

Abstract
The mechanism of cyclohexyne insertion into a C(O)-Cα bond of cyclic ketones, explored experimentally by the Carreira group, has been investigated using density functional theory. B3LYP and M06-2X calculations were performed in both gas phase and THF (CPCM, UAKS radii). The reaction proceeds through a stepwise [2+2] cycloaddition of cyclohexyne to the enolate, followed by three disparate ring opening possibilities of the cyclobutene alkoxide to give the product: 1) thermally-allowed conrotatory electrocyclic ring opening, 2) thermally-forbidden disrotatory electrocyclic ring opening, or 3) non-pericyclic C-C bond cleavage. Our computational results for the model alkoxide and potassium alkoxide systems show that the thermally-allowed electrocyclic ring opening pathway is favored by less than 1 kcal/mol. In more complex systems containing a potassium alkoxide (e–f), the barrier of the allowed conrotatory ring opening is disfavored by 4–8 kcal/mol. This suggests that the thermodynamically more stable disrotatory product can be formed directly through a “forbidden” pathway. Analysis of geometrical parameters and atomic charges throughout the ring opening pathways provides evidence for a non-pericyclic C-C bond cleavage, rather than a thermally-forbidden disrotatory ring opening. A true forbidden disrotatory ring opening transition structure was computed for the cyclobutene alcohol; however, it was 19 kcal/mol higher in energy than the allowed conrotatory transition structure. An alternate mechanism in which the disrotatory product forms via isomerization of the conrotatory product was also explored for the alkoxide and potassium alkoxide systems.
Introduction
Arynes and cycloalkynes are potentially useful building blocks in organic synthesis.1–3 Stoltz and Tambar have reported a mild direct aryne insertion into C-C bonds.1 They obtained an unexpected C-C addition product (boxed; Scheme 1) in comparable yield with the expected product.
Scheme 1.

Acyl-Alkylation of Benzyne into a β-ketoester
Carreira and co-workers recently reported the formal addition of cyclohexyne to C-C bonds of cyclic ketones to yield medium-sized fused ring systems.2 This process involves simultaneous formation of an enolate from a cyclic ketone and formation of cyclohexyne via base-induced elimination from an iodonium salt of cyclohexene. In some cases, a tricyclic product containing a cyclobutene ring is produced, suggesting the intermediacy of this [2+2] cycloaddition product in formation of the cycloheptenone, as shown in Scheme 2.
Scheme 2.
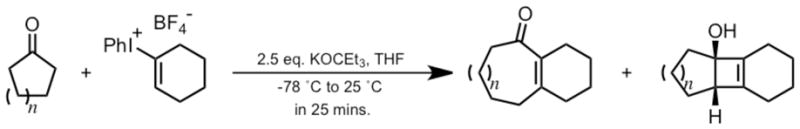
Cycloinsertion Reaction of Cyclohexyne into Cyclic Ketones
Carreira and Gampe have applied this reaction to the total synthesis of guanacastepenes,3 and a comprehensive review of their applications of this ring expansion reaction has recently been documented.4 Comandini and Brezinsky computationally explored the potential energy surface of a radical/π-bond addition mechanism of o-benzyne to cyclopentadiene. One of the multiple pathways they studied involved the disrotatory electrocyclic ring opening of a strained cyclobutene; however, it seems that the allowed conrotatory ring opening was not considered.5 To determine the details of this cycloalkyne insertion mechanism a computational study using density functional theory (DFT) calculations was initiated. Of prime interest in this study was the nature of the electrocyclic ring opening step.
Computational Methods
Intermediate and transition state geometry optimizations were performed with Gaussian 09 using the hybrid functional B3LYP with the 6-31G(d) basis set or LANL2DZ for potassium.6 In addition, the anions were optimized with the diffuse 6-311++G(d,p) basis set. A stability check of the restricted wavefunction was performed to insure that open-shell wavefunctions are not more stable.7 Analytical frequencies were calculated for each optimized structure using the corresponding level of theory. Single point energy calculations were performed with M06-2X/6-311++G(d,p) on B3LYP/6-31G(d) geometries and thermally corrected using the B3LYP/6-31G(d) vibrational data in order to report enthalpies. Such calculations have been shown to give improved thermodynamics for C-C bond forming reactions.8 All optimized saddle points were explored by Intrinsic Reaction Coordinate (IRC)9,10 calculations, ensuring that they connect the appropriate reactants and products on the potential energy surface.
The effects of THF solvation on the reaction energetics were evaluated using the conductor-like polarizable continuum solvation model (CPCM).11 The solute surface was defined by UAKS radii.12
Results and Discussion
1. Reaction of cyclohexyne and cyclopentanone enolate
The cycloaddition step was confirmed by our calculations to be stepwise, in accordance with the Woodward-Hoffmann rules. The energetics of cycloaddition are summarized in Figure 1.
Figure 1.

Stationary points on the energy surface for cycloaddition of cyclohexyne and cyclopentanone enolate. B3LYP/6-311++G(d,p) thermal energies, ΔH, with thermally corrected M06-2X/6-311++G(d,p)//B3LYP/6-31G(d) energies, ΔH, in parentheses, shown in kcal/mol.
Nucleophilic attack on cyclohexyne is the first step of the reaction and occurs without an activation barrier. Examples of nucleophilic additions to cycloalkynes without a barrier of activation have been reported elsewhere in the literature.13 The addition is exceedingly exothermic, forming 3a with a ΔH of −36 to −48 kcal/mol. Our calculations show that the alkyne (1) is distorted from linearity by 48°, which is in agreement with past theoretical studies of cyclohexyne using Hartree-Fock, DFT, or Møller-Plesset perturbation theory methods.14–16 The C-C and C-O bond lengths in cyclopentanone enolate (2a) are representative of enolate character. Figure 2 shows the geometries of cyclopentanone enolate nucleophilic addition to cyclohexyne optimized in the gas phase with B3LYP/6-31G(d) and B3LYP/6-311++G(d,p).
Figure 2.

Optimized geometries of cyclohexyne insertion into cyclopentanone enolate. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles with B3LYP/6-311++G(d,p) in parentheses, are shown.
Ring closure of vinyl anion 3a to form cyclobutene alkoxide 5a has an activation barrier of only 2–5 kcal/mol and is exothermic by only 6–7 kcal/mol. Structural deviations from normal cyclobutene geometries are immediately apparent in the bond lengths of 5a. A computational study by Houk and Rondan showed that donor substituents like alkoxide groups interact strongly with the C3–C4 σ* orbital in cyclobutene, thereby lowering the barrier to ring opening.17 This hyperconjugative effect results in an increase of the C3–C4 bond length as well as a decrease of the C3-O bond length relative to unsubstituted cyclobutene and is evident in the C3–C4 bond length of 1.69 Å and the C3-O bond length of 1.31 Å.
Electrocyclic ring opening of 5a is the next step in the pathway, and the energetics are summarized in Figure 3. The experimental enthalpy of activation for the electrocyclic ring opening of cyclobutene is 32.5 ± 0.5 kcal/mol, and the reaction is exothermic by 10.8 kcal/mol.18 According to the Woodward-Hoffmann rules, cyclobutene undergoes a symmetry-allowed conrotatory ring opening under thermal conditions. Numerous experimental and computational studies have confirmed this preference.17–21 Specifically, experiments by Brauman and Golden estimated that the allowed conrotatory process is 15.0 kcal/mol more favorable than the forbidden disrotatory process.21 The results of our computational study showed the enthalpy of activation for the allowed conrotatory electrocyclic ring opening of 5a is 4.8 (6.0) kcal/mol, and the reaction is exothermic by 8.0 kcal/mol by B3LYP, but slightly endothermic by M06-2X. Non-pericyclic C-C bond cleavage gave an enthalpy of activation of 5.5 (8.0) kcal/mol and an exothermicity of 34 kcal/mol, only slightly higher than the allowed conrotatory pathway. We use the term “non-pericyclic” since there appears to be no cyclic delocalization, and this transition state appears to be different from pseudopericyclic transition states of electrocyclization reactions. The term “pseudopericyclic” was first used by Lemal to describe reactions that are characterized by the exchange of roles between orthogonal bonding and nonbonding atomic orbitals.22 Computational investigations have provided evidence that psuedopericyclic transition states are typically planar with no rotation of the terminal π-bonds. For instructive reviews of the subject, the reader is directed to articles of Kappe et al.,23 Birney et al.,24 and references therein. Isomerization of 7a via 8a forms the more stable product 7a′. The difference between the enthalpies of reaction in the conrotatory and non-pericyclic cases is due to formation of a strained cis, trans-cycloheptadiene skeleton in the former and a relatively stable cis, cis-cycloheptadiene skeleton in the latter.
Figure 3.

Stationary points on the energy surface for electrocyclic ring opening and cis-trans isomerization. B3LYP/6-311++G(d,p) thermal energies, ΔH, with thermally corrected M06-2X/6-311++G(d,p)//B3LYP/6-31G(d) energies, ΔH, in parentheses, shown in kcal/mol.
The optimized geometries of conrotatory and non-pericyclic ring opening are shown in Figure 4. Previous work has shown that the skeleton of the conrotatory transition structure of cyclobutene is nonplanar (φ≈15°), while the disrotatory transition structure is planar.25 It was also found that constraining planarity in the conrotatory transition structure reduces overlap between the σ orbital of the breaking bond and the π orbital present in cyclobutene. It was concluded that this reduction in stabilizing orbital overlap lowers the preference for the allowed conrotatory pathway. As seen in Figure 4, the C3C2C1C4 dihedral angle of the conrotatory transition structure is 5°, considerably more planar than the transition state of cyclobutene itself.
Figure 4.

Optimized geometries of conrotatory (left) and non-pericyclic (right) ring opening of cyclobutene alkoxide 5a. B3LYP/6-31G(d) selected bond distances (Å) and dihedral angles, with B3LYP/6-311++G(d,p) in parentheses, are shown.
A number of investigations in the literature provide evidence that cis-trans isomerization can occur in similarly strained systems, directly connecting conrotatory products to disrotatory products on the potential energy surface.26–29 No such transition structure was found using spin-restricted B3LYP; however, use of broken symmetry, spin-unrestricted B3LYP successfully yielded structure 8a (Figure 5). This result clearly indicates diradical character, which is known to exist in transition structures of π-bond rotation. Elongated C1–C4 and C2–C3 bond lengths relative to 6a indicate more single- bond character and provide further evidence for the diradical nature of 8a. The decreased C1–C2 bond length also supports this idea. It is worthwhile to note that the C3–C4 distance has increased almost to its final value of 3.20 Å as seen in 7a′, while a comparatively small amount of outward rotation of C4-H has occurred. The enthalpy of activation for cis-trans isomerization was calculated as 12 kcal/mol.
Figure 5.

Optimized transition structure of cis-trans isomerization of alkoxide intermediate 7a to 7a′. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles, with B3LYP/6-311++G(d,p) in parentheses, are shown.
2. Reaction of cyclohexyne and cyclopentanone potassium enolate in THF
An equivalent analysis of the reaction mechanism and energetics was performed with cyclohexyne and cyclopentanone potassium enolate. While potassium will be solvated in solution, this model provides a picture of the extreme effect of ion pairing on the reaction pathway. The energetics of cycloaddition are summarized in Figure 6. The addition yields an intermediate with a ΔH of −40 kcal/mol. For ring closure, an activation barrier of 3 kcal/mol was computed and the reaction is exothermic by 8 kcal/mol.
Figure 6.

Stationary points on the energy surface for cycloaddition of cyclohexyne and cyclopentanone potassium enolate. B3LYP/6-31G(d) thermal energies, ΔH, shown in kcal/mol.
Figure 7 shows the geometries of cyclohexyne insertion into cyclopentanone potassium enolate optimized in THF with B3LYP/6-31G(d), using LANL2DZ for potassium. As shown by the bond distances in 3b and 4b, the potassium ion is stabilizing the development of charge on both the oxygen of the cyclopentanone enolate and C2 of cyclohexyne. A C3–C4 bond length of 1.64 Å and a C3-O bond length of 1.35 Å in 5b both indicate less donation of electron density of oxygen into the σ* orbital of C3–C4 when compared to the bare enolate. As a direct consequence, the barrier to electrocyclic ring opening of 5b is 10 kcal/mol greater than the electrocyclic ring opening of 5a (Figures 3 and 8).
Figure 7.

Optimized geometries in the cyclohexyne insertion into cyclopentanone potassium enolate. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown. LANL2DZ was used for potassium.
Figure 8.

Stationary points on the energy surface for electrocyclic ring opening and cis-trans isomerization. B3LYP/6-31G(d) thermal energies, ΔH, shown in kcal/mol.
The electrocyclic ring opening step favors the allowed conrotatory process by about 1 kcal/mol; however, unlike the case of cyclopentanone enolate, the barrier of cis-trans isomerization is greater than both the barrier for subsequent ring closing (reversion) and the barrier for non-pericyclic ring opening. The energetics of ring opening are summarized in Figure 8. Optimized geometries of conrotatory and non-pericyclic ring opening are shown in Figure 9. The C3C2C1C4 dihedral angles of the conrotatory and non-pericyclic transition structures are 18° and −4°, respectively, which closely resemble 19° and ~0°,25 those of the ring opening transition structures of cyclobutene itself. There is an apparent interaction between the potassium and C4 in the non-pericyclic transition structure (6b′), but the preference for the allowed conrotatory ring opening is less than 1 kcal/mol, about the same as in the free alkoxide. The reaction leading to the conrotatory product 7b is endothermic by 8 kcal/mol. As in the case where potassium is absent, the large thermodynamic difference between 7b and 7b′ is due to the presence of a trans double bond in the 7-membered conrotatory product.
Figure 9.


Optimized geometries of conrotatory (left) and non-pericyclic (right) ring opening of cyclobutene potassium alkoxide 5b. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown. LANL2DZ was used for potassium.
A transition structure connecting the cis/trans product 7b to the cis/cis product 7b′ is shown in Figure 10. Like the free alkoxide, cis-trans isomerization by direct π-bond rotation occurs here, necessitating the use of open-shell DFT to locate this transition structure. The C3–C4 bond elongates from 1.38 Å to 1.50 Å, and the C2–C3 bond elongates from 1.42 Å to 1.46 Å. The C3–C4 bond length of 3.15 Å and C3C2C1C4 dihedral angle of −4° in 8b are both essentially the final values observed in 7b′. Interestingly, the C4C3O bond angle has increased to 175°, more than that seen in the disrotatory product, while the C3C4H bond angle has only increased by 10° relative to 7b. The activation enthalpy of cis-trans isomerization was calculated as 14 kcal/mol.
Figure 10.

Optimized transition structure of isomerization of potassium alkoxide intermediate 7b to 7b′. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown.
A better model for the experimental reaction conditions would be a fully THF-solvated potassium enolate, but this would be computationally highly challenging. The results are expected to be somewhere between the results for the potassium-coordinated and free anions.
3. Ring opening of the cyclobutene alcohol
We have also explored the extreme of coordination to the cyclobutene alkoxide, namely the corresponding alcohol. These results provide an assessment of allowed vs. forbidden processes on a more conventionally substituted cyclobutene. Stationary points on the energy surface of electrocyclic ring opening computed with B3LYP/6-31G(d) are shown in Figure 11. Thermally corrected M06-2X/B3LYP-6311++G(d,p) single point energies on B3LYP/6-31G(d) geometries are shown in parentheses. There is a preference of 19–21 kcal/mol for the allowed conrotatory ring opening of the cyclobutenol, in spite of the 30 kcal/mol energetic preference for the less strained product that would result from forbidden disrotatory opening. This indicates that the normal preference for the allowed pathway is operating, in spite of large thermodynamic differences in the reactions.
Figure 11.

Stationary points on the energy surface for electrocyclic ring opening. B3LYP/6-31G(d) thermal energies, ΔH, with thermally corrected M06-2X/6-311++G(d,p)//B3LYP/6-31G(d) energies, ΔH, in parentheses, shown in kcal/mol.
The optimized geometries of conrotatory and disrotatory ring opening of cyclobutenol are shown in Figure 12. The C3–C4 bond length of 1.60 Å in 5c is unusually long, but less stretched than the corresponding alkoxide. The bond angles and dihedral angles in the ring opening transition structures are not appreciably different from those in the previous cases, but the three C-C bond lengths in the ring are nearly identical, showing significant delocalization that was not observed in the other cases. Conrotatory electrocyclic ring opening is endothermic by 17–18 kcal/mol, while disrotatory ring opening is exothermic by 12–14 kcal/mol. A summary of computed enthalpies for the model reaction including all basis sets and solvent models used is shown in Table 1. An analogous table reporting Gibbs free energies is given in the Supporting Information (Table S1).
Figure 12.


Optimized geometries of conrotatory (left) and disrotatory (right) ring opening of cyclobutene alcohol 5c. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown.
Table 1.
Summary of computed enthalpies, ΔH, for the model alkoxide, potassium alkoxide, and alcohol systems.
| Gas Phase
|
THF
|
||||||
|---|---|---|---|---|---|---|---|
| a
|
c
|
a
|
b
|
||||
| B3LYP/6-31G(d) | B3LYP/6-311++G(d,p) | M06-2X | B3LYP/6-31G(d) | B3LYP/6-31G(d) | B3LYP/6-311++G(d,p) | B3LYP/6-31G(d) | |
| 1 + 2 | 0.0 | 0.0 | 0.0 | - | 0.0 | - | 0.0 |
| 3 | −43.6 | −35.9 | −46.2 | - | −33.8 | - | −39.9 |
| 4 | −41.8 | −34.2 | −41.3 | - | −31.2 | - | −36.9 |
| 5 | −48.0 | −41.6 | −52.6 | 0.0 | −41.5 | 0.0 | −48.3 |
| 6 | −45.5 | −36.8 | −46.6 | 28.3 | −36.9 | 14.4 | −33.7 |
| 6′ | −44.4 | −36.1 | −43.8 | 47.5 | −35.9 | 16.2 | −33.0 |
| 7 | −58.4 | −49.6 | −51.5 | 16.8 | −48.5 | 3.1 | −40.6 |
| 7′ | −85.7 | −75.3 | −85.7 | −13.9 | −75.5 | −22.5 | −72.5 |
| 8 | −44.0 | −37.8 | −47.7 | − | - | - | −26.5 |
4. Ring opening of an alkoxide-substituted guanacastepene precursor
Next, we expanded the investigation of ring opening energetics to more complex systems incorporated by Carreira.3 The electrocyclic ring opening of anion 5d was shown to favor the allowed conrotatory process; however, the observed preference of less than 1 kcal/mol is essentially negligible when considering error in B3LYP/6-31G(d) calculations. The thermally corrected M06-2X/6-311++G(d,p)//B3LYP/6-31G(d) single point energies show the energy gap to be 2 kcal/mol. Conrotatory product 7d is less stable than cyclobutene enolate 5d by 1 kcal/mol, while disrotatory product 7d′ is more stable than 5d by 32 kcal/mol. The energetics of ring opening are shown in Figure 13.
Figure 13.

Stationary points on the energy surface for electrocyclic ring opening of 5d. B3LYP/6-31G(d) thermal energies, ΔH, with thermally corrected M06-2X/6-311++G(d,p)//B3LYP/6-31G(d) energies, ΔH, in parentheses, shown in kcal/mol.
Optimized geometries of conrotatory and non-pericyclic ring opening are shown in Figure 14. The C3C4 bond length in 5d is 1.68 Å, shorter than the 1.73 Å distance seen in the B3LYP/6-31G(d) optimized structure of cyclobutene enolate 5a. This difference can be attributed to a decrease in donation of the C3-O nonbonding orbital into the C3C4 σ* orbital due to electrostatic interactions of the alkoxide with hydrogens on an adjacent ring (5d, Figure 14), an effect qualitatively similar to coordination of the alkoxide with K+. This interaction is observed throughout the process of ring opening. The C3C4 distances are very similar in the two transition structures 6d and 6d′, 2.39 Å and 2.43 Å respectively, unlike those seen when comparing the allowed and forbidden transition structures 6a–c and 6a′–c′. The C3C2C1C4 dihedral angles of 6d and 6d′ are −17° and 13° respectively. As mentioned previously, the disrotatory ring opening transition structure of cyclobutene is planar,25 so an observed dihedral angle of 13° in 6d′ is indicative of non-pericyclic character. Interestingly, the positive dihedral angles in 6d′ become negative in 7d′. As a result, the carbon adjacent to C4 in the cycloheptadiene ring has flipped from the concave to the convex side of the molecule.
Figure 14.


Optimized geometries of conrotatory (left) and non-pericyclic (right) ring opening of 5d. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown.
5. Ring opening of a potassium alkoxide-substituted guanacastepene precursor
The thermally allowed conrotatory electrocyclic ring opening of potassium alkoxide 5e was disfavored by 8 kcal/mol, in sharp contrast to all other systems studied. Conrotatory product 7e is less stable than 5e by 9 kcal/mol, while 7e′ is more stable than 5e by 22 kcal/mol. Stationary points detailing the energetics of ring opening of 5e are presented in Figure 16.
Figure 16.

Stationary points on the energy surface for electrocyclic ring opening of 5e. B3LYP/6-31G(d) thermal energies, ΔH, shown in kcal/mol.
Optimized geometries of conrotatory and non-pericyclic ring opening are shown in Figure 17. The transition structure of isomerization from 7e to 7e′ is shown in Figure 18. All relevant geometrical information is presented with the same interpretations as provided before; however, there is one important difference. In this molecule, there are other oxygen atoms to which the potassium atom can coordinate. Specifically in 6e′, the coordination of the potassium to C4 and to another oxygen is likely responsible for the stabilization of this transition structure relative to 6e. With that idea in mind, a slight modification was made to replace the two oxygens in the adjacent ring with methylene carbons to form 5f. Ring opening structures are shown in Figure 19, while Table 2 summarizes the ring opening energies of 5d–f. Although replacing the oxygens in the ring with methylene groups did lower the energy difference between 6f and 6f′ relative to 6e and 6e′, Table 2 indicates a preference of 4 kcal/mol still remains for non-pericyclic ring opening over conrotatory electrocyclic ring opening. See Table S2 in the Supporting Information for Gibbs free energies.
Figure 17.


Optimized geometries of conrotatory (left) and non-pericyclic (right) ring opening of 5e. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown.
Figure 18.
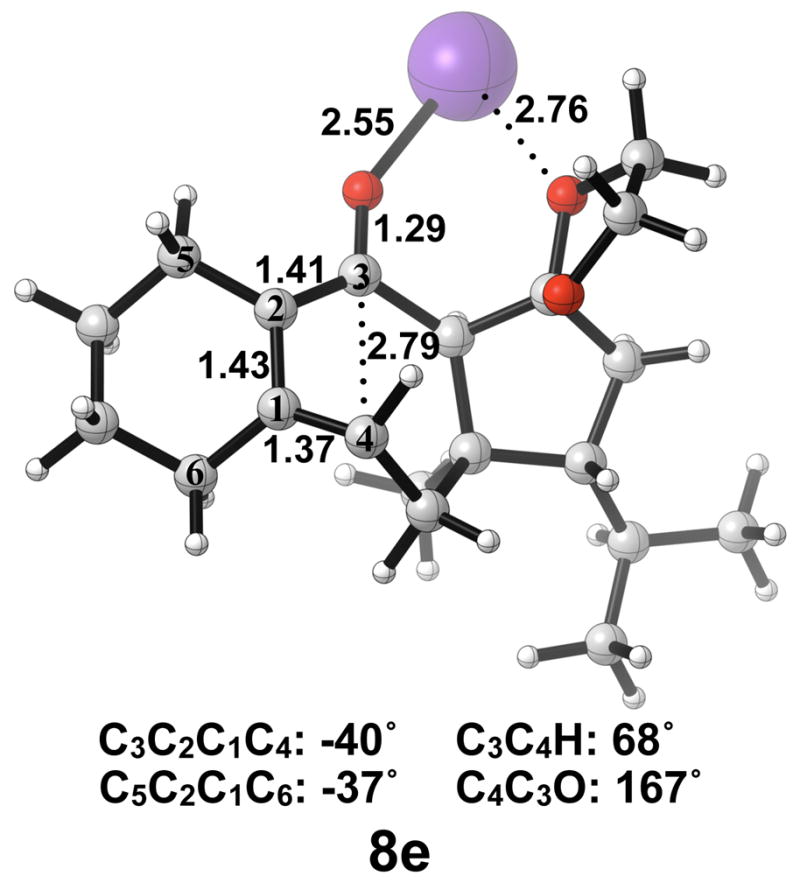
Optimized transition structure of isomerization of potassium alkoxide intermediate 7e to 7e′. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown.
Figure 19.

Optimized geometries of conrotatory (left) and non-pericyclic (right) ring opening of 5f. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown.
Table 2.
Summary of computed enthalpies, ΔH, for the more complex alkoxide and potassium alkoxide guanacastepene precursors.
| Gas Phase
|
THF
|
|||
|---|---|---|---|---|
| d
|
e
|
f
|
||
| B3LYP/6-31G(d) | M06-2X | B3LYP/6-31G(d) | B3LYP/6-31G(d) | |
| 5 | 0.0 | 0.0 | 0.0 | 0.0 |
| 6 | 10.9 | 13.4 | 18.7 | 17.4 |
| 6′ | 11.3 | 15.5 | 10.7 | 13.7 |
| 7 | 0.9 | 1.9 | 9.2 | - |
| 7′ | −32.1 | −30.5 | −22.3 | - |
| 8 | 2.7 | 4.0 | 9.9 | - |
6. ChelpG calculations on the mechanism of ring opening
ChelpG (charges from electrostatic potentials using a grid based method)28 calculations on B3LYP/6-31G(d) optimized geometries were used to characterize the electronic structure of all ring opening systems. Unlike the conrotatory transition structure of cyclobutene, the conrotatory transition structures 6a–f possess unequal charge distribution throughout the ring due to electronic effects originating from the oxygen on C3. The magnitude of negative charge on the oxygens in the non-pericyclic transition structures is close to that in the intermediates. This is uncharacteristic of electronic structure changes in pericyclic reactions, in which atomic charges gradually change throughout ring opening. C4 develops a full negative charge in the anionic systems, decreasing from −0.1 in alkoxide 5 to −1.1 in transition state 6′. In the potassium alkoxide systems, this charge buildup on C4 is stabilized by coordination of the potassium cation. Together, the trends in electronic structure of oxygen and C4 through 5–6′–7′ provide evidence against a pericyclic disrotatory ring opening. Instead, the mechanism resembles carbonyl formation in the transition state with anion migration to C4 upon C-C bond cleavage (Scheme 4). Ring opening of 5c is the only system that displays true forbidden disrotatory nature, as shown by the magnitude of difference in energies between the two pathways and the plots of atomic charges. Figure 20 shows the atomic charges of the four carbons in the cyclobutene ring and of the oxygen on C3 throughout both ring opening pathways of 5a. Corresponding plots of all systems presented in this paper and numerical tables of charges are given in the Supporting Information.
Scheme 4.
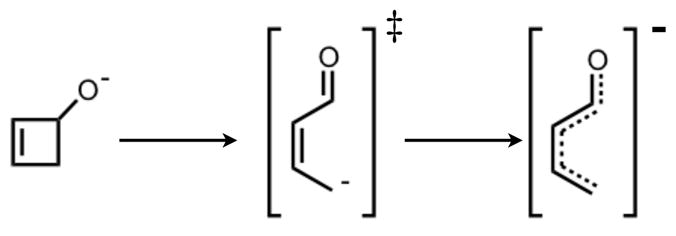
Schematic Representation of Non-Pericyclic Ring Opening
Figure 20.


Atomic charges on the four carbons and the oxygen substituent in the cyclobutene ring of 5a throughout the two ring opening pathways.
Conclusions
The large preference for allowed conrotatory ring opening in the cyclobutene alcohol becomes very small in the alkoxide, presumably due to the very early transition states arising from the very large exothermicity of the reaction. The C1–C4 and C2–C3 bonds in 6a, 6a′, 6b, and 6b′ show primarily single bond character (1.46–1.52 Å), which, along with the fact that the C1–C2 bonds are 1.34–1.36 Å, indicates a very early transition state.
In the gas phase reaction of the free alkoxide, the mechanism is a conrotatory electrocyclic ring opening pathway that is preferred by 1–3 kcal/mol, followed by cis-trans isomerization with a barrier of 6–12 kcal/mol to give the experimentally observed product (Figure 3). In the reaction model where oxygen is coordinated with potassium in THF, as seen in Figure 8, conrotatory ring opening is also preferred by about 1 kcal/mol; however, cis-trans isomerization is unfeasible at low temperatures as this barrier is 7 kcal/mol higher than the barrier for electrocyclic ring closing. This suggests a mechanism in which there is equilibration of conrotatory ring opening and closing with a percentage of non-pericyclic ring opening to give the thermodynamic product. In the gas phase reaction of the free alkoxide 5d, the mechanism is an allowed conrotatory ring opening followed by cis-trans isomerization to give 7d′. For the ring openings of potassium alkoxides 5e and 5f in THF, the mechanism follows a non-pericyclic ring opening that is 8 kcal/mol and 4 kcal/mol lower than conrotatory ring opening, respectively. This appears to be the mechanism studied by the Carreira group. ChelpG calculations of the ring opening pathways help show that the forbidden “disrotatory” mechanism is, in fact, non-pericyclic in nature.
Supplementary Material
Figure 15.

Optimized transition structure of isomerization of alkoxide intermediate 7d to 7d′. B3LYP/6-31G(d) selected bond distances (Å), angles, and dihedral angles are shown.
Scheme 3.

Proposed Mechanism of Cycloinsertion Reaction
Acknowledgments
We are grateful to the National Institutes of Health for financial support (R01 GM036700), and to Erick M. Carreira for helpful discussions.
Footnotes
Cartesian coordinates, computed total energies of all optimized structures, imaginary frequencies of all transition structures, and plots of ChelpG atomic charges. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tambar UK, Stoltz BM. J Am Chem Soc. 2005;127:5340–5341. doi: 10.1021/ja050859m. [DOI] [PubMed] [Google Scholar]
- 2.Gampe CM, Boulos S, Carreira EM. Angew Chem Int Ed. 2010;49:4092–4095. doi: 10.1002/anie.201001137. [DOI] [PubMed] [Google Scholar]
- 3.Gampe CM, Carreira EM. Angew Chem Int Ed. 2011;50:2962–2965. doi: 10.1002/anie.201007644. [DOI] [PubMed] [Google Scholar]
- 4.Gampe CM, Carreira EM. Angew Chem Int Ed. 2012;51:3766–3778. doi: 10.1002/anie.201107485. [DOI] [PubMed] [Google Scholar]
- 5.Comandini A, Brezinsky K. J Phys Chem A. 2012;116:1183–1190. doi: 10.1021/jp208368a. [DOI] [PubMed] [Google Scholar]
- 6.Frisch MJ, et al. Gaussian 09, revision A.02. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 7.Staroverov VN, Davidson ER. J Am Chem Soc. 2000;122:7377–7385. [Google Scholar]
- 8.Pieniazek SN, Clemente FR, Houk KN. Angew Chem, Int Ed. 2008;47:7746–7749. doi: 10.1002/anie.200801843. [DOI] [PubMed] [Google Scholar]
- 9.Fukui K. J Phys Chem. 1970;74:4161–4163. [Google Scholar]
- 10.Deng L, Ziegler T. Int J Quantum Chem. 1994;52:731–765. [Google Scholar]
- 11.Barone V, Cossi M. J Phys Chem A. 1998;102:1995–2001. [Google Scholar]
- 12.Takano Y, Houk KN. J Chem Theory Comput. 2005;1:70–77. doi: 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
- 13.Cheong PHY, Paton RS, Bronner SM, Im GYJ, Garg NK, Houk KN. J Am Chem Soc. 2010;132:1267–1269. doi: 10.1021/ja9098643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olivella S, Pericàs MA, Riera A, Solé A. J Org Chem. 1987;52:4160–4163. [Google Scholar]
- 15.Johnson RP, Daoust KJ. J Am Chem Soc. 1995;117:362–367. [Google Scholar]
- 16.Yavari I, Nasiri F, Djahaniani H, Jabbari A. Int J Quantum Chem. 2005;106:697–703. [Google Scholar]
- 17.Rondan NG, Houk KN. J Am Chem Soc. 1985;107:2111–2121. [Google Scholar]
- 18.Cooper W, Walters WD. J Am Chem Soc. 1958;80:4220–4224. [Google Scholar]
- 19.Dolbier WR, Jr, Koroniak H, Houk KN, Sheu C. Acc Chem Res. 1996;29:471–477. [Google Scholar]
- 20.Breulet J, Schaefer HF., III J Am Chem Soc. 1984;106:1221–1226. [Google Scholar]
- 21.Brauman JI, Golden DM. J Am Chem Soc. 1968;90:1920–1921. [Google Scholar]
- 22.Ross JA, Seiders RP, Lemal DM. J Am Chem Soc. 1976;98:4325–4327. [Google Scholar]
- 23.Fabian WMF, Bakulev VA, Kappe CO. J Org Chem. 1998;63:5801–5805. doi: 10.1021/jo980238u. [DOI] [PubMed] [Google Scholar]
- 24.Ji H, Li L, Xu X, Ham S, Hammad LA, Birney DM. J Am Chem Soc. 2009;131:528–537. doi: 10.1021/ja804812c. [DOI] [PubMed] [Google Scholar]
- 25.Lee PS, Sakai S, Hörstermann P, Roth WR, Kallel EA, Houk KN. J Am Chem Soc. 2003;125:5839–5848. doi: 10.1021/ja028963g. [DOI] [PubMed] [Google Scholar]
- 26.López CS, Faza ON, de Lera AR. Chem Eur J. 2007;13:5009–5017. doi: 10.1002/chem.200601151. [DOI] [PubMed] [Google Scholar]
- 27.Qin C, Davis SR. J Org Chem. 2003;68:9081–9087. doi: 10.1021/jo035168s. [DOI] [PubMed] [Google Scholar]
- 28.Johnson RP, Daoust KJ. J Am Chem Soc. 1996;118:7381–7385. [Google Scholar]
- 29.Inoue Y, Hagiwara S, Daino Y, Hakushi T. J Chem Soc Chem Commun. 1985;19:1307–1309. [Google Scholar]
- 30.Breneman CM, Wiberg KB. J Comput Chem. 1990;11:361–373. [Google Scholar]
- 31.Guner V, Khuong KS, Leach AG, Lee PS, Bartberger MD, Houk KN. J Phys Chem A. 2003;107:11445–11459. [Google Scholar]
- 32.Houk KN. Acc Chem Res. 1975;8:361–369. [Google Scholar]
- 33.Woodward RB, Hoffmann R. Angew Chem Int Ed. 1969;8:781–853. [Google Scholar]
- 34.Evans DA, Golob AM. J Am Chem Soc. 1975;97:4765–4766. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.