through the tail vein and lung metastases were assessed 6 weeks after injection. Sections of the lungs were stained with antibody against human vimentin (a) and quantification revealed that MYC significantly reduced lung metastasis (b, n=9, p<0.0049). Assessment of Ki-67 (c) revealed increased proliferation in the MYC-expressing cells. Scale bars: 200 μm. (d) Luminescence detection shows comparable luminescence per cell for both vector and MYC over-expressing cells. (e) MYC-expressing cells show more rapid growth at the primary tumor site, assessed by quantitative in vivo imaging (n=5 for each group). (f) Sample images from in vivo imaging of primary tumors for vector and MYC (week 6). (g) Increased size of primary tumors from MYC-expressing cells (week 6; n=5 for each group). (h-i) Decreased metastatic burden in lungs of mice implanted with MYC-expressing cells, assessed by luminescence (week 6; h, sample images, scale bar 1 cm; i, quantification of lung luminescence; n=5 for each group; differences between cells expressing vector alone and those expressing MYC were not statistically significant). (j) Images of lung metastases, stained for human cytokeratins (top), and with H&E (bottom). scale bar=200 μm. (k-l) Quantification of number (k) and size (l) of lung metastases indicates that MYC cells form much fewer metastases but grow to larger size (n=5 for each group). (m-n) Decreased metastatic burden in livers of mice implanted with MYC-expressing cells (week 6; m, sample images, scale bar 1 cm; n, quantification of liver luminescence; differences between conditions were not statistically significant; n=6 for vector, n=5 for MYC). (o) Images of liver metastases, stained for human cytokeratins (top), and with H&E (bottom). Scale bar=200 μm. (q-r) Quantification of number (p) and size (q) of liver metastases indicates that MYC-expressing cells form fewer metastases that grow to larger size (n=5 for each group). Results are expressed as mean± SEM. *, p<0.05; **, p<0.01; ***, p<0.005.
MYC modulates cell interaction with the extracellular matrix
Motility is a dynamic process associated with major changes in cellular phenotype including spreading, actin polymerization, formation of actin-rich protrusions at the leading edge of migrating cells, and creation and dissolution of focal adhesions during cell translocation32,33. We found that exogenous overexpression of MYC in both MDA-MB-231 and RPE cells (Fig. 3a,b) reduced cell spreading, decreased stress fiber and focal adhesion formation, and increased cortical actin (Fig. 3c,d). Since formation of focal adhesions requires the interaction of cell surface integrins with their ligands in the extracellular matrix (ECM), we hypothesized that the effect of overexpression of MYC on focal adhesion might reflect a defect in the interaction of cells with specific ECM ligands. To test this hypothesis, we assessed cell attachment to purified human ECM ligands and found that overexpression of MYC in MDA-MB-231 or RPE cells reduced their attachment to vitronectin (Fig. 3e,f). Such defects could in turn impair cellular motility. To explore this possibility, we measured chemotaxis towards purified vitronectin in Boyden chamber assays, and found that overexpression of MYC in MDA-MB-231 and RPE cells decreased migration of both cell types (Fig. 3g,h).
Figure 3.
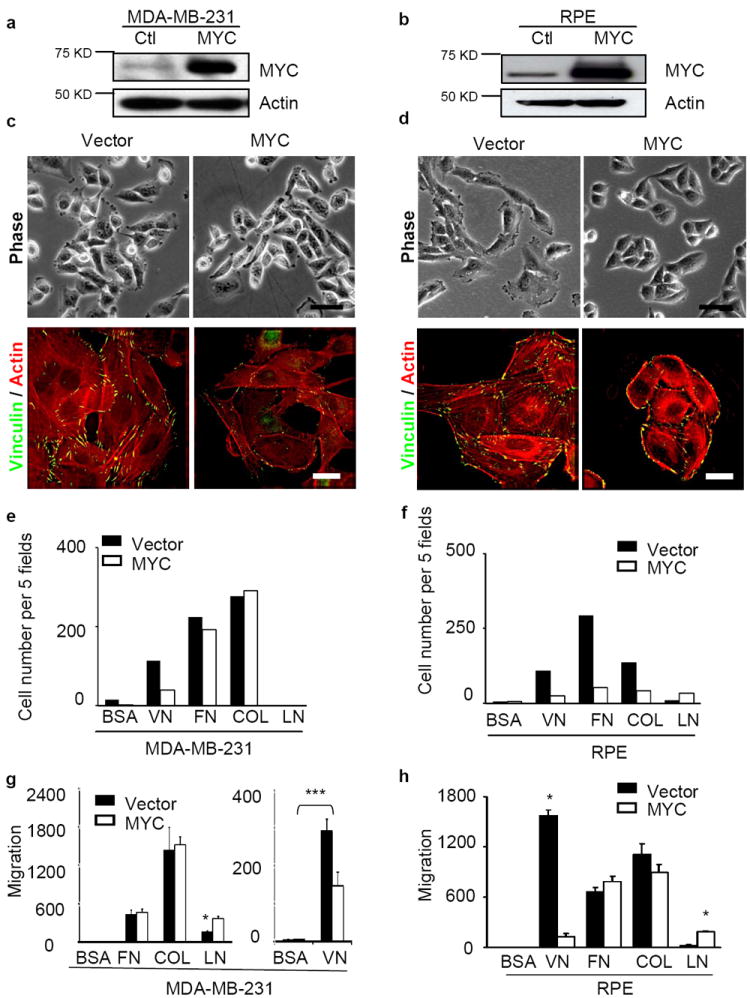
MYC modulates cell shape, actin cytoskeleton, focal adhesion formation, adhesion to and migration towards ECM. (a-b) Overexpression of ectopic MYC in MDA-MB-231 (a) and RPE (b) cells assessed by western blot. (c-d) MYC overexpression inhibits cell spreading, stress fiber and focal adhesion formation. MDA-MB-231 (c) and RPE (d) cells expressing vector or ectopic MYC were cultured for 24 hr, and then stained with anti-vinculin antibody (green) and Texas-Red-conjugated Phalloidin (red). Scale bars: 20 μm for phase contrast and 5 μm for immunofluorescence staining. (e-f) MYC overexpression reduces cell adhesion to ECM. MDA-MB-231 (e) and RPE (f) cells in serum-free medium were plated into 96-well plates coated with purified matrix proteins (VN: vitronectin, FN: fibronectin, Col I: collagen I, LN: laminin; n=2). After 45 minutes, adhered cells were counted in five fields. (g-h) Increased MYC expression blocks cell migration towards vitronectin in a Boyden-chamber assay of MDA-MB-231 (g) and RPE (h) cells (n=3). The results of cell adhesion assay are expressed as mean± SD, *, p<0.05.
MYC inhibits expression of αv and β3 integrins
To explore the mechanism by which MYC impedes invasion and motility, we measured the expression of subunits of αvβ3 integrin, which is a well-known cell surface receptor for vitronectin34. Overexpression of MYC in RPE, MDA-MB-231 and BT549 cells was accompanied by substantial reductions in both integrin subunits in all three cell lines, and less consistent reductions of α5, β1 and β5 subunits (Fig. 4a). Conversely, when the high levels of endogenous MYC in MCF7 and T47D cells were knocked down by siRNA, the levels of αv, α5, β1 and β5 integrin subunits rose (Fig. 4a and data not shown). Irrespective of the level of MYC expression, β3 integrin could not be detected by either RT-PCR or western blots in MCF7 cells35 (Fig. 4a, and data not shown), nor in T47D cells (see below, Fig. 6c). Hence, both cell lines appear to be intrinsically deficient in β3.
Figure 4.
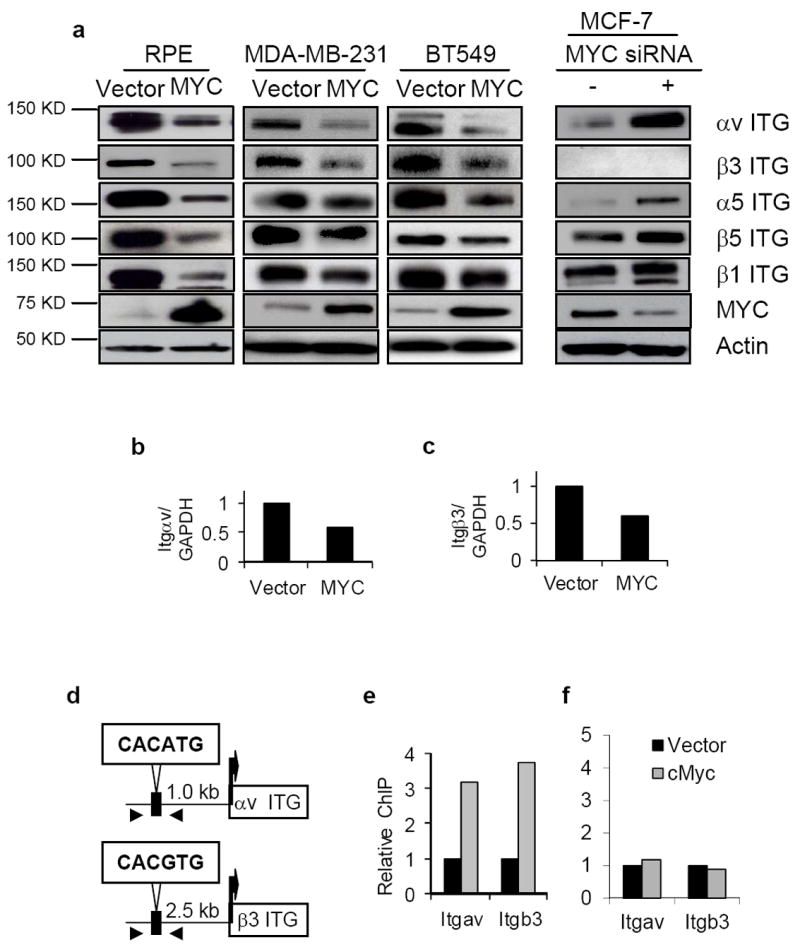
MYC down-regulates the expression of αv and β3 integrin genes through binding to their proximal promoters. (a) MYC modulates the abundance of αv and β3 integrins. Total lysates of the indicated cells were fractionated and immunoblotted with the antibodies against the integrins as shown. (b-c) Quantitative PCR assessment of integrin αv (Itgav; b) and β3 (Itgb3; c) expression in response to MYC expression in MDA-MB-231 cells (n=2 for each experiment). (d) Schematic illustration of the E-box motif upstream of αv and β3 integrin genes; arrowheads locate the designed primers for ChIP assay. (e-f) MYC binding to the proximal promoter of αv and β3 integrin genes as determined by ChIP assay. Cross-linked nuclear extracts of MDA-MB-231 transduced with vector only (black columns) or MYC (grey columns) were immunoprecipitated by either anti-MYC antibody or a control IgG. The regions of the MYC binding sites (e) or nonspecific sites (f) upstream of αv or β3 integrin genes were quantified and normalized to IgG pulldown (n=2 for each experiment). Data are expressed as mean± SEM. *, p<0.05; **, p<0.01; ***, p<0.005.
Figure 6.
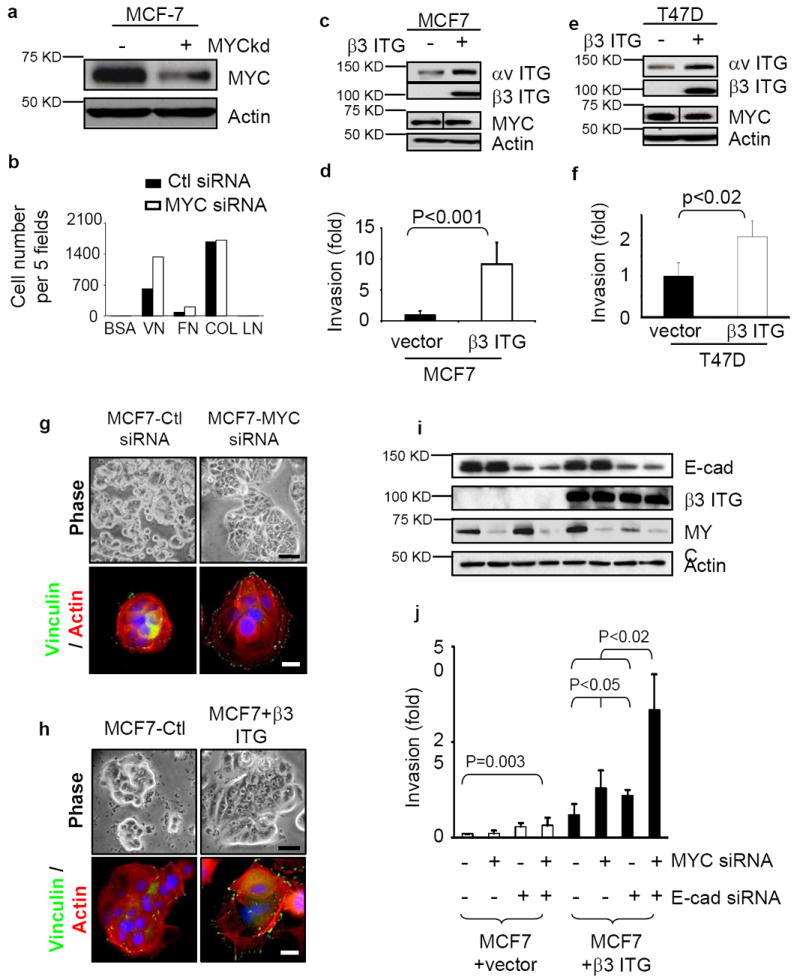
MYC and E-cadherin can prevent β3 integrin-induced invasion. (a) Suppression of MYC expression by siRNA in MCF7 cells assessed by western blot. (b) Suppression of MYC by siRNA augments cell adhesion to vitronectin and fibronectin. The cells in serum-free medium were plated into 96-well plates coated with purified matrix proteins (VN: vitronectin, FN: fibronectin, Col I: collagen I, LN: laminin). After 45 minutes, adhered cells were counted in five fields, n=2. (c-f) Ectopic expression of β3 integrin in MCF-7 (c) and T47D (e) cells, assessed by Western blot, enhanced invasiveness (d,f; n=3), as assessed by Boyden chamber assay. (g-h) Cell spreading, stress fiber and focal adhesion formation are enhanced in MCF7 cells when MYC is depleted by siRNA (g) or when β3 integrin is exogenously expressed (h). Changes in cell shape, actin cytoskeleton and focal adhesion formation were demonstrated by phase contrast (top) and staining with anti-vinculin (green) and Texas red Phalloidin (bottom). Scale bar: 20 μm for phase contrast and 5 μm for immunofluorescence staining. (i-j) Decreased expression of MYC and E-cadherin by siRNA increased the invasiveness of MCF7 cells only when β3 integrin was expressed. The depletion of MYC and E-cadherin was assessed by western blot (i) and invasiveness was assessed by Boyden chamber assays (j; n=3). Results are expressed as mean± SD.
Overexpression of MYC in MDA-MB-231 cells caused a reduction of RNA for both αv and β3 (Fig. 4b,c). A canonical E-box binding site for MYC is located upstream of the transcription initiation sites for both αv (CACATG) and β3 (CACGTG) integrins in human DNA (Fig. 4d). Quantitative chromatin immunoprecipitation assays (ChIP) showed that MYC bound directly to the E-box region of both these genes in MDA-MB-231 cells (Fig. 4e,f) and in RPE cells (not shown), but not to non-specific sequences in the same domain. Although MYC was originally viewed as a transcriptional activator, it can serve also as a transcriptional repressor36. Our results show that overexpression of MYC represses transcription of integrin genes involved in migration and metastasis.
Inhibition of invasion and metastasis by MYC depends upon αv and β3 integrin modulation
We found that the invasive and migratory phenotype of MDA-MB-231 and RPE cells was dependent on αvβ3 integrin (Fig. 5a,b, and data not shown) as was the invasive growth of MDA-MB-231 cells in lrECM (Fig. 5c), and that defects in cell invasion and migration caused by overexpression of MYC could be partially or completely rescued by expressing αv and β3 integrins in MDA-MB-231 (Fig. 5d), RPE (Fig. 5e), or BT549 cells (Fig. 5f). Invasive growth of MDA-MB-231 in 3D lrECM and cell spreading, focal adhesion and stress fiber formation in RPE cells were rescued as well (Fig. 5g). To evaluate whether exogenous overexpression of αv and β3 integrins was sufficient to reconstitute metastatic capability in cells also overexpressing MYC, we generated MYC-expressing MDA-MB-231 cells that also expressed integrin αv, and integrin β3 using exogenous promoters unaffected by MYC (Fig. 5h). When these cells were implanted orthotopically, ectopic expression of integrins αv and β3 substantially increased lung metastasis (Fig. 5i,j). These results indicate that suppression of integrin αvβ3 expression is the key mechanism by which MYC inhibits breast cancer cell metastasis.
Figure 5.
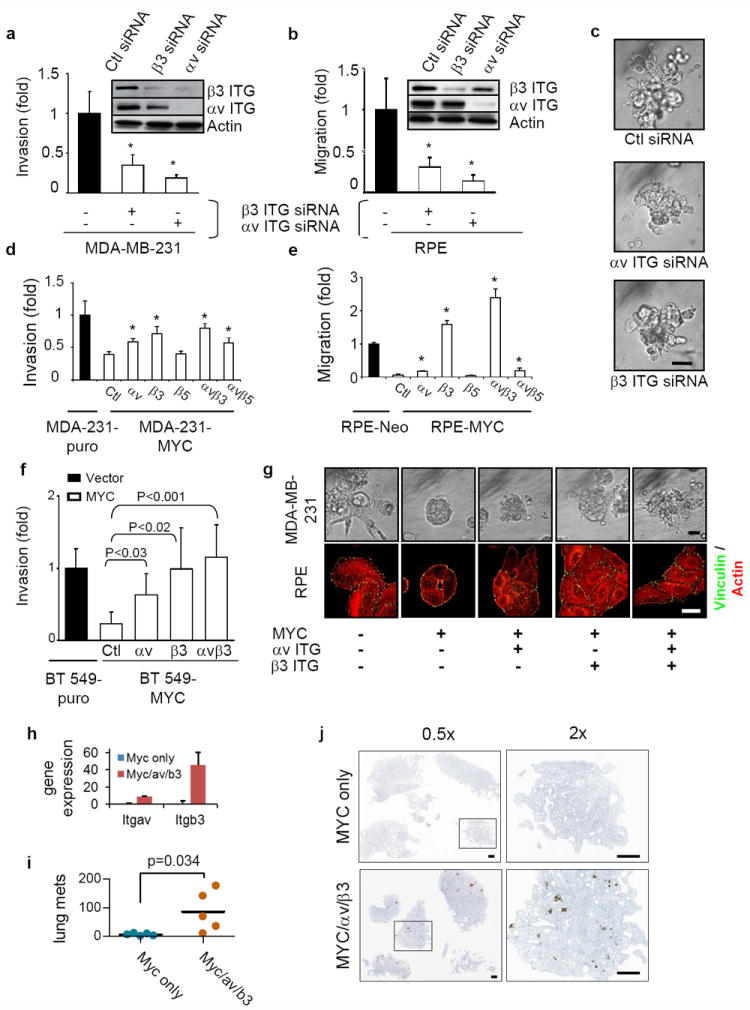
MYC affects breast cancer cell invasiveness by suppressing integrin αv and β3 subunits. (a-b) Knockdown of αv or β3 integrin inhibited invasion (a) and migration (b) in a Boyden-chamber assay, n=3. Knockdown of the integrins by siRNA was confirmed by western blots. (c) Knockdown of αv or β3 integrin inhibits the invasiveness of MDA-MB-231 cells grown in a 3D Matrigel assay for 6 days, n=3. Scale bar: 50μm. (d-e) αv and β3 integrin rescues the compromised migration (d) and invasiveness-(e) elicited by high MYC expression, n=3 for each. MDA-MB-231 and RPE cells overexpressing MYC were transiently transfected with vector, αv, β3, or β5 integrin constructs. Cell invasiveness and migration were assessed by Boyden chamber assay. (f) Inhibition of cancer cell invasiveness by MYC over-expression can be rescued by exogenous expression of αv and β3 integrin subunits in BT549 cells (n=3). (g) Expression of exogenous αv and β3 integrin partially rescued actin cytoskeleton, focal adhesion formation of RPE cells grown on 2D tissue culture plastic dishes, and the compromised invasiveness of MDA-MB-231 cells in a 3D Matrigel assay. RPE or MDA-MB-231 cells, stably expressing the indicated constructs, were plated on cell culture dishes for 24 hours or in 3D Matrigel for 6 days. RPE cells were then stained with anti-vinculin antibody (green) and Texas-Red-conjugated Phalloidin (red). Images of 3D Matrigel culture were obtained by phase contrast microscopy. Scale bars: 50 μm for phase contrast and 5 μm for immunofluorescence staining. (h) Quantitation of Itgav and Itgb3 transcripts by quantitative PCR in MDA/MYC and MDA/MYC/αv/β3 cells, n=3. (i) Quantitation of increased lung metastases in mice orthotopically implanted with MDA/MYC/αvb3 cells as compared to MDA/MYC cells, n=5 for each. (j) Images of lungs of mice orthotopically implanted with MDA/MYC/αvβ3 or MDA/MYC cells. Results are expressed as mean± SD. *, p<0.05.
We noticed that depletion of either αv or β3 integrin resulted in a parallel reduction of the other (Figure 5a,b); conversely, exogenous expression of β3 integrin increased the amount of αv integrin (see below, Fig. 6a,b, and Supplementary Figs. 1 and 2). Moreover, exogenous expression of either integrin at least partially rescued the phenotype suppressed by MYC (Fig. 5d-g). These findings could be explained by mass action, wherein an increase in either integrin alone can augment the formation of heterodimers and stabilize both components. We explored this possibility by co-immunoprecipitation to detect the formation of heterodimers. We found that transfection of αv and β3, either individually or together, substantially increased the amount of heterodimers in extracts of MDA-MB-231 cells (Supplementary Fig. 1). The relative amounts of heterodimer correlated with the extent to which exogenous expression of the integrins rescued a defect in invasion caused by MYC (compare Fig. 5d with Supplementary Fig. 1). We conclude that the defects in motility and invasion elicited by overexpression of MYC are due to reduced expression of the αv and β3 integrin subunits, which in turn reduces formation of the heterodimer.
MYC prevents β3 integrin-induced cell invasion
We also explored the effect of depleting the high level of MYC in MCF-7 cells with RNAi (Fig. 6a). Depletion of MYC significantly reduced cellular proliferation as expected (data not shown), whereas it increased cell adhesion to vitronectin and fibronectin (Fig. 6b), as well as cell spreading, focal adhesion and stress fiber formation (Fig. 6g). However, knockdown of MYC in these cells neither increased cell motility (data not shown) nor invasiveness (Fig. 6j), implicating the absence of β3 integrin in MCF7 cells and the consequent inability to form αvβ3 heterodimers. Accordingly, exogenous expression of β3 integrin in MCF-7 cells (Fig. 6c,i) increased invasiveness (Fig. 6d,j) as well as cell spreading (Fig. 6h); similar effects were seen with exogenous expression of β3 integrin in T47D cells (Fig. 6e,f and data not shown).
MCF7 cells grow in a cuboidal morphology with tight cell-cell junctions that restrict individual cell motility. In order to isolate the inhibitory effect of MYC on cell motility, we simultaneously knocked down E-cadherin and MYC, and expressed exogenous β3 integrin in MCF7 cells (Fig. 6i). This combination greatly increased invasion (Fig. 6j). We conclude that when cell-cell interactions are abrogated, inhibition of αv and β3-integrin expression is the primary barrier to cell invasion in MCF7 and possibly other human breast cancer cells that overexpress MYC.
DISCUSSION
MYC has been implicated extensively in tumor growth and cell transformation through studies in cultured cells6,37, targeted expression in mice38-40 and retrospective analyses of MYC expression in human tumors8. MYC is highly pleiotropic and plays multiple biological roles in driving tumorigenesis. Examples include the ability to initiate tumorigenesis in mice7, destabilization of the genome4 and a requirement for maintenance of established tumors41. To this list we add here a previously unrecognized effect of MYC: ability to inhibit metastasis through inhibition of transcription of an integrin involved in metastasis.
The possibility that MYC could act to inhibit metastasis could have been surmised by the observations that overexpression of the gene is sometimes dissociated from the tendency of tumors to metastasize13,15,18. Why this may be the case, however, had not been explored. In the case of human breast cancer, distal metastases may express MYC at the level of cognate normal cells, even though the gene is over-expressed in the primary tumor13. Similarly, mammary carcinoma and other tumors induced by MYC in mice frequently fail to metastasize. Examples include murine mammary cancer elicited by MYC under the control of the mouse mammary tumor virus (MMTV) promoter15,17 and aggressive breast tumors elicited by retroviral transduction of both MET and MYC into normal breast epithelial cells18.
Here we explored the impact of MYC on metastasis by using cell lines derived from carcinomas of the human breast. Two of these lines express low levels of MYC and display a metastatic phenotype, whereas the other two overexpress the gene but do not metastasize. Irrespective of the reason for this variation, the differences in MYC expression allowed reciprocal studies on induction or repression of the metastatic phenotype by manipulation of MYC expression and its downstream targets. Clearly human breast cancers that overexpress MYC may still metastasize if other factors override its function. In fact our in vivo assays demonstrated that expression of MYC will support increased growth of those few metastatic cells that escape the inhibitory function of MYC by means of other mutations or by changes in gene expression (Fig. 2m,r).
Given this complexity, we sought direct experimental demonstration that MYC can indeed inhibit metastasis. Using MDA-MB-231, the human cell line that expresses only low levels of MYC and is used extensively for metastatic studies, we confirmed their highly metastatic potential, by both tail vein injection (Fig. 2c,d) and orthotopic implantation (Fig. 2i-r) in mouse models. However, when exogenous MYC was over-expressed in these cells, their ability to metastasize was greatly reduced. Further, the behavior of breast cancer cell lines was remarkably malleable when the level of MYC was modulated: it was possible to reduce the metastatic properties by over-expressing MYC, or to augment those properties by reducing MYC levels. At the same time, MYC could increase proliferation even in overtly malignant cells (Fig. 1b). We reported previously an apparent dichotomy between stimulation of proliferation and inhibition of other malignant properties of breast cancer cells also for the oncogene, AKT1 (protein kinase B1)42-44.
Our finding that MYC overexpression leads to inhibition of cell motility and invasiveness through direct downmodulation of αv and β3 integrin subunits supports reports that integrins with these subunits are involved in cellular motility, invasiveness, adhesion, and transmigration through endothelium, and are associated with metastasis22,24,35,45,46. There are reports also that MYC may be involved in controlling the expression of integrin genes using cells from different tissues26,28,29. However, none of these reports has linked the effect of MYC on integrin expression to cellular components of metastasis, and none has implicated the αvβ3 heterodimer directly in the cellular changes elicited by MYC. Our results demonstrate that overexpression of MYC represses transcription from the promoters of the αv and β3 integrin genes, which in turn reduces properties that are essential for metastasis. These data, however, do not mean that MYC affects invasion and metastasis only through regulation of integrins αv and β3, nor that MYC is the sole and only regulator of integrin αvβ3 function in invasion and metastasis. The fact that MYC siRNA increases invasion when MCF7 cells are forced to have high levels of integrins αv and β3 (Fig. 6j) suggests that there may be mechanism(s) other than integrin αvβ3 by which MYC can inhibit invasion and metastasis. Nevertheless, since exogenous expression of these integrins is sufficient to bypass the repressive effect of MYC in invasion (Fig. 6d,f,j) and metastasis (Fig. 5h,j), it is apparent that a primary effect of MYC on invasion in breast cancer cells is mediated through its inhibition of integrins αv and β3.
Tumor progression often culminates in metastatic disease indicating that there must be selection for antidotes to the MYC inhibition of the metastatic phenotype. One obvious possibility would be selection against overexpression of MYC during the course of tumor progression. Indeed, the two breast cancer cell lines used in the present study that express relatively low levels of MYC both originated from metastatic tumors: MDA-MB-231 from pleural effusion and BT-549 from an invasive ductal tumor in regional lymph nodes. Other possibilities include loss of β3 integrin functions, as in the case of MCF-7 and T47D cell lines. Constitutive signaling from active Ras or overexpression of Bcl-XL can also override the inhibition of metastasis by MYC (unpublished results of H.L and D.Y). These complexities might account for the fact that in some experimental circumstances, MYC may appear to favor metastasis47-49. The findings reported here prompt a cautionary note about therapeutic strategies involving MYC. The frequency with which MYC is over-expressed, and the variety of tumors in which that over-expression occurs, have made MYC a seemingly advantageous therapeutic target50. Our findings raise the possibility that inhibition of MYC in human tumors might at times be contraindicated since its suppression may indeed promote metastasis.
Supplementary Material
Transfection of αv and β3 integrins augments formation of αvβ3 integrin heterodimers. MDA-MB-231 cells overexpressing MYC were transiently transfected with constructs expressing either αv or β3 integrin, or both. The transfected constructs are indicated at the base of the figure. The lysates of transfectants were immunoprecipitated by antibodies against αv integrin or αvβ3 integrin heterodimers, separated by SDS-PAGE electrophoresis and immunoblotted with the indicated antibodies.
Exogenous expression of integrin β3 in MCF7 cells leads to increased generation of αvβ3 heterodimers even if MYC is not knocked down. MCF-7 cells with control vector or stably over-expressing β3 integrin were transfected with negative control siRNA or 2 different oligoes of MYC siRNA. The lysates of transfectants were immunoprecipitated by antibodies against αv integrin or αvβ3 integrin heterodimers, separated by SDS-PAGE electrophoresis and immunoblotted with the indicated antibodies.
Acknowledgments
We thank David Cheresh, Filippo G. Giancotti, Andrei Goga, and Dean Sheppard for kindly providing DNA constructs and cell lines. We are grateful to Chih-Ying Chen, Linda Prentice, and Brandy Edenfeld for help with histology and imaging. We thank Davitte Khauv for work with the ChIP assay, Erin Miller for work with the orthotopic animal assay, Magdalena Cichon for cloning constructs, and Melody Stallings-Mann for work with analysis of experiments. We also thank members of the Bishop, Bissell and Radisky laboratories for their constructive discussion and help. This work was funded initially by the George Williams Hooper Foundation (JMB). The founder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Additional support came from NIH (P50 CA091956) and the DoD (PC094054) (to E.S.R.); NCI CA122086 and the Mayo Clinic Breast Cancer SPORE grant CA116201 (to D.C.R.); the DoD (W81XWH0810736, NIH/NCI (R37CA064786, U01CA143233, U54CA143836, and U54CA126552) and the Department of Energy OBER Low Dose Radiation Program (contract no. DE-AC02-05CH1123),), (to M.J.B.).
Footnotes
AUTHOR CONTRIBUTIONS
H.L., D.C.R., D.Y., E.S.R., M.J.B. and J.M.B designed the research; H.L., D.C.R., R.X., E.S.R., and D.Y. performed research; H.L., D.Y., D.C.R., E.S.R., M.J.B and J.M.B. analyzed data; H.L., D.C.R., M.J.B. and J.M.B. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 2.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 3.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuttler F, Mai S. c-Myc, Genomic Instability and Disease. Genome Dyn. 2006;1:171–190. doi: 10.1159/000092507. [DOI] [PubMed] [Google Scholar]
- 5.Oster SK, Ho CS, Soucie EL, Penn LZ. The myc oncogene: MarvelouslY Complex. Adv Cancer Res. 2002;84:81–154. doi: 10.1016/s0065-230x(02)84004-0. [DOI] [PubMed] [Google Scholar]
- 6.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 7.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 8.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 9.Alitalo K, Schwab M. Oncogene amplification in tumor cells. Adv Cancer Res. 1986;47:235–281. doi: 10.1016/s0065-230x(08)60201-8. [DOI] [PubMed] [Google Scholar]
- 10.Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell. 2004;6:577–586. doi: 10.1016/j.ccr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7:143–164. doi: 10.1677/erc.0.0070143. [DOI] [PubMed] [Google Scholar]
- 12.Pompetti F, et al. Oncogene alterations in primary, recurrent, and metastatic human bone tumors. J Cell Biochem. 1996;63:37–50. doi: 10.1002/(SICI)1097-4644(199610)63:1%3C37::AID-JCB3%3E3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 13.Shiu RP, Watson PH, Dubik D. c-myc oncogene expression in estrogen-dependent and -independent breast cancer. Clin Chem. 1993;39:353–355. [PubMed] [Google Scholar]
- 14.D’Cruz CM, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–239. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 15.Leder A, Pattengale PK, Kuo A, Stewart TA, Leder P. Consequences of widespread deregulation of the c-myc gene in transgenic mice: multiple neoplasms and normal development. Cell. 1986;45:485–495. doi: 10.1016/0092-8674(86)90280-1. [DOI] [PubMed] [Google Scholar]
- 16.Sinn E, et al. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–475. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 17.Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell. 1984;38:627–637. doi: 10.1016/0092-8674(84)90257-5. [DOI] [PubMed] [Google Scholar]
- 18.Welm AL, Kim S, Welm BE, Bishop JM. MET and MYC cooperate in mammary tumorigenesis. Proc Natl Acad Sci U S A. 2005;102:4324–4329. doi: 10.1073/pnas.0500470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frye M, Gardner C, Li ER, Arnold I, Watt FM. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development. 2003;130:2793–2808. doi: 10.1242/dev.00462. [DOI] [PubMed] [Google Scholar]
- 20.Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet. 2001;28:165–168. doi: 10.1038/88889. [DOI] [PubMed] [Google Scholar]
- 21.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 22.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 23.Lu P, Weaver VM, Werb Z. The extracellular matrix: A dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Felding-Habermann B. Integrin adhesion receptors in tumor metastasis. Clin Exp Metastasis. 2003;20:203–213. doi: 10.1023/a:1022983000355. [DOI] [PubMed] [Google Scholar]
- 25.Mizejewski GJ. Role of integrins in cancer: survey of expression patterns. Proc Soc Exp Biol Med. 1999;222:124–138. doi: 10.1177/153537029922200203. [DOI] [PubMed] [Google Scholar]
- 26.Gebhardt A, et al. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J Cell Biol. 2006;172:139–149. doi: 10.1083/jcb.200506057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Nigris F, et al. Cooperation between Myc and YY1 provides novel silencing transcriptional targets of alpha3beta1-integrin in tumour cells. Oncogene. 2007;26:382–394. doi: 10.1038/sj.onc.1209804. [DOI] [PubMed] [Google Scholar]
- 28.van Golen CM, Soules ME, Grauman AR, Feldman EL. N-Myc overexpression leads to decreased beta1 integrin expression and increased apoptosis in human neuroblastoma cells. Oncogene. 2003;22:2664–2673. doi: 10.1038/sj.onc.1206362. [DOI] [PubMed] [Google Scholar]
- 29.Wilson A, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18:2747–2763. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao J, Jethanandani P, Ziober BL, Kramer RH. Regulation of alpha7 integrin expression during muscle differentiation. J Biol Chem. 2003;278:49780–49788. doi: 10.1074/jbc.M308542200. [DOI] [PubMed] [Google Scholar]
- 31.Husemann Y, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 32.Ridley AJ, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 33.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 34.Carman CV, Springer TA. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr Opin Cell Biol. 2003;15:547–556. doi: 10.1016/j.ceb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Bauer K, Mierke C, Behrens J. Expression profiling reveals genes associated with transendothelial migration of tumor cells: a functional role for alphavbeta3 integrin. Int J Cancer. 2007;121:1910–1918. doi: 10.1002/ijc.22879. [DOI] [PubMed] [Google Scholar]
- 36.Wanzel M, Herold S, Eilers M. Transcriptional repression by Myc. Trends Cell Biol. 2003;13:146–150. doi: 10.1016/s0962-8924(03)00003-5. [DOI] [PubMed] [Google Scholar]
- 37.Small MB, Hay N, Schwab M, Bishop JM. Neoplastic transformation by the human gene N-myc. Mol Cell Biol. 1987;7:1638–1645. doi: 10.1128/mcb.7.5.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellwood-Yen K, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 39.Jamerson MH, Johnson MD, Dickson RB. Of mice and Myc: c-Myc and mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2004;9:27–37. doi: 10.1023/B:JOMG.0000023586.69263.0b. [DOI] [PubMed] [Google Scholar]
- 40.Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16:313–317. doi: 10.1016/j.semcancer.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 42.Liu H, Radisky DC, Bissell MJ. Proliferation and polarity in breast cancer: untying the Gordian knot. Cell Cycle. 2005;4:646–649. doi: 10.4161/cc.4.5.1674. [DOI] [PubMed] [Google Scholar]
- 43.Liu H, Radisky DC, Wang F, Bissell MJ. Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells. J Cell Biol. 2004;164:603–612. doi: 10.1083/jcb.200306090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu H, et al. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc Natl Acad Sci U S A. 2006;103:4134–4139. doi: 10.1073/pnas.0511342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liapis H, Flath A, Kitazawa S. Integrin alpha V beta 3 expression by bone-residing breast cancer metastases. Diagn Mol Pathol. 1996;5:127–135. doi: 10.1097/00019606-199606000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Desgrosellier JS, et al. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med. 2009;15:1163–1169. doi: 10.1038/nm.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma L, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rapp UR, et al. MYC is a metastasis gene for non-small-cell lung cancer. PLoS One. 2009;4:e6029. doi: 10.1371/journal.pone.0006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolfer A, et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc Natl Acad Sci U S A. 2010;107:3698–3703. doi: 10.1073/pnas.0914203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transfection of αv and β3 integrins augments formation of αvβ3 integrin heterodimers. MDA-MB-231 cells overexpressing MYC were transiently transfected with constructs expressing either αv or β3 integrin, or both. The transfected constructs are indicated at the base of the figure. The lysates of transfectants were immunoprecipitated by antibodies against αv integrin or αvβ3 integrin heterodimers, separated by SDS-PAGE electrophoresis and immunoblotted with the indicated antibodies.
Exogenous expression of integrin β3 in MCF7 cells leads to increased generation of αvβ3 heterodimers even if MYC is not knocked down. MCF-7 cells with control vector or stably over-expressing β3 integrin were transfected with negative control siRNA or 2 different oligoes of MYC siRNA. The lysates of transfectants were immunoprecipitated by antibodies against αv integrin or αvβ3 integrin heterodimers, separated by SDS-PAGE electrophoresis and immunoblotted with the indicated antibodies.