

SUMMARY
Promiscuous mutant EcoRI endonucleases bind to the canonical site GAATTC more tightly than does the wild-type endonuclease, yet cleave variant (EcoRI*) sites more rapidly than does wild-type. The crystal structure of the A138T promiscuous mutant homodimer in complex with a GAATTC site is nearly identical to that of the wild-type complex, except that the Thr138 side chains make novel packing interactions with bases in the 5′-flanking regions outside the recognition hexanucleotide, while excluding two bound water molecules seen in the wild-type complex. Molecular dynamics simulations confirm exclusion of these waters. The structure and simulations suggest multiple possible reasons why binding of A138T protein to the GAATTC site has ΔS° more favorable and ΔH° less favorable than for wild-type endonuclease binding. The novel interactions of Thr138 with flanking bases may permit A138T, unlike wild-type enzyme, to form complexes with EcoRI* sites that structurally resemble the specific wild-type complex with GAATTC.
INTRODUCTION
In order to understand how site-specific DNA binding proteins achieve functional specificity, we require an understanding of the structural and energetic rules that govern protein-DNA recognition. Restriction endonucleases must have a high level of sequence discrimination in order to carry out their function of inactivating unmethylated foreign DNA, while avoiding potentially lethal cleavage at related sites in the resident genomic DNA. For example, the EcoRI restriction endonuclease, which targets GAATTC, also cleaves at so-called EcoRI* sites that differ by one base-pair from GAATTC (e.g., AAATTC), but with a second order rate constant (taking into account both binding and cleavage) as much as 1011-fold lower (Jen-Jacobson, 1997). Nonspecific DNA (≥ 2 base pair change from GAATTC) is not cleaved (Lesser et al., 1990).
Crystal structures revealed the contacts between the protein and DNA that likely stabilize the specific complex (Kim et al., 1990; Grigorescu, 2003; Grigorescu, 2004), but structures of EcoRI* and nonspecific complexes have remained elusive, presumably due to the high degree of structural heterogeneity in the co-crystalline complexes. Using probes designed to assess phosphate contacts (Lesser et al., 1990), water release (Cao, 2002) and the ΔH°, ΔS° and ΔC°p (Jen-Jacobson et al., 2000a; Grigorescu, 2003) associated with binding to the three classes of DNA sites, Jen-Jacobson and colleagues have inferred that the specific, EcoRI* and non-specific complexes are profoundly different in structure. It was postulated that when binding to EcoRI* sites, wild-type endonuclease does not form a highly specific protein-DNA interface, but instead “adapts” structurally to optimize binding free energy. This structural adaptation, roughly similar for the various EcoRI* sites, comes at the cost of greatly diminished ability to reach the transition state for phosphodiester bond cleavage (Lesser et al., 1990; Jen-Jacobson, 1997).
Attempts to change the specificity of any restriction endonuclease have been uniformly unsuccessful. Exhaustive site-directed mutagenesis of the amino acid residues directly involved in EcoRI-DNA recognition did not yield variants with altered specificity, but rather mutants that have lost (or diminished) ability to cleave the cognate site with no concomitant gain in EcoRI* site cleavage activity (Alves et al., 1989; Needels et al., 1989; Heitman and Model, 1990a; Osuna et al., 1991; Fritz et al., 1998; Ivanenko et al., 1998). These results exemplify the difficulty of the problem: Recognition is not simply the sum of one-to-one interactions between protein side chains and DNA bases, but includes redundant interactions with both members of a base-pair, and conformational factors determined by sequence motifs larger than a single base-pair.
A genetic screen (Heitman and Model, 1990b) for relaxed-specificity mutations of EcoRI endonuclease produced point mutants A138T, A138V, E192K, H114Y and Y193H. The purified mutant proteins in vitro (Heitman and Model, 1990b) make double-strand cuts at EcoRI* sites in plasmid and bacteriophage DNA more readily than does the wild-type enzyme, and also retain normal levels of activity towards the canonical GAATTC recognition site. To further determine the thermodynamic and kinetic basis for promiscuity, we characterized promiscuous mutant proteins A138T, E192K and H114Y, with respect to binding affinities and first-order cleavage rate constants towards the three classes of DNA sites: specific, EcoRI* and nonspecific (Sapienza et al., 2005). The naive expectation was that a promiscuous mutant enzyme would have reduced affinity for the canonical recognition site GAATTC, and/or greater affinity for incorrect EcoRI* sites, but this was not the case. The mutant enzymes in vitro not only bind more tightly than the wild type enzyme to the specific recognition site GAATTC, but also have a greater preference for binding to the cognate site relative to EcoRI* sites. Crucially, since the mutations do not affect non-specific binding affinity, the preference for EcoRI* over non-specific DNA is increased. In addition, the mutant enzymes cleave the specific GAATTC site at a normal rate, but cleave EcoRI* sites faster than does the wild-type enzyme. Thus, the mutant enzymes are promiscuous for two related reasons: (1) Binding to EcoRI* sites is more probable than for wild type enzyme because nonspecific DNA is less effective as a competitive inhibitor; (2) The combination of increased affinity and faster cleavage at EcoRI* sites makes double strand cleavage of these sites a more probable outcome than it is for the wild type enzyme. The three mutant proteins therefore used a common set of altered binding and cleavage properties to achieve varying degrees of relaxed specificity. Interestingly, each of the mutated residues lies within or at the boundary of a subdomain of EcoRI endonuclease known as the arm (Figure 1B). Crystallographic studies showed that the arm is disordered in the absence of DNA and undergoes a folding transition coupled to specific DNA binding (Grigorescu, 2003). These observations led to the suggestion that the conformational change is involved in the discrimination mechanism of EcoRI and that the promiscuous mutants have a perturbed conformational change (Grigorescu, 2003).
Figure 1. Structures of wild type and mutant endonuclease-DNA complexes.

(A) DNA oligonucleotide used in crystallographic and molecular dynamics studies. The GAATTC recognition site is in red. (B) Cartoon rendering of the wild type EcoRId(TCGCGAATTCGCG) complex (Grigorescu, 2003). The main domains of the two enzyme subunits are colored red (foreground) and grey (background). The arms of both subunits are pink. The residues implicated in relaxed specificity by a genetic screen (Heitman and Model, 1990b) are rendered as sticks in the foreground subunit only. The labels identify the amino acid changes that lead to relaxed specificity. These amino acids all reside within the arms or at the junction between the arms and the main domain. (C) Superposition of the wild type (red) and A138T (blue) x-ray models. The two structures are aligned with protein backbone, DNA all-atom and all non-solvent atom root mean square deviations of 0.3 Å, 0.4 Å and 0.8 Å respectively. Thr138 side chain in both subunits of the mutant is highlighted in yellow. (D-E) DNA recognition by the EcoRI extended chain motif (residues 137-142). 2Fo-Fc simulated annealing omit maps are contoured at 1.5 σ in both panels with mesh covering protein in red and DNA in blue. (D) wild type complex; (E) A138T mutant complex. Hydrogen bonds are depicted by black dashes. All hydrogen bonds made by amino acids of the extended chain motif in the wild type model are conserved in the mutant structure.
While these findings provided a satisfying explanation for relaxed specificity in vivo, we are ultimately interested in understanding how the mutations affect molecular recognition. We present here a structural characterization of the complex of the A138T promiscuous mutant protein with its specific DNA site. We chose A138T because it exhibited the most severe relaxed specificity phenotype (i.e., most severe lethality) in the genetic screen (Heitman and Model, 1990b), showed the greatest departure from the wild type enzyme in equilibrium association constants and first-order cleavage rate constants with EcoRI* sites (Sapienza et al., 2005), and because we have the largest body of data on the interactions of this mutant with non-cognate DNAs. We use x-ray crystallography, molecular dynamics simulations and dissection of thermodynamic parameters to gain insight into the physical basis for the observed increase in specific DNA binding affinity exhibited by the A138T promiscuous mutant. The data allow us to address the following questions: (1) Are the structural differences between the wild type and mutant complexes consistent with measurements of changes in enthalpy (ΔH°), entropy (ΔS°), and heat capacity (ΔC°P) for the specific DNA binding reactions? (2) How does the structure of the A138T specific complex help us to understand the basis for relaxed DNA sequence specificity of this enzyme?
RESULTS
Choice of oligonucleotide substrates
To facilitate comparison between structural and energetic data, we used similar DNA substrates for the crystallographic, molecular dynamics, and van't Hoff studies. The flanking sequence around the GAATTC recognition site, which significantly affects binding free energy ΔG° (Jen-Jacobson, 1997) as well as ΔH° and ΔS° (Jen-Jacobson et al., 2000a), is maintained constant in all DNA oligonucleotides used in this work unless otherwise specified. The only difference in the constructs is that we used a 24 base pair oligonucleotide for the van't Hoff experiments and a 12-bp oligonucleotide with a 5'- unpaired thymine for growing crystals as well as for the molecular dynamics simulations. The difference in DNA length is insignificant, as the EcoRI endonuclease binds equally well to the 12 and 24 base pair oligonucleotides containing the specific site (M. Kurpiewski, A. Grigorescu and L. Jen-Jacobson, unpublished).
Crystal structure and molecular dynamics simulations of the A138T promiscuous enzyme complexed with specific DNA
We determined the crystal structure of the A138T protein in complex with a 12-bp DNA, to a resolution of 1.95Å (See Experimental Procedures for data collection and refinement statistics). Prior to initiation of this work, a 1.87Å crystallographic model of the wild type EcoRI complex was submitted to the Protein Data Bank (accession code 1CKQ, M. Horvath and J.M. Rosenberg, unpublished data). The wild type model we used for comparison was a highly refined version of 1CKQ (Grigorescu, 2003). This model is superior to 1CKQ based upon reductions in R (0.21 vs. 0.24) and Rfree (0.25 vs. 0.29), and decreases in real space R factors throughout the model (Grigorescu, 2003). The same DNA oligonucleotide was used in the wild type and A138T complexes; the sequence and the numbering scheme of the DNA bases are shown in Figure 1A.
Global features of the A138T and wild type EcoRI-DNA complexes are very similar. As is the case with each of the other EcoRI endonuclease structures (PDB accession numbers: 1CKQ, 1QPS, 1ERI, 1QRH, 1QRI, and 1CL8), the N-terminal 16 amino acids of each subunit are not visible in electron density maps and hence not included in the model of the A138T complex. The two structures in Figure 1C can be superposed with protein backbone, DNA all-atom and all non-solvent atom root mean square deviations of 0.3 Å, 0.4 Å and 0.8 Å respectively. The secondary structure assignments based on the STRIDE algorithm (Heinig and Frishman, 2004) are the same for all amino acids in the wild type and mutant models. In addition, the two nucleic acids exhibit the same types of deviations from idealized B-DNA in the form of the previously described EcoRI kink (Kim et al., 1990; Kumar et al., 1994; Grigorescu, 2003) (Local base pair and base step parameters assigned with 3DNA (Lu and Olson, 2003) are in Supplemental Data, Tables S1-S3).
Residue 138 is located within the ‘extended chain motif’ (Kim et al., 1990), comprising 137 to 142. This element contains multiple groups that make site-specific contacts to the recognition sequence (Figure 1D). The ß-branched threonine side chain is accommodated within the extended chain motif without any significant structural adaptations (rmsd = 0.16 Å for residues 137-142) and the ϕ, ψ angles for residue 138 remain the same within 2-3°. Indeed, all contacts between residues of the extended chain motif and the GAATTC bases are intact in the A138T complex (Figure 1E), as are the remaining contacts to the bases within the recognition site. There are no novel contacts made by the A138T enzyme to the GAATTC bases, showing that this is not the reason for the improved binding of the mutant enzyme. The observation that the GAATTC recognition interfaces are similar is consistent with our previous finding (Sapienza et al., 2005) that first-order cleavage rate constants are the same for the wild type and A138T enzymes at the GAATTC site, since these rate constants are exquisitely sensitive not only to the local geometry of the reacting groups but also to the global protein-DNA interactions that stabilize the transition state (Lesser et al., 1990; Jen-Jacobson, 1997).
The effects of the A138T mutation on specific DNA binding are at least in part due to changes in solvent mediated contacts to bases upstream of the recognition site. (We built water molecules into all solvent peaks observed in the wild type and mutant electron density maps according to strict criteria given in Experimental Procedures. The identification of the critical molecules as waters rather than other small ions is supported by the occupation of these positions by explicit waters from bulk solvent in our molecular dynamics simulations; see below. We refer to these water molecules by their PDB file residue identification numbers.) In the wild type model ((Grigorescu, 2003), water41 bridges the side chain of Arg200 and Ile197-carbonyl to the 4-amino group of the cytosine immediately upstream of the recognition site (C−4) (Figure 2A). Water41 thus serves to couple flanking sequence interactions to GAATTC base recognition, inasmuch as Arg200 also contacts water1, which bridges Arg203 to N7 and to O6 of G−3, the first base of the recognition site (Figure 3A) (Grigorescu, 2003). (Ala139 also hydrogen bonds with both Arg200 and Ile197 to complete the network.) Water41 is well ordered, as its corresponding electron density is visible in omit maps, and the B-factor is similar to those associated with waters that bridge amino acid moieties to DNA functional groups within the recognition site.
Figure 2. Effects of the A138T mutation on water mediated contacts to bases flanking the GAATTC recognition site.
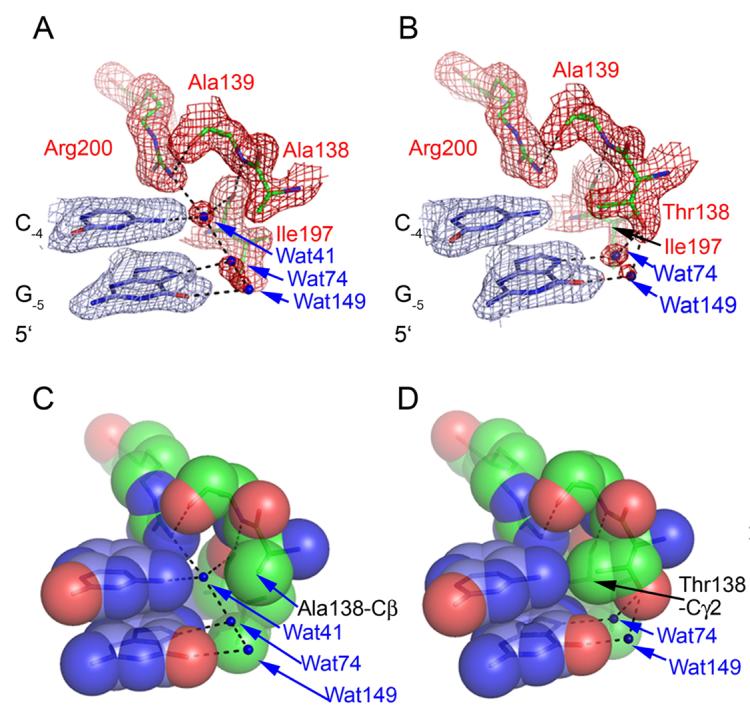
2Fo-Fc simulated annealing omit maps covering the wild type (A) and A138T (B) structures in the vicinity of G-5C-4. Maps are contoured at 1.5s in both panels with mesh covering protein in red and DNA in blue. Water molecules are blue spheres and hydrogen bonds are depicted as black lines in all panels. (C) and (D) show spacefill renderings of the same views given in (A) and (B). Note the novel water mediated interactions between Thr138 and G-5, the packing interactions of the Thr138 g-methyl group with C-4 -N4 and G-5-N7, and the absence of water41 in the mutant model.
Figure 3. Water network at the 5'-side of the recognition site.
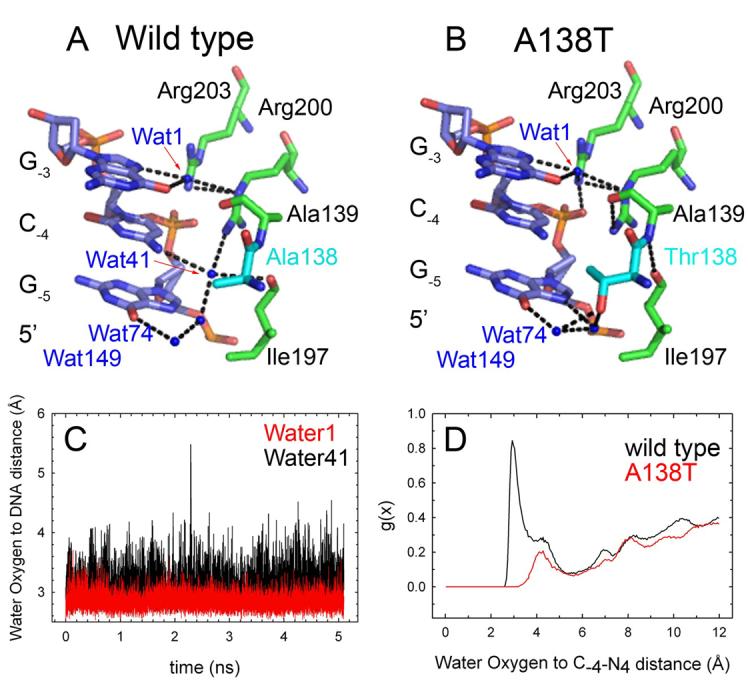
(A) Wild type model. Coupling between water1, which contacts G-3 in the recognition site, and water41, which contacts the flanking base C-4. Note that Arg200-Nε interacts with water1 and Arg200-NH1 with water41. Water1 is involved in similar contacts in the A138T structure. Arg200-NH1 is hydrogen bonded to Asn199 and Gly196 carbonyls (not shown). (B) A138T model. The position and contacts of water1, water74 and water149 are conserved despite the absence of water41. (C) Water oxygen to DNA functional group distances calculated over the course of a 5 ns MD simulation of the wild type-d(TCGCGAATTCGCG) complex at 300K. The plots refer to sites occupied by water molecules in positions analogous to water41 (black) and water1 (red) in the X-ray model. The plot shows that 1) The water41 binding site occupied in the 100K model is also occupied in the 300K complex, and 2) The residence time of water41 is similar to that of a solvent molecule that is buried at the GAATTC recognition site. (D) Radial distribution functions (g(x)) for water oxygens relative to the 4-amino group of C-4. Data are from wild type (black) and A138T (red) simulations. The A138T plot shows no peak within hydrogen bonding distance, so this binding site is not occupied during the 300K simulation of the A138T complex. Plots (B) and (C) are for one half-site only, but results are typical of both half-sites.
To ask if features of the average structures determined from diffraction data determined from crystals quenched to 100K are typical of the structural ensemble at room temperature, we performed 5 ns MD simulations of the wild type and A138T complexes in the presence of explicit solvent at 300K. In the wild-type simulation at 300K, one water molecule remains hydrogen bonded to the 4-amino group of C−4 over the entire simulation; this long residence time is typical of other solvent molecules buried at the interface (Figure 3C). The other protein-solvent and protein-protein hydrogen bonds that stabilize the water41 network observed in the cryostructure (Figures 2A and 3A) are also reproduced throughout the simulation (Supplementa Data, Figure S1). In a separate MD simulation at 300K, we stripped water41 away in silico prior to initiation of simulation, and observed that this position in both subunits was rapidly filled by water from bulk solvent. This position showed 100% occupancy throughout a 6 ns simulation, and after an initial period (1-3 ns) of exchange with bulk solvent became stably occupied by single water molecules with residence times up to 5 ns (Supplemental Data, Figure S2). (There was no significant difference in the stripped and unstripped starting structures.) Simulations of the Trp repressor-operator complex (Wibowo et al., 2004) also showed that water molecules with multi-ns residence times participate in contacts that are critical to complex stability as deduced from site-directed mutagenesis studies (Joachimiak et al., 1994).
In contrast to the wild type complex, electron density maps of the mutant complex show no density corresponding to water41 (Figure 2B). In its stead, the γ-methyl group of the Thr138 side chain packs against the 4-amino group of C−4 such that the water molecule hydrating the 4-amino group in the free DNA must be released upon DNA binding (Figure 2D). Superposition of the wild-type and A138T crystal structures shows that steric clash with the Thr138 side chain would result if a solvent molecule assumed the position occupied by water41 in the wild-type complex. It is important that despite the absence of water41 in the A138T-DNA interface, the mutant complex retains (compare Figures 3A and 3B) the protein-protein buttressing contacts and protein-solvent contacts that hold Arg200 in place, and stabilize the network of hydrogen bonds connecting Arg200 to Ala139, Ile197 to Ala139, and Arg200 to water1. Other members of the water network (water74 and water149) also remain in place. The absence of water41 leaves only one hydrogen (Arg200-guanidino) without a hydrogen bonding partner, but this contacts Thr138-γ-CH3 (Figures 2B, 2D, 3A, 3B).
Molecular dynamics simulations of the A138T complex also support the absence of the solvent molecule in the complex at 300K. Figure 3C shows a large peak in the water to C−4-N4 radial distribution function at the appropriate hydrogen bonding distance over the wild type simulation, whereas there is no water peak at this position during the simulations of the mutant complex. Furthermore, we “mutated” the wild-type complex to A138T in silico (leaving water41 in place) prior to simulated annealing and MD simulation at 300K. We observed that water41 was ejected whenever Thr138 assumed the rotamer conformation (χ1 ≈ −60°) observed in the crystal structure, and a solvent molecule returned when Thr138 later transiently sampled the χ1 ≈ +60° rotamer (Supplemental Data, Figure S3). As an adventitious internal control, Thr138 in the other subunit never visited the χ1 ≈ −60° rotamer during the 3.4 ns simulation, and here the water41 position remained occupied by a succession of solvent molecules with residence times on the order of 1-1.5 ns. These parallel observations establish a strong correlation between Thr138 rotation and the presence or absence of water41.
One more base away from the GAATTC recognition site, at G−5, there are additional differences between the wild type and mutant structures. First, the γ-methyl group (which contacts C−4-N4, see above) also packs against the N7 atom of G−5 (Figure 2D), which nevertheless remains solvated by water74 (Figures 2B and 2D). Whereas the wild type enzyme achieves shape complementarity with the flanking bases G−5 and C−4 only through reliance on ordered solvent molecules to match the surfaces of the two macromolecules, the A138T enzyme contacts these bases directly. Second, the Thr138 side-chain hydroxyl makes water-mediated contacts, via water74 to G−5-N7 and water149 to G−5-O6 (Figure 2B). Both these guanine functional groups are hydrated in the wild type complex, but no amino acid moiety is close enough to bridge the solvent molecule(s) to the DNA (Figure 2A). Omit maps support the presence of both water molecules in the mutant model; however, the molecule solvating the N7 group has stronger associated density than the molecule bridging the interaction with O6. This suggests that the solvent binding site at N7 may have a higher fractional occupancy in the crystalline protein-DNA complexes or that this solvent molecule is less mobile than the one associated with O6.
To ask if the contact bridged by the higher occupancy solvent molecule is responsible for the improved binding of the mutant enzyme, we replaced G−5 with 7-deazaguanine (7cG), in which a carbon atom replaces the N7 atom. If the contact were responsible for the improved binding of the mutant protein, one would predict for the 7cG substrate a reduction in the binding enhancement for A138T protein relative to the wild type. Surprisingly, both enzymes bind more favorably to the substrate containing the 7cG modification than to the unmodified DNA (Table 1) and this effect is greater for the mutant enzyme. (As a control, a 7cG substitution at a position more distal to the recognition site has no effect on binding of either protein (Table 1)). Thus, taking away the water mediated contact enhances rather than diminishes the binding advantage of A138T over wild-type enzyme, and argues that this contact does not play a key role in the binding affinity enhancement observed for A138T protein with the unmodified DNA. Molecular dynamics simulations also suggest that the novel water-mediated interactions between Thr138 and G−5 do not play a dominant role in the differences in specific binding, based on the aggregate of the following observations: 1) The N7 and O6 functional groups are hydrogen-bonded to water molecules throughout the duration of both wild type and A138T simulations. 2) Whereas only two water molecules visit the binding sites that bridge contacts to bases in the recognition sequence during the course of the 5 ns MD simulations, 50 waters visit each of the G−5 binding sites. 3) There is no significant difference between the wild type and A138T simulations in water residence times at the G−5-N7 binding site (Figure S4).
Table 1.
Effect of 7-deazaguaninea substitutions in flanking DNA on EcoRI specific binding
| Specific Sequence b | KAc (M−1) |
Substitution effect ΔΔG°bindd (kcal/mol) |
Mutation effect ΔΔG°binde (kcal/mol) |
|---|---|---|---|
| Wild type | |||
| GGGCGGGCGCgaattcGCGGGCGC | |||
| CCCGCCCGCGcttaagCGCCCGCG | 7.9 (± 0.4)×108 | 0 | 0 |
| GGGCGGGC7cGCgaattcG CGGGCGC | |||
| CCCGCCCG CGcttaagC7cGCCCGCG | 1.2 (± 0.1)×1010 | −1.6 ± 0.1 | 0 |
| GGGC7cGGGCGCgaattcGCGGG CGC | |||
| CCCG CCCGCGcttaagCGCCC7cGCG | 7.5 (± 0.3)×108 | 0 ± 0.1 | 0 |
| A138T | |||
| GGGCGGGCGCgaattcGCGGGCGC | |||
| CCCGCCCGCGcttaagCGCCCGCG | 9.4 (± 0.4)×109 | 0 | −1.5 ± 0.1 |
| GGGCGGGC7cGCgaattcG CGGGCGC | |||
| CCCGCCCG CGcttaagC7cGCCCGCG | 8.8 (± 1.2)×1011 | −2.7 ± 0.1 | −2.5 ± 0.1 |
| GGGC7cGGGCGCgaattcGCGGG CGC | |||
| CCCG CCCGCGcttaagCGCCC7cGCG | 7.3 (± 0.2)×109 | 0.2 ± 0.1 | −1.3 ± 0.1 |
The abbreviation, 7cG, denotes 7-deaza-Guanine.
Complete sequence of 24 base-pair oligonucleotide. The specific recognition site, gaattc, is underlined.
Equilibrium association constants were measured in binding buffer plus 0.28 M KCl (pH 7.3, 21°C); means ± std. dev. of ≥ 3 determinations for each sequence.
Thermodynamic dissection of ΔG° for specific DNA binding
Each of several promiscuous mutant EcoRI endonucleases is characterized by improved binding to the specific recognition sequence, relative to the wild type endonuclease (Sapienza et al., 2005). Implicit in these differences in ΔG°bind is that the constituent ΔH° and ΔS° must differ as well. We extracted thermodynamic parameters from the temperature dependence of the equilibrium association constant using two methods: The first uses a multiparametric fit that assumes that ΔC°P for the binding reaction is independent of temperature (see Experimental Procedures). The second method of analysis, developed by Clarke and Glew (Clarke and Glew, 1966; Dec and Gill, 1985) assumes only that KA and the thermodynamic parameters ΔG°, ΔH°, ΔS° and ΔCp are continuous functions of T (see Experimental Procedures). Importantly, we obtained the same values of ΔH°, ΔS°, and ΔC°P from both methods (Table 2), and the latter confirmed that ΔC°P is temperature-independent in the range of our experiments. Further, the mutation-induced changes we note below are significant as the differences in ΔH° and TΔS° derived from both fitting methods are at least twice the propagated error.
Table 2.
Thermodynamic parameters for specific EcoRI binding at 25°Ca
| Wild type | A138T | |||
|---|---|---|---|---|
| Multiparametric fitb |
Clarke and Glewc |
Multiparametric fitb |
Clarke and Glewc |
|
| ΔG° (kcal/mol) | −13.1 ± 0.1 | −13.0 ± 0.01 | −14.6 ± 0.1 | −14.6 ± 0.02 |
| ΔH° (kcal/mol) | −15.4 ± 0.4 | −15.1 ± 0.5 | −12.8 ± 0.3 | −12.9 ± 0.6 |
| TΔS° (kcal/mol) | −2.3 ± 0.2 | −2.1 ± 0.5 | 1.8 ± 0.3 | 1.7 ± 0.6 |
| ΔC°p (kcal/mol•K) | −1.8 ± 0.1 | −1.9 ± 0.1 | −1.90 ± 0.1 | −1.9 ± 0.1 |
| dΔC°p/dT (kcal/mol•K2) | - | −0.01 ± 0.04 | - | 0.0 ± 0.02 |
| ΔΔG° (kcal/mol)d | 0 | 0 | −1.5 ± 0.1 | −1.6 ± 0.1 |
| ΔΔH° (kcal/mol) | 0 | 0 | 2.6 ± 0.5 | 2.2 ± 0.8 |
| Δ(TΔS°) (kcal/mol) | 0 | 0 | 4.1 ± 0.4 | 3.8 ± 0.8 |
| ΔΔC°p (kcal/mol•K) | 0 | 0 | −0.1 ± 0.1 | 0.0 ± 0.1 |
| Δ(dΔC°p/dT) (kcal/mol•K2) | - | 0 | - | 0.01 ± 0.03 |
Equilibrium association constants (KA) were measured as a function of temperature in 10mM cacodylate, 0.27M KCl, pH 7.3, using the oligonucleotide: 5'GGGCGGGCGCGAATTCGCGGGCGC (specific recognition sequence underlined).
See Experimental Procedures
See Experimental Procedures
All difference parameters (e.g., ΔΔG°) are referenced to the wild-type.
Formation of the A138T-GAATTC complex has a less favorable ΔH° than does wild type binding by +2.6 kcal/mol at 25°C (Figure 4, Table 2). This loss of favorable ΔH° is more than compensated by a +4.1 kcal/mol increase in TΔS° for mutant binding, such that net ΔG°bind is more favorable for the mutant protein. Both the ΔΔH° and TΔΔS° reported at a single temperature in Table 2 are constant over the experimental temperature range because the temperature dependence of ΔH° and ΔS° (that is, ΔC°P) is the same for both enzymes (Figures 4B and 4C). The identical values of ΔC°P further imply that the major factors that influence ΔC°P (water release from polar and nonpolar surfaces, conformational-vibrational restriction in the complexes, molecular strain (Jen-Jacobson et al., 2000a,b) must be nearly the same for the two complexes.
Figure 4. Temperature dependence of wild type and A138T EcoRI endonuclease binding to specific DNA.

(A) van't Hoff plot showing the effect of temperature on wild type (filled circles) and mutant (open circles) binding to 5'-GGGCGGGCGCGAATTCGCGGGCGC. (B) and (C) show the contributions to binding free energy of ΔH° and TΔS°, respectively, for wild type (filled circles) and A138T (open circles) binding over the experimental temperature range. See Experimental Procedures for data fitting methods.
The ΔC°P for wild type endonuclease binding reported here (−1.8 kcal/mol•K in 0.27M KCl) is less negative than that measured previously (Jen-Jacobson et al., 2000a) for binding to the same sequence (−2.5 kcal/mol•K in 0.18M NaCl), reflecting the fact that the two measurements were made at different salt concentrations. Salt affects both the ΔH° and ΔC°P associated with DNA binding (Kozlov and Lohman, 1998; Kozlov and Lohman, 2000; Holbrook et al., 2001; Poon and Macgregor, 2004) (P. Sapienza, T. Niu and L. Jen-Jacobson, unpublished), such that ΔC°P becomes less negative with increasing salt concentration.
Role of flanking sequence recognition in A138T relaxed specificity
Heitman and Model first noticed, in multiple turnover DNA cleavage experiments, that the A138T promiscuous mutant exhibited a preference for cutting phage f1 EcoRI* sites that are flanked by a 5' thymine (Heitman and Model, 1990b). When we looked more closely at these preferentially cleaved EcoRI* sites, we observed that every one of the non-cognate half sites is abutted by a 5'-RT dinucleotide motif (R = purine). This impelled us to a more systematic comparison of the influence of flanking context on cleavage of an EcoRI* site (AAATTC) by both wild-type and A138T enzymes. The wild-type endonuclease shows substantial variation of EcoRI* cleavage rate constant with the flanking context, but the A138T enzyme (Figure 5) shows two distinct groups of flanking context effects: (a) Sites where an RY motif flanks the recognition site (i.e., RYAAATTC) are cleaved most rapidly. There is only a weak dependence of kcleave on the particular nucleotides of the RY motif, and the third nucleotide from the site (i.e., X in XRYAAATTC) has only a slight modulating effect. (The slow cleavage of the site flanked by GGC is an unexplained anomaly.) (b) For those contexts that do not have the flanking RY, there is a much more pronounced dependence of kcleave on flanking context (Figure 5), in this respect resembling the behavior of the wild-type enzyme with EcoRI* sites. Notably, the values of kcleave are 20- to 50-fold greater for the flanking contexts with the RY motif than for those that lack the flanking RY. The division of the flanking dinucleotides into two classes reflects the favorable packing of T138 with the flanking RY motif (see Discussion).
Figure 5.

Effect of flanking sequence on first-order cleavage rate constants for an EcoRI* site. Bars show first order rate constants (note log scale) for cleavage of the AAATTC site, embedded in different flanking contexts. (A) k1 represents cleavage in the unmodified GAA half site. (B) k2, cleavage in the modified AAA half site. Black bars in each pair denote wild type and open bars denote A138T.
DISCUSSION
In this work we described the crystal structure of a mutant EcoRI endonuclease with relaxed cleavage specificity, in complex with its cognate DNA recognition sequence. The structure shows that the A138T mutation affects contacts to the DNA bases flanking the GAATTC site. The fact that the wild type and A138T structures have such high global similarity permits us to focus our attention on these changes in the flanking regions of the complex, which are proximal to the site of mutation. The principal questions to consider are: (a) How do these localized changes in structure of the specific complexes inform us about the origins of the observed differences in the thermodynamic parameters for binding? (b) What insights do the structures provide into the causes of the relaxed DNA sequence specificity of the A138T enzyme?
It is usually impossible to associate observable net differences in thermodynamic parameters with specific differences between two structures, primarily because even a perturbation as simple as a single amino acid substitution causes multiple structural changes, which often have opposing thermodynamic effects. Nevertheless, we can often understand whether any given structural feature influences ΔH° and/or ΔS° and, if so, the sign of the contribution(s). Rough estimates of these contributions can sometimes be made based on studies with model compounds. The existence of imperfect entropy-enthalpy compensation in the interactions of macromolecules with each other and with solvent (Jen-Jacobson et al., 2000b) means that the effect of any given structural feature on the observed net ΔG° is nearly impossible to evaluate.
Relationship between structural and thermodynamic data
The promiscuous A138T mutant protein binds more strongly (ΔΔG° = −1.5 kcal/mol) than wild-type protein to the specific recognition site GAATTC. Our thermodynamic analyses showed that this advantage arises because A138T binding to DNA is entropically more favorable (TΔΔS° = +4.1 kcal/mol at 298K) and enthalphically less favorable (ΔΔH° = +2.6 kcal/mol) relative to the wild type protein. Inspection of a “thermodynamic pseudocycle” (Jen-Jacobson, 1995) (Figure 6) for these binding reactions shows that
where the subscript “mut” refers to the hypothetical free energy effect of making the A138T mutation. It follows that the experimental difference in binding free energies
is given by
Corresponding equations can also be written for the state variables ΔH° and ΔS° associated with each process. In principle, the A138T mutation may affect the enthalpy, entropy and free energy of the protein-DNA complex, of the free protein, or of both. The crystal structures of the two complexes may inform us about structural factors that alter ΔG°mut,complex, but absent a crystal structure of the free A138T protein, we can only conjecture about how the mutation might affect ΔG°mut,free. We consider below the structural factors (Table 3) that may influence the thermodynamic parameters. It will be seen that all these factors are in fact various aspects of the differences between Ala138 and Thr138 in interacting with solvent, with other parts of the free protein and with the DNA.
Figure 6. Thermodynamic pseudocycle for the A138T mutation.

The arrows denote the free energy effects of the A138T mutation on the free protein (vertical arrow left) and on the protein-DNA complex (vertical arrow right), and the relation of these to the observable binding free energy changes (horizontal arrows) for mutant and wild-type. proteins. This is a “pseudocycle” because the vertical arrows do not correspond to observable physical processes.
Table 3.
Predicted effect of structural differences on thermodynamics of complex formationa
| Structural difference: A138T vs. wild type a |
Predicted effect on ΔΔS°bind | Predicted effect on ΔΔH°bind |
|---|---|---|
| Desolvation of nonpolar surface of larger Thr side chain |
More favorable | Little effect b |
| Reduced conformational entropy of arms in free protein? |
More favorable | Little effect |
| Loss of water41: desolvation of polar DNA and protein groups c |
More favorable | More unfavorable |
| Thr138 adopts single χ1 rotamer | More unfavorable d | Little effect |
| Novel packing interactions between Thr138 and C−4G−5 |
Rotamer restriction as above, plus unfavorable vibrational restriction of interacting DNA groups |
More favorable |
| Two new water mediated contacts to G−5 |
More unfavorable e | Little effect e |
Desolvation of nonpolar groups contributes little to ΔH° at room temperature.
The polar groups are C−4-N4, Arg200-guanidino and Ile197-carbonyl.
This will have no effect if the rotamer in the free protein is similarly restricted (see text).
Molecular dynamics simulations and equilibrium binding studies with the base analog 7-deazaguanine (Table 1) do not support a role for these water mediated contacts in stabilizing the A138T complex.
The absence of water41 from the mutant complex
Water41 in the wild type complex (Figure 3A) hydrogen bonds directly to Arg200-guanidino, Ile197-carbonyl, C−4–N4, and another solvent molecule (water74) that interacts directly with G−5; however, water41 is absent in the A138T complex. The local network of bases, sidechains and waters 74 and 149 remains otherwise intact (Figures 3A, 3B). How does releasing these two waters (one from each half site) from the complex affect ΔG°mut,complex and its constituent ΔH° and ΔS° terms? Dunitz (Dunitz, 1994) compared the standard entropies of crystalline hydrates with the standard entropies of the corresponding anhydrous salts, and from this estimated that the gain in entropy associated with the release of a “frozen” water molecule into the bulk solvent had an upper limit of TΔS° = +2 kcal/mol at 300 K. Water41 in the wild-type EcoRI-DNA complex is highly immobilized due to its participation in an extensive hydrogen bond network (Figure 3A) involving an amino acid side chain, two main-chain atoms, DNA and another solvent molecule. Thus, the immobilization of water41 in the wild-type complex may exact a free energy penalty (of entropic origin) up to +2 kcal/mol per water, or +4 kcal/mol for the two half-sites. It follows that the absence of water41 from the A138T complex could make a favorable entropic contribution up to −4 kcal/mol to ΔG°mut,complex.
At the same time, the absence of water41 at the A138T interface may contribute unfavorable ΔH°mut,complex from loss of the water mediated contact and desolvation of the polar groups C−4-N4, Arg200-guanidino and Ile197-carbonyl. This is consistent with our observation that A138T binding is more enthalpically unfavorable than wild type binding. However, dissecting this contribution from the observed ΔΔH° of +2.6 kcal/mol is not straightforward, because the water-mediated contact found in the wild-type complex is replaced in the mutant complex by an induced dipole-dipole interaction between the γ-methyl of Thr138 and 4-amino of C−4 (see below). Computational studies of the electrostatics of GCN4-DNA interaction (Hendsch and Tidor, 1999) indicate that the net of these factors is likely to be an unfavorable contribution to ΔΔH°mut,complex.
Entropic effects of the Thr138 side chain
The introduction of the bulkier Thr138 sidechain may have two distinct effects on net entropy change ΔS°: (a) A reduction of sidechain rotamer entropy in the mutant complex, as evident in the MD simulations; (b) A possible decrease in conformational entropy of the peptide backbone in the arms of the free protein. Note that these two roles would have opposite effects on the net binding entropy ΔS°. Detailed considerations are as follows:
(a) It is clear from our molecular dynamics simulations of the A138T-DNA complex that the mutation leads to a small loss in side chain conformational entropy relative to the wild type. We find that the Thr138 side chain adopts a single χ1 rotamer throughout the entire 5 ns simulation (Supplemental Data, Figure S5), evidently because the rotamer is maintained in place by the packing interactions between the Thr138 side chain and the G−5C−4 bases. The hydrogen bond between the main chain carbonyl of amino acid 138 and the 4-amino group of C3 will probably fix the conformation of the main chain. A backbone dependent rotamer library (Dunbrack and Cohen, 1997) shows that 95% of threonines with the ϕ,ψ backbone angles characteristic of residue 138 (these differ little between wild type and mutant complexes) adopt the same χ1 conformation that we observe in the crystal structure and during the simulations of the A138T complex. Modeling with a tripeptide that maintains the backbone conformation found in the crystal structures shows that this is the only χ1 rotamer that avoids steric clashes between the Thr138 sidechain and the peptide backbone. Therefore, rotation of the threonine χ1 dihedral angle is unlikely. By contrast, the Ala138 side chain explores all three gauche χ1 configurations during our simulations (Supplemental Data, Figure S5), as required by symmetry. This reduction in the number of side-chain degrees of freedom could lead to an entropic loss for each Thr of as much as Rln(1/3) = −2.2 cal/mol•K , or a contribution for two Thr of the EcoRI dimer of +1.3 kcal/mol (at 298K) to ΔG°mut,complex. Note, however, that this effect has an opposite sign from that of the experimentally observed ΔΔS°bind.
We also point out that this contribution will be realized if and only if the same rotamer restriction does not exist in the free mutant protein. Residues 116-140 are invisible in the crystal structure of the free wild-type enzyme except where stabilized by lattice contacts (Grigorescu, 2003), which may mean that they are disordered. If, as seems likely, disorder allows Thr138 more conformational freedom than it has in the complex (i.e., access to other ϕ,ψ angles) and consequently more rotamer choices, then the mutation may have little effect on ΔS°mut,free and the entropic difference between the complexes due to rotamer restriction (ΔS°mut,complex) would be fully realized in ΔΔS°bind and ΔΔG°bind. On the other hand, if Thr138 suffers rotamer restriction in the free enzyme as it does in the complex (this is less likely, but cannot be excluded by the invisibility of this region in the crystal structure of the free protein), this would cancel the rotamer entropy difference between the complexes and there would be little significant contribution to ΔΔS°bind or to ΔΔG°bind.
(b) An entropic effect of distinct origin and opposite sign might arise because the bulkier Thr138, lying at a hinge region between the arms and the ordered core, restricts peptide backbone conformations in the free enzyme. Residues 138 of the two subunits lie in symmetrical “inner arms” that extend from the main domains of the endonuclease and, in both wild-type and A138T complexes, enfold the DNA such that the 138 side chains are directed toward the DNA. The arms of the wild-type protein may become fully ordered only upon DNA binding (Grigorescu, 2003). Grigorescu et al., proposed that in the nascent specific complex, such boundary regions make protein-DNA contacts that restrict their conformational freedom, enabling them to serve as nucleation points for further ordering of the arms (Grigorescu et al., 2004). This nucleation potential would be enhanced by the additional protein-DNA contacts in the mutant complex. If the bulkier, ß-branched Thr138 side chain reduces the conformational entropy of the arms of the free A138T protein (Table 3), this would make ΔS°mut,free more unfavorable, with the consequence that ΔΔS°bind would be entropically more favorable, in accord with the thermodynamic observations. A significant role for the ß-branch is consistent with the fact that mutant A138V also has promiscuous properties (Heitman and Model, 1990b).
Direct and water-mediated interactions of Thr138 and G−5C−4
The γ-CH3 group of Thr138 packs against G−5-N7 and C−4-N4. The additional packing contacts in the mutant complex would make a favorable contribution to ΔH°mut,complex, but may also cause a reduction in the conformational-vibrational entropy of the interacting groups, resulting in an unfavorable contribution to ΔS°mut,complex.
The crystal structure of the mutant complex also shows two water-mediated contacts between Thr138-OH and the O6 and N7 atoms of G−5. These bridging interactions are not present at the wild type interface, since the Ala138 side chain is not large enough to reach the DNA. However, experiments with the 7cG base-analog (Table 1) do not support a role of the contact to G−5-N7 in stabilizing the mutant complex. Furthermore, it is important to note that these DNA functional groups are solvated in the free DNA (Shui et al., 1998) as well as in the crystal structures of both the wild type and A138T complexes. Molecular dynamics simulations do not show longer residence times for these water molecules in the mutant simulations relative to those of the wild type complex. Thus, we do not believe that these water mediated contacts contribute significantly to ΔG°mut,complex.
Solvation-desolvation of nonpolar groups of the Thr138 side chain
The packing interactions described above have the effect of excluding the ß-methylene and γ-methyl groups of Thr138 from solvent. Using NACCESS (Hubbard and Thornton, 1993), we have calculated that upon DNA binding, Thr138 contributes 22Å2 more buried non-polar surface than does Ala138. This has a favorable effect on ΔΔS°bind, but little or no effect on ΔΔH°bind (Table 3).
Balancing favorable and unfavorable factors
To summarize, the various structural consequences of the A138T mutation, as well as effects on the dynamics of both the free protein and the protein-DNA complex, may all contribute (Table 3) to the somewhat unexpected thermodynamic differences, in which ΔS°bind is more favorable and ΔH°bind less favorable for the A138T mutant protein relative to wild-type. By examining the structures of the mutant and wild-type complexes, attention is easily drawn to the novel packing interactions of Thr138 with the DNA, which taken in isolation might contribute favorable ΔH°bind from the dipole induced-dipole interactions, and unfavorable ΔS°bind due to the restricted motion of Thr138 and its partner groups on the DNA. However, we must also take into account the release of two additional waters during binding of the A138T mutant protein to DNA, the removal from solvent of the larger Thr side chain, and possibly a reduction in conformational entropy of the free A138T mutant protein. The thermodynamic profile tells us that the latter factors predominate. The existence of compensating factors emphasizes the hazard in relying on the enthalpy-centric view to which one becomes predisposed while examining a crystal structure that represents equilibrium atomic positions.
Role of flanking sequence recognition in the relaxed specificity of A138T
Figure 5 shows that an EcoRI* site is most rapidly cleaved by the A138T mutant enzyme when abutted by a 5' RY motif (i.e., RYAAATTC). For such sites, the third base away from the central hexanucleotide exerts only a slight modulating influence. The pronounced preference of the A138T enzyme for this subset of sites suggests the possibility that the novel packing interactions between the Thr138 side chain and the flanking bases may stabilize the transition state for cleavage of EcoRI* sites. Such recognition of the 5' flanking bases might well be degenerate, as Thr138-γ-CH3 could make equivalent interactions with the N7 of either A or G in the −5 position, and with the sterically similar C-N4 or T-O4 in the −4 position. Consistent with this hypothesis, mutation of A138 to valine, the side chain of which is nearly isosteric with that of threonine, also produces an enzyme that has relaxed specificity, the same preference for an RY abutting EcoRI* sites (Heitman and Model, 1990b), and improved binding to the canonical GAATTC site (H. Dayal & L. Jen-Jacobson, unpublished). During molecular dynamics simulations of the A138V complex with GAATTC, Val138 makes the same packing contacts with the G−5C−4 bases that we see in the A138T crystal structure (Supplemental Data, Figure S6).
These packing interactions stabilize the ground-state complex of A138T with oligonucleotides containing the GAATTC site, but lie about 15Å from the scissile phosphate. How then can these packing interactions have such a dramatic effect on EcoRI* site cleavage?
For macromolecular substrates, enzyme-substrate interactions far from the reaction center can be preserved as the complex progresses from the ground state to the transition state, with the consequence that binding free energy is utilized efficiently on the path to the transition state (Jen-Jacobson, 1997). Much evidence was adduced (Jen-Jacobson, 1997; Kurpiewski et al., 2004) to show that this is the case for wild-type EcoRI endonuclease interaction with DNA containing the specific recognition site GAATTC. The ground-state GAATTC complex was dubbed a “pre-transition-state” complex to emphasize this close structural resemblance between the ground-state and transition-state complexes. Flanking sequence variation affects equally the stability of the ground-state and transition-state complexes (Jen-Jacobson, 1997), and thus the first-order cleavage rate constant (and ΔG°‡) does not change as the flanking trinucleotide is varied.
The situation is quite different for cleavage of EcoRI* sites, for which the ground-state complexes show evidence of “adaptive” structural differences from the canonical ground-state complex (Lesser et al., 1990), but transition-state complexes must presumably be more stringently constrained to a precise (local) configuration required to support catalysis. Consequently, flanking-sequence variations preferentially affect the stability of the transition state rather than the ground state, and therefore modify the activation free energy ΔG°‡ (Figure 5).
For the promiscuous mutant A138T cleavage of the EcoRI* site AAATTC, the cleavage rate constants kcleave (and ΔG°‡) are most favorable when flanked by an RY motif (Figure 5). If there is an RY motif, there is relatively little distinction between particular nucleotides; this resembles a characteristic property of the wild-type enzyme with the canonical GAATTC site (Jen-Jacobson, 1997), where kcleave is completely independent of flanking context. By contrast, if there is no RY motif, kcleave varies considerably with flanking triplet (Figure 5); this resembles the behavior of the wild-type enzyme with EcoRI* sites, where kcleave is very sensitive to flanking context. Furthermore, A138T cleavage of the RY-flanked AAATTC is much faster than wild-type cleavage of the same site and faster than A138T cleavage of the non-RY-flanked sites (Figure 5).
These observations suggest that the additional binding energy conferred by the packing interactions of Thr138 with the RY flanking dinucleotide may be sufficient to partially overcome the unfavorable factors associated with binding to an AAATTC site, and to drive the A138T-AAATTC complex into a structural configuration that more nearly resembles the wild-type “specific” complex with GAATTC than it does the wild-type EcoRI* complex. If this hypothesis is correct, it follows that stabilizing factors that appear in the ground state specific complex, as seen in protein-GAATTC co-crystals, can be used to infer how the A138T enzyme manages to cleave EcoRI* sites faster than the wild type enzyme. We will provide elsewhere (P. Sapienza, T. Niu and L. Jen-Jacobson, in preparation) a variety of experimental data that support the “specific-like” nature of the A138T-EcoRI* complexes, which in comparison to wild-type endonuclease-EcoRI* complex show improved contacts to the DNA phosphate backbone, increased affinity for the Mg2+ cofactor, and ΔC°P values that are much more negative.
EXPERIMENTAL PROCEDURES
Enzyme expression and purification
Plasmids encoding the EcoRI endonucleases were transformed into the E. coli strain ER2566 (=fhuA2 [lon] ompT lacZ::T7 genel gal sulAll Δ(mcrC-mrr)l 14::IS10 R(mcr-73::miniTn10--Tets)2 R(zgb-210::Tn10--Tets) end A1 [dcm]) (New England Biolabs), already containing the EcoRI methylase plasmid (pAXU22-8, a gift from New England Biolabs). We expressed and purified the endonucleases (Cheng et al., 1984; Sapienza et al., 2005) to >99% homogeneity, based on SDS PAGE gels stained with Coomassie Blue. Absolute protein concentrations were determined by SDS gel electrophoresis and Coomassie staining, using a standard curve generated with an EcoRI endonuclease preparation whose absolute protein concentration was determined by direct amino acid analysis (Jen-Jacobson et al., 1983).
Crystallization and data collection
The protein-DNA co-crystals were grown by vapor diffusion at 4°C. 1 ml of reservoir solution (40 mM BTP, 16% v/v PEG 400, pH 6.5) was added to the reservoir compartment. 7μl DNA (10 mg/ml), 6 μl enzyme (6 mg/ml in 40 mM bis-tris propane, 0.5 M NH4Cl, 1 mM EDTA, 15% v/v dioxane, pH 8.5) and 3μl of precipitant solution (40 mM BTP, 40% v/v PEG 400, pH 7.0) were added to the sitting drop in the order listed. Crystals of approximate dimensions 400 μm × 200 μm × 100 μm grew within a month. For cryoprotection, the crystals were soaked for three minutes in reservoir solution supplemented with 10% glycerol and then transferred to reservoir solution with 15% glycerol and allowed to soak for another three minutes. Crystals were then flash frozen in loops under liquid nitrogen.
Diffraction data were collected from a single crystal at the X6A beam-line of the National Synchrotron Light Source. Data were collected at a single X-ray wavelength of 0.979 Å using the rotation/oscillation method (Dauter, 1999; Minor et al., 2000). The crystal temperature was maintained at 100K throughout the experiment. The diffraction data were indexed, integrated and scaled using the DENZO/SCALEPACK software package (Otwinowski, 1997). Statistics for data collection and data processing are shown in Table 4.
Table 4.
Data collection and refinement statistics for the A138T-DNA complex
| Data collection | |
|---|---|
| Temperature | 100 K |
| Space group | P321 |
| Cell dimensions a=b, c (Å) | 116.5, 48.5 |
| Resolution (Å) | 99-1.95 (2.01-1.95)a |
| Unique reflections | 27,856 (2,283) |
| Completeness (%) | 99 (100) |
| Redundancy | 17.0 (10.2) |
| Rmerge (%)b | 8 (44) |
| I/σ(I) | 30.2 (4.2) |
| Refinement | |
| Asymmetric unit | One enzyme monomer + one DNA strand |
| Resolution limits | 12-1.95 |
| Reflections used (working set) | 22,743 |
| Cutoff for structure factors | 2σ |
| Rwork/Rfreec | 0.20/0.24 |
| No. of atoms | |
| Protein | 2053 |
| DNA | 263 |
| Solvent | 137 |
| B-factors (Å2) | |
| Entire structure | 40.1 |
| Protein | 40.7 |
| DNA | 35.8 |
| Solvent | 40.1 |
| Rmsd bond lengths | 0.009 Å |
| Rmsd bond angles | 1.3° |
| Ramachandrandran Plot | |
| Residues in most favored region | 90% |
| Residues in disallowed regions | 0% |
Numbers in parenthesis correspond to values for the highest resolution shell
, where I is the integrated intensity of a given reflection
. Rfree was calculated with 9.1% of the data excluded from refinement
Structure determination and refinement
Crystals of the A138T-d(TCGCGAATTCGCG) complex were found to be isomorphic to crystals of the wild type complex, so we were able to carry out rigid body refinement using the wild type structure (further refinement of PDB entry 1CKQ) (Grigorescu, 2003) as the initial model. The Crystallography and NMR System (CNS) (Brünger et al., 1998) was utilized for all stages of refinement. 10% of the reflections were randomly chosen to be used in the cross validation set for calculating Rfree (Brünger, 1992) during refinement. After initial rigid body refinement, several rounds of simulated annealing torsion angle refinement and Cartesian refinement with maximum likelihood target were carried out. Engh and Huber bond and angle parameters were used for the protein (Engh, 1991), and parameters from Parkinson et al. were used for the DNA (Parkinson, 1996). 2F0-Fc and F0-Fc difference maps were calculated in CNS with σa weights for refitting the model. To reduce model bias, simulated annealing omit maps and composite omit maps were calculated. The resultant maps and real space R-factors (Jones et al., 1991) were used as guides for making adjustments to the atomic model from within the program O (Jones et al., 1991). We used CNS to build solvent molecules into residual electron density that was not assigned to the macromolecules according to the following criteria: Putative solvent peaks must exist above the 3Å contour level in difference maps and have good hydrogen bonding geometry with other solvent molecules, the protein, or the DNA. Waters were deleted if their corresponding electron density could be interpreted as an alternative position of a side chain, if the peak was not visible at the 1Å level in subsequent 2Fo-Fc maps, or if the B-factor for a given solvent atom rose above 75 Å2 during subsequent refinements. After five rounds of water peak picking, refinement, and map examination, 136 bound water molecules satisfying the above criteria were added to the atomic model. Finally, an anisotropic B-factor correction was applied, and individual atom restrained B-factor refinement was carried out in CNS. All molecular renderings were created in pyMOL (DeLano, 2002).
Equilibrium binding, cleavage kinetics, and thermodynamic parameters
Equilibrium binding constants were determined by the direct binding method on nitrocellulose filters as described previously (Engler et al., 1997; Lesser et al., 1992). First-order cleavage rate constants were measured in single-turnover experiments (Lesser et al., 1990; Lesser et al., 1992). The effect of temperature on the equilibrium association constant KA was measured by direct filter binding in a temperature controlled room over a temperature range of 3°C to 37°C ± 0.2°C. Binding reactions were conducted in 10 mM Cacodylate, 0.27M KCl, 1mM EDTA, 0.01% NaN3, 100μM dithiothreitol, 100μg/ml bovine serum albumin, pH 7.3. The pH of the buffer was checked at each individual temperature, and no additions of acid or base were required to keep the pH at 7.3 over the entire temperature range. A minimum of three measurements of the binding constant were made at each temperature. In order to confirm that changes in the binding constants with temperature were due to changes in the binding equilibrium, and not to enzyme denaturation, binding reactions were incubated at the high temperature extreme, subsequently transferred to 21°C, equilibrated at this temperature and filtered. The binding constants determined from these reactions were the same as those determined from reactions incubated at 21°C only.
Thermodynamic parameters ΔC°p, TH, and TS were extracted from the temperature-dependence of ln Kobs in two ways. First, we used a multiparametric fit to equation (1):
| (1) |
ΔC°p is the observed heat capacity change at constant pressure for the binding reaction, TH is the temperature at which ΔH° = 0, TS is the temperature where ΔS° = 0 and R is the universal gas constant. The standard free energy (ΔG°), enthalpy (ΔH°) and entropy (ΔS°) changes for the binding reaction over the experimental temperature range (3° to 37°C) were then calculated from the following equations assuming a constant ΔC°P over this temperature range.
| (2) |
| (3) |
| (4) |
Extensive comparisons in our laboratory, primarily with BamHI endonuclease (Jen-Jacobson et al., 2000a); P. Mehta, L. Engler and L. Jen-Jacobson, unpublished), have shown that the values of ΔH° and ΔC°P derived by this method closely coincide with direct measurements by isothermal titration calorimetry.
Second, we extracted the thermodynamic parameters by the method of Clarke and Glew (Clarke and Glew, 1966; Dec and Gill, 1985), which assumes only that the equilibrium association constant and thermodynamic parameters are continuous functions of temperature. We used this second method because dΔC°p/dT can be evaluated and because the fitted thermodynamic parameters and their associated errors are independent of one another; the latter is not always the case for the multiparametric fit method (Dec and Gill, 1985; Jen-Jacobson et al., 2000a). Clarke and Glew showed that the values of the thermodynamic parameters ΔH°, ΔS°, ΔG° and ΔC°P, at temperature T, can be expressed as perturbations of their values at a reference temperature, θ, using the Taylor's series expansion:
| (5) |
which we truncate after taking the first temperature derivative of the heat capacity change. In eqn. (5), and are the free energy change, the enthalpy change, and the heat capacity change, respectively at θ. Equation (5) can be re-written as:
| (6) |
with
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
We therefore calculated the temperature dependent coefficients, ti (i=1 to 4), around a reference temperature of θ = 298.15 K and fit equilibrium association constants to eqn. (6) in order to obtain the bi partial regression coefficients. Propagation of errors in these coefficients were obtained from the diagonal elements of the error matrix as described (Clarke and Glew, 1966; Bevington, 1969; Dec and Gill, 1985).
Molecular dynamics simulations
A highly refined model (Grigorescu, 2003) based on PDB file 1CKQ was used as the starting point for simulations of the wild type complex and the coordinates (PDB accession code 2OXV) generated in this work were used for the A138T-specific complex. Hydrogen atoms were built onto the protein, DNA and crystallographic water molecules using the AMBER 8.0 package (Case et al., 2004). TIP3 water molecules were added to extend 10 Å beyond any atoms of the EcoRI-DNA complex, creating a 70,000 Å3 octahedral water box (about 17,000 water molecules total). 92 Na+ and Cl− ions were added to give a salt concentration of 0.22 M supplemented by an additional 22 Na+ ions to achieve electroneutrality. The all atom force field of Duan et al. (Duan et al., 2003) was used for the simulations. SHAKE was used to restrain bonds involving hydrogens (tolerance 0.0001Å). Equations of motion were integrated using a 0.0015-ps time step in an NPT ensemble. Electrostatic interactions were treated with an Ewald sum (de Leeuw, 1980), and the temperature was regulated using the bath coupling method (Berendsen, 1984). Positions of the added salt and water atoms were refined through energy minimization followed by successive rounds of molecular dynamics to equilibrate T and V. In order to escape any local minima, we heated the system to 373K and annealed back to 300K and allowed P, V and ρto equilibrate. Production dynamics were then initiated on the Cray XT3 at the Pittsburgh Supercomputing Center. The A138V simulation was performed as above except the Thr138 side chains in the A138T crystallographic model were changed to valines in pyMOL (DeLano, 2002). The backbone dependent χ1 rotamer (Dunbrack and Cohen, 1997) observed in 95% of valines in similar ϕψ contexts was then imposed on Val138 prior to simulation.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by an NIH MERIT grant 5R37GM029207 (to L.J.-J), allocations of advanced computing resources supported by NSF (MCB050027P and MCB060029P) to L.J.J, and an NIH grant 5R01RR014477 to J.M.R. X-ray diffraction data were collected at the X6A beam line, National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy under contract No.DE-AC02-98CH10886. X6A is funded by NIH/NIGMS under agreement Y1 GM-0080-03. Computations were performed on the Cray XT3 MPP system at the Pittsburgh Supercomputing Center. We thank A. Grigorescu for providing the coordinates of the refined wild type model and assistance with the Clarke and Glew analysis of thermodynamic data, I. Kuo for HPLC purification of DNA oligonucleotides used in crystallography, and W. Jack for providing the EcoRI methylase plasmid and the E. coli strain carrying T7 RNA polymerase.
Footnotes
Accession Numbers
Coordinates and structure factors for the A138T-DNA complex have been deposited in the Protein Data Bank with accession code 2OXV.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alves J, Ruter T, Geiger R, Fliess A, Maass G, Pingoud A. Changing the hydrogen-bonding potential in the DNA binding site of EcoRI by site-directed mutagenesis drastically reduces the enzymatic activity, not, however, the preference of this restriction endonuclease for cleavage within the site-GAATTC. Biochemistry. 1989;28:2678–2684. doi: 10.1021/bi00432a047. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- Bevington PR. Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill; New York: 1969. [Google Scholar]
- Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cao DF. Co-solute effects as probes of the role of water release in DNA binding and catalysis by three restriction endonucleases. University of Pittsburgh; Pittsburgh, PA: 2002. Ph.D. Thesis thesis. [Google Scholar]
- Case DA, Darden TA, Cheatham TE, III., Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell J, Ross W, Kollman PA. AMBER. Vol. 8. University of California; San Francisco: 2004. W., S. [Google Scholar]
- Cheng SC, Kim R, King K, Kim SH, Modrich P. Isolation of gram quantities of EcoRI restriction and modification enzymes from an overproducing strain. J Biol Chem. 1984;259:11571–11575. [PubMed] [Google Scholar]
- Clarke ECW, Glew DN. Evaluation of thermodynamic functions from equilibrum constants. Trans. Faraday Soc. 1966;62:539–547. [Google Scholar]
- Dauter Z. Data-collection strategies. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 10):1703–1717. doi: 10.1107/s0907444999008367. [DOI] [PubMed] [Google Scholar]
- de Leeuw SW, Perram JW, Smith ER. Ewald summations and dielectric constants. Proc R Soc Lond. 1980;A373:27–56. [Google Scholar]
- Dec SF, Gill SJ. Temperature dependence of errors in parameters derived from van't Hoff studies. Journal of Chemical Education. 1985;62:879–881. [Google Scholar]
- DeLano WL. The pyMOL Molecular Graphics System. DeLano Scientific; 2002. [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Dunbrack RL, Jr., Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunitz JD. The Entropic Cost of Bound Water in Crystals and Biomolecules. Science. 1994;264:670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- Engh RA, Huber R. Accurate bond and angle parameters for X-ray structure refinement. Acta Cryst. A. 1991;47:392–400. [Google Scholar]
- Engler LE, Welch KK, Jen-Jacobson L. Specific binding by EcoRV endonuclease to its DNA recognition site GATATC. J Mol Biol. 1997;269:82–101. doi: 10.1006/jmbi.1997.1027. [DOI] [PubMed] [Google Scholar]
- Fritz A, Kuster W, Alves J. Asn141 is essential for DNA recognition by EcoRI restriction endonuclease. FEBS Lett. 1998;438:66–70. doi: 10.1016/s0014-5793(98)01274-5. [DOI] [PubMed] [Google Scholar]
- Grigorescu A. Structural and Energetic Determinants of the Binding Specificity of EcoRI Endonuclease. University of Pittsburgh; Pittsburgh, PA: 2003. Ph.D. Thesis thesis. [Google Scholar]
- Grigorescu A, Horvath M, Wilkosz PA, Chandrasekhar K, Rosenberg JM. The integration of recognition and cleavage: X-ray structures of pre-transition state complex, post reactive complex and the DNA-free endonuclease. In: Pingoud AM, editor. Restriction Endonucleases. Springer; Berlin: 2004. pp. 137–173. [Google Scholar]
- Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32:W500–502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J, Model P. Substrate recognition by the EcoRI endonuclease. Proteins. 1990a;7:185–197. doi: 10.1002/prot.340070207. [DOI] [PubMed] [Google Scholar]
- Heitman J, Model P. Mutants of the EcoRI endonuclease with promiscuous substrate specificity implicate residues involved in substrate recognition. Embo J. 1990b;9:3369–3378. doi: 10.1002/j.1460-2075.1990.tb07538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendsch ZS, Tidor B. Electrostatic interactions in the GCN4 leucine zipper: substantial contributions arise from intramolecular interactions enhanced on binding. Protein Sci. 1999;8:1381–1392. doi: 10.1110/ps.8.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook JA, Tsodikov OV, Saecker RM, Record MT., Jr. Specific and non-specific interactions of integration host factor with DNA: thermodynamic evidence for disruption of multiple IHF surface salt-bridges coupled to DNA binding. J Mol Biol. 2001;310:379–401. doi: 10.1006/jmbi.2001.4768. [DOI] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM. NACCESS: Department of Biochemistry and Molecular Biology. University College London; 1993. [Google Scholar]
- Ivanenko T, Heitman J, Kiss A. Mutational analysis of the function of Met137 and Ile197, two amino acids implicated in sequence-specific DNA recognition by the EcoRI endonuclease. Biol Chem. 1998;379:459–465. doi: 10.1515/bchm.1998.379.4-5.459. [DOI] [PubMed] [Google Scholar]
- Jen-Jacobson L, Kurpiewski M, Lesser D, Grable J, Boyer HW, Rosenberg JM, Greene PJ. Coordinate ion pair formation between EcoRI endonuclease and DNA. J Biol Chem. 1983;258:14638–14646. [PubMed] [Google Scholar]
- Jen-Jacobson L. Structural-perturbation approaches to thermodynamics of site-specific protein-DNA interactions. Methods Enzymol. 1995;259:305–344. doi: 10.1016/0076-6879(95)59050-1. [DOI] [PubMed] [Google Scholar]
- Jen-Jacobson L. Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers. 1997;44:153–180. doi: 10.1002/(SICI)1097-0282(1997)44:2<153::AID-BIP4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Jen-Jacobson L, Engler LE, Ames JT, Kurpiewski MR, Grigorescu A. Thermodynamic parameters of specific and nonspecific protein-DNA binding. Supramolecular Chemistry. 2000;12:143–160. [Google Scholar]
- Jen-Jacobson L, Engler LE, Jacobson LA. Structural and thermodynamic strategies for site-specific DNA-binding proteins. Structure. 2000;8:1015–1023. doi: 10.1016/s0969-2126(00)00501-3. [DOI] [PubMed] [Google Scholar]
- Joachimiak A, Haran TE, Sigler PB. Mutagenesis supports water mediated recognition in the trp repressor-operator system. Embo J. 1994;13:367–372. doi: 10.1002/j.1460-2075.1994.tb06270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kim YC, Grable JC, Love R, Greene PJ, Rosenberg JM. Refinement of Eco RI endonuclease crystal structure: a revised protein chain tracing. Science. 1990;249:1307–1309. doi: 10.1126/science.2399465. [DOI] [PubMed] [Google Scholar]
- Kozlov AG, Lohman TM. Calorimetric studies of E. coli SSB protein-single-stranded DNA interactions. Effects of monovalent salts on binding enthalpy. J Mol Biol. 1998;278:999–1014. doi: 10.1006/jmbi.1998.1738. [DOI] [PubMed] [Google Scholar]
- Kozlov AG, Lohman TM. Large contributions of coupled protonation equilibria to the observed enthalpy and heat capacity changes for ssDNA binding to Escherichia coli SSB protein. Proteins Suppl. 2000;4:8–22. doi: 10.1002/1097-0134(2000)41:4+<8::aid-prot20>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Kumar S, Duan Y, Kollman PA, Rosenberg JM. Molecular dynamics simulations suggest that the Eco RI kink is an example of molecular strain. J Biomol Struct Dyn. 1994;12:487–525. doi: 10.1080/07391102.1994.10508757. [DOI] [PubMed] [Google Scholar]
- Kurpiewski MR, Engler LE, Wozniak LA, Kobylanska A, Koziolkiewicz M, Stec WJ, Jen-Jacobson L. Mechanisms of coupling between DNA recognition specificity and catalysis in EcoRI endonuclease. Structure. 2004;12:1775–1788. doi: 10.1016/j.str.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Lesser DR, Kurpiewski MR, Jen-Jacobson L. The energetic basis of specificity in the Eco RI endonuclease--DNA interaction. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- Lesser DR, Grajkowski A, Kurpiewski MR, Koziolkiewicz M, Stec WJ, Jen-Jacobson L. Stereoselective interaction with chiral phosphorothioates at the central DNA kink of the EcoRI endonuclease-GAATTC complex. J Biol Chem. 1992;267:24810–24818. [PubMed] [Google Scholar]
- Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Tomchick D, Otwinowski Z. Strategies for macromolecular synchrotron crystallography. Structure Fold Des. 2000;8:R105–110. doi: 10.1016/s0969-2126(00)00139-8. [DOI] [PubMed] [Google Scholar]
- Needels MC, Fried SR, Love R, Rosenberg JM, Boyer HW, Greene PJ. Determinants of EcoRI endonuclease sequence discrimination. Proc Natl Acad Sci U S A. 1989;86:3579–3583. doi: 10.1073/pnas.86.10.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuna J, Flores H, Soberon X. Combinatorial mutagenesis of three major groove-contacting residues of EcoRI: single and double amino acid replacements retaining methyltransferase-sensitive activities. Gene. 1991;106:7–12. doi: 10.1016/0378-1119(91)90559-t. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Parkinson G. New parameters for the refinement of nucleic acid-containing structures. Acta Crystallogr D Biol Crystallogr. 1996;52:57–64. doi: 10.1107/S0907444995011115. [DOI] [PubMed] [Google Scholar]
- Poon GM, Macgregor RB., Jr. A thermodynamic basis of DNA sequence selectivity by the ETS domain of murine PU.1. J Mol Biol. 2004;335:113–127. doi: 10.1016/j.jmb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- Sapienza PJ, Dela Torre CA, McCoy WH, III, Jana SV, Jen-Jacobson L. Thermodynamic and kinetic basis for the relaxed DNA sequence specificity of “promiscuous” mutant EcoRI endonucleases. J Mol Biol. 2005;348:307–324. doi: 10.1016/j.jmb.2005.02.051. [DOI] [PubMed] [Google Scholar]
- Shui X, McFail-Isom L, Hu GG, Williams LD. The B-DNA dodecamer at high resolution reveals a spine of water on sodium. Biochemistry. 1998;37:8341–8355. doi: 10.1021/bi973073c. [DOI] [PubMed] [Google Scholar]
- Wibowo FR, Rauch C, Trieb M, Wellenzohn B, Liedl KR. Water-mediated contacts in the trp-repressor operator complex recognition process. Biopolymers. 2004;73:668–681. doi: 10.1002/bip.20023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.