

Summary
Kinase-substrate recognition depends on the chemical properties of the phosphorylatable residue as well as the surrounding linear sequence motif. Detailed knowledge of these characteristics increases the confidence of linking identified phosphorylation sites to kinases, predicting phosphorylation sites, and designing optimal peptide substrates. Here, we present a mass spectrometry-based approach for determining linear kinase substrate motifs by elaborating the positional and chemical preference of the kinase for a phosphorylatable residue using libraries of naturally-occurring peptides which are amenable to peptide identification by commonly used proteomics platforms. We applied this approach to a structurally and functionally diverse set of purified kinases, which recapitulated their previously described substrate motifs and discovered novel ones, including preferences of certain kinases for phosporylatable residues adjacent to peptide termini. Furthermore, we identify specific and distinguishable motif elements for the four members of the polo-like kinase (Plk) family and verify novel members of these motif elements for Plk1 in vivo.
Introduction
Protein phosphorylation is a ubiquitous post-translational modification implicated in nearly all cellular signal transduction processes. Approximately 30% of all cellular proteins are phosphorylated by one or more of the 518 putative kinases encoded in the human genome (Cohen, 2000; Manning et al., 2002). To identify their specific substrates in the vast pool of potential candidates, kinases utilize a range of mechanisms, including direct protein-protein interactions of kinase and substrate or though scaffolding proteins, localization to subcelluar structures, and interactions of residues in the kinase active site with linear sequence elements surrounding the phosphorylatable residue (Ubersax and Ferrell, 2007; Yaffe et al., 2001). Selective recognition of and interaction with linear sequence motifs in substrates by the kinase are due to structural characteristics of the kinase active site. Hydrophobicity, charge, and depth are the main factors that determine this interaction and are often complementary between kinase and substrates (Ubersax and Ferrell, 2007). In addition, other contextual factors such as the action of regulatory subunits have been shown to play a major role in connecting kinases and their substrates in cells (Linding et al., 2008).
The identification of linear sequence motifs was first described using oriented synthetic peptide libraries and Edman sequencing (Songyang et al., 1994). In this approach, a large oriented peptide libraries containing only one serine or tyrosine per peptide in a position seven were synthesized and incubated for a brief period with the kinase of interest. Phosphopepitdes were separated from non phosphorylated peptides using a ferric-IDA column and peptide sequences were determined by Edman sequencing, The introduction of the positional scanning peptide library approach eliminate the need for phosphopeptide purification as well as Edman sequencing and proved to be powerful for determining kinases consensus sequences in higher throughput (Alexander et al., 2011; Hutti et al., 2004; Miller et al., 2008; Mok et al., 2010). Here, 198 peptide libraries were synthesized that contained a fixed serine or threonine as well as second fixed amino acid in one of the flanking positions, while all other positions were degenerate in their amino acids composition. In addition, peptides were tagged with biotin at the C-terminus, which allows for efficient capture on avidin-coated membranes after an in-solution kinase assay with gamma 32P-labeled adenosine triphosphate (ATP). Linear sequence motifs were determined based on incorporation of radiolabeled phosphate. Surface capture after solution-phase phosphorylation reactions eliminated problems observed with solid phase based methods such as unspecific binding of radioactive phosphate or the kinase to the solid phase matrix, which had resulted in artifacts (Yaffe, 2004) and improved the prediction of in vivo observed linear sequences motifs. However, linear sequence motifs generated by this approach represent an average of all motifs generated by a given kinase and without connectivity. In cases were several amino acids in the same or in neighboring positions are selected independently of each other by a given kinase, averaging of the signal reduces sensitivity.
Here, we present a mass spectrometry-based approach for determining multiple independent linear sequence motifs that are individually recognized by a specific kinase. In contrast to synthetic peptide libraries which require de novo or Edman sequencing for peptide identification, libraries of naturally-occurring peptides are amenable to sequence determination by standard high though-put LC-MS/MS-based proteomics technologies that identify peptide sequences from translated genomic databases. Importantly, the identification of individual phosphorylated peptide sequences via this approach allows for decoupling of individual motif elements based on the specific amino acids that are found in connection with one another. In the present work, we show the flexibility of our approach in identifying statistically significant motifs that are chemically distinct from “averaged” motifs generated by other high-throughput approaches.
Results and Discussion
Mass spectrometry- based linear kinase motif assay
First, naturally-occurring peptide substrates were generated by protease digestion of HeLa whole cell lysates, followed by exhaustive peptide dephosphorylation by alkaline and λ-phosphatases (Figure 1A). To reduce the complexity of this peptide pool, the digest was separated into 12 fractions by strong cation-exchange chromatography. Standardized aliquots of these fractions were then used as substrate libraries and phosphorylated in vitro by treatment with the kinase of interest, followed by phosphopeptide isolation. These phosphopeptides were then analyzed by LC-MS/MS, database searched, and filtered to both an identification false discovery rate (FDR) of less than 1% using target-decoy methods (Elias and Gygi, 2007) and a phosphorylation site localization probability of 0.99 or higher using PhosphoRS (Taus et al., 2011). Separately, each peptide substrate fraction was subjected to replicate mock kinase reactions containing buffer and peptides but lacking kinase, to allow for correction of residual phosphopeptides that remained in each pool due to incomplete dephosphorylation. After removal of these background contaminants, statistically significant motifs were extracted from the collection of individually-sequenced phosphopeptide results online using our in-house developed motif algorithm GrMFPh (Greedy Motif Finder for Phosphopeptides) (http://www.cs.dartmouth.edu/~cbk/grmfph/). Details of the algorithm can be found in Experimental Procedures. To exclude artifacts in motif generation due to phosphopeptide isolation conditions, we compared motifs for an acidophilic (Plk1) and basophilic (Pim1) kinase generated via phosphopeptide enrichment with two common phosphopeptide enrichment methods: titanium dioxide microspheres (Kettenbach and Gerber, 2011; Pinkse et al., 2004; Sugiyama et al., 2007) and immobilized metal affinity chromatography (Fe-IMAC; (Villen and Gygi, 2008). No significant differences in the motifs generated for either class of kinases as a function of enrichment method were observed (Figures S1 and S2), although TiO2 consistently led to ~25% greater number of phosphopeptide identifications than Fe-IMAC. Because of this increase in sensitivity, we used titanium dioxide microspheres for phosphopeptide purification in all subsequent analyses.
Figure 1. Workflow and results of kinase substrate motif assay.
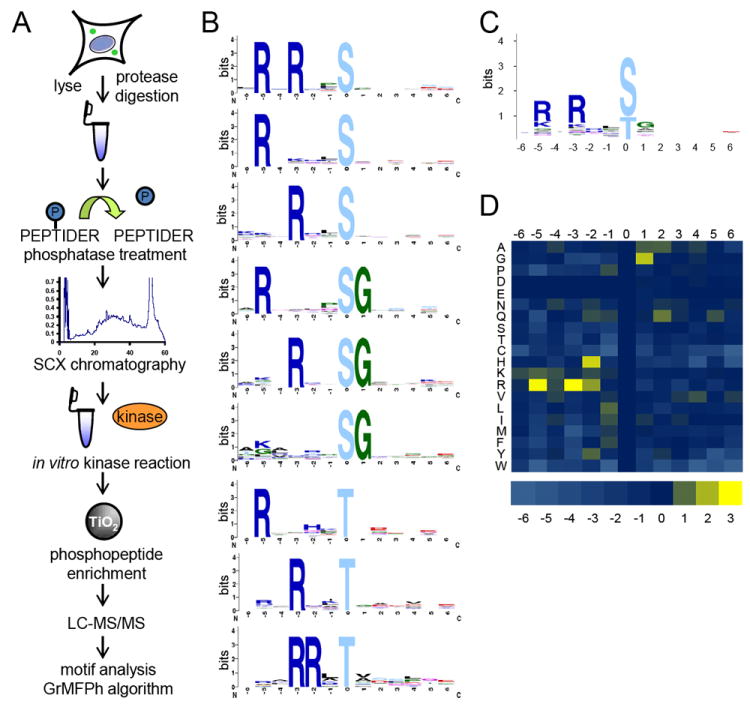
(A) Depiction of peptide kinase assay workflow. HeLa cells were lysed, digested, dephosphorylated, and separated into 12 fractions by strong cation exchange (SCX) chromatography. Select fractions were used in in vitro kinase reactions. Phosphopeptides were purified from non-phosphopeptides using titanium dioxide (TiO2) microspheres and analyzed by LC-MS/MS. Statistically significant motifs were extracted from the identified phosphopeptides by the in-house developed motif algorithm GrMPh. (B) Linear kinase motifs from Pim1 in vitro kinase reaction. (C) Averaged motif of all motif-containing peptides in (B). (D) Heat map representation of the log2 values of the ratio of foreground to background amino acid frequencies of motif-containing peptides in (A). See also Figures S1 – S9, S11 – S15, and Tables S1 and S2.
We began by interrogating kinases (Pim1, Pim3, Clk3, Dyrk1A) that had been previously studied by other high-throughput methods (Miller et al., 2008), in an effort to validate our approach. For example, it was shown previously that Pim1 and Pim3 prefer to phosphorylate serines and threonines with basic residues in the -3 and -5 position, and to a lesser degree a histidine or basic residue in the -2 position ([R/K]X[R/K][H/R]Xp[S/T]) (Hutti et al., 2004; Miller et al., 2008). Using our mass spectrometry-based approach, we also observed this preference for basic residues in the -3 and -5 position, as well as a basic residue in the -2 position (Figure 1B, Figure S3A and S4A). However, the motifs we identified contained these motif elements in very specific combinations. We only observed motif-specific amino acids in either the -3 and -5 positions, or in the -2 and -3 positions, but never in all three positions, underscoring the utility of individual over averaged motifs. In fact, we frequently observed phosphopeptide substrates with only one of these motif determinants in a peptide sequence at a time (either the -5 or -3 position). Averaged motifs are helpful for rapid viewing of all key amino acids that comprise a specific kinase motif, and for high-level summarization of the fundamental aspects of a given kinase motif. However, we note that in this case (as well as others described later), the “averaged” motif we generated for Pim1 kinase based on all motif-containing peptides (Figure 1C, Figure S3B and S4B) resulted in a disconnect of motif information for those significant motifs that contained only one basic residue in either the -3 or -5 positions, or a basic residue in each of the -2 and -3 positions. Furthermore, based on the averaged motif, it is nearly impossible to distinguish between the motif preferences of Pim1 and Pim3 (Figure 1C, Figure S3B, Figure S4B), while the individual motifs highlight a preference of Pim3 for linear sequences containing not only arginine, but also lysines. Both Pim1 and Pim3 displayed a further preference for glycine in the +1 position (Bullock et al., 2005). Finally, to demonstrate over- as well as under-representation of all amino acids in motif-containing peptides for a specific kinase, we plotted the background corrected amino acid occurrence in motif-containing peptides as a heatmap (Figure 1D, Figure S3C, Figure S4C) (Schilling and Overall, 2008).
For Clk3, we found a preference for arginine and lysine in the -3 position and to a lesser degree in the -2 position (Figure S5A), which is similar to the motif previously established for Clk kinases (Miller et al., 2008). In addition, we found proline in the +1 position to be favored by Clk3. We also investigated Dyrk1A, for which a motif is only available from detailed studies of peptide and protein substrates, but not from high-throughput approaches. In agreement with those analyses (Campbell and Proud, 2002; Himpel et al., 2000), we found that Dyrk1A preferentially phosphorylated serine and threonine with an arginine in the -3 and a proline in the +1 position (Figure S6A). Taken together, the data from these four kinases clearly validate our method via recapitulation of motifs generated previously by other researchers. However, our approach allow us to further extend these motifs and generate important contextual information regarding the connectivity and relationships between individual motif elements. In contrast to synthetic library-based methods (Hutti et al., 2004), which yield only one averaged motif per kinase, the identification of multiple independent motifs leads to increased sensitivity by allowing for sub- stoichiometric yet highly statistically significant motif elements to be identified, which might otherwise be lost in assays with an averaged readout.
Chemical and positional kinase preference
Next, we examined kinases for which no consensus motif existed, including Haspin, Camkk2b, and Bmpr2. We found that Haspin preferentially phosphorylated threonines with a basic residue in the -1 position, as well as a histidine in the -1 or a lysine in the +1 or -1 position (Figure S7A). The Camkk2b consensus motif contained a hydrophobic amino acid (F, Y, M, or L) in the -2 position (Figure S8A). The kinase Bmpr2 phosphorylated serines and threonines with an acidic residue (E or D) in the -2 position, or tyrosines with an aspartic acid in the -1 position (Figure S9A). We searched the literature for known targets and found only one substrate with a known phosphorylation site for Haspin (Dai et al., 2005) and Camkk2b (Hurley et al., 2005), each of which conforms to a motif identified in our analysis.
Because our library was derived from the human proteome, the kinases were presented with a wide diversity of peptide sequences; thus, preferences for serine, threonine or tyrosine residues could be determined in one reaction and likely reflect the endogenous preference of the kinases. The analysis of Haspin and Camkk2b consensus motifs revealed a preference of both kinases for threonine as the phosphorylated residue (Figure 2A). While many serine-threonine kinases phosphorylate both amino acids, a general preference for serine over threonine has been shown (Ubersax and Ferrell, 2007). This is consistent with the observation that in large-scale phosphoproteomics analysis (Kettenbach et al., 2011b) and in vivo 32P labelling (Hunter and Sefton, 1980), the ratios of phosphorylated serine to phosphorylated threonine are 5:1 and 9:1, respectively, while the overall ratio of serine to threonine in the human proteome is approximately 1.5:1 (Echols et al., 2002). In addition, we found that Bmpr2 not only phosphorylated serine and threonine, but also tyrosine residues (Figure 2A).
Figure 2. Chemical preference of phosphorylatable substrate residues.
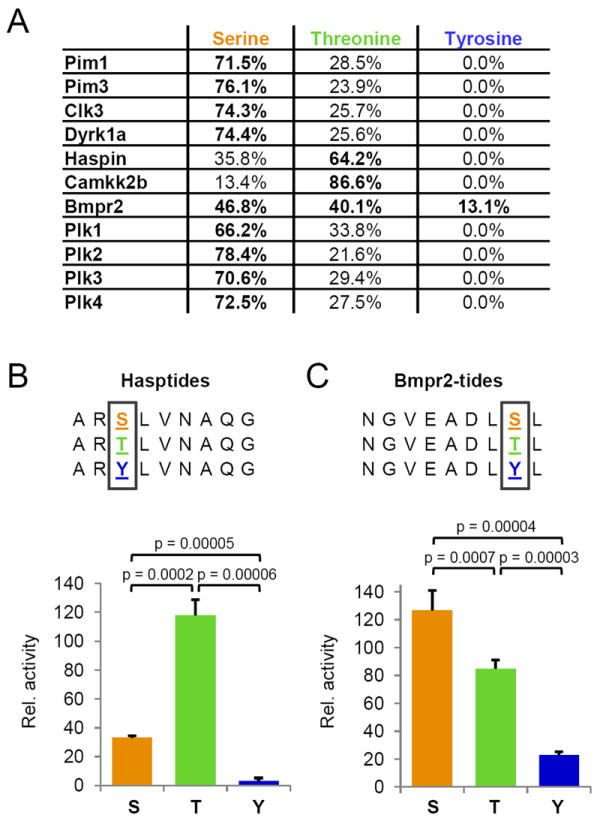
(A) Table depicting the preferences for phosphorylatable residues in motif-containing peptides (serine, threonine, or tyrosine) by the investigated kinases. (B) Validation of Haspin preference for phosphorylating threonine residues. Three different synthetic peptide substrates containing unique phosphorylatable residues were assayed for initial reaction kinetics. (C) Validation of Bmpr2 preference for phosphorylating serine and threonine residues, and capacity to phosphorylate tyrosines. Three different synthetic peptide substrates containing unique phosphorylatable residues were assayed for initial reaction kinetics as for Haspin. Note that unlike Haspin, Bmpr2 readily phosphorylated tyrosine residues, albeit at a reduced rate (~25%) relative to serine. See also Figures S7, S9 and S10.
We sought to confirm the observed chemical preferences of Haspin for threonines over serines and of Bmpr2 for tyrosine in addition to serines and threonines through the design and interrogation of synthetic peptide substrates (Hasptides and Bmpr2tides). The primary template peptide was synthesized such that it contained an optimal linear substrate motif for the respective kinase, including the preference for the phosphorylatable residue; two additional peptide substrates for each kinase were also synthesized and differed in sequence only at the position of phosphorylation (serine, threonine, or tyrosine). We then used these purified synthetic peptides in individual in vitro kinase assays that monitored the initial rates of conversion of ATP to ADP (< 10% conversion) to determine the relative activity of Haspin and Bmpr2 for the respective phosphorylatable residues. (Figure 2 B and C). Consistent with the results from the motif assay, we found that Haspin exhibited 4-fold greater kinetics in phosphorylating threonines over serines; in contrast, we detected no significant activity of Haspin on the tyrosine-containing Hasptide. Analogously, we found that Bmpr2 initial phosphorylation velocities were ~50% higher when using serine-containing versus threonine-containing Bmpr2-tides. In addition, Bmpr2 readily phosphorylated tyrosine-containing peptides, albeit at a significantly reduced rate relative to serine (~20%), which is consistent with the relative frequency with which we observed phosphotyrosine-containing peptides in our motif-generation assay. Finally, to further support our finding that Bmpr2 phosphorylates tyrosine residues, we developed selected ion monitoring (SIM) methods for phosphotyrosine-containing peptides identified in Bmp2 kinase reactions (e.g. LDVTSVEDpYK, VDDDFTAQDpYR, etc.), and used these methods and high mass accuracy LTQ-Orbitrap extracted ion chromatograms (+/- 2.5 ppm) to monitor Bmpr2 kinase reactions and mock controls for the relative abundance of these putative reaction products. While the ion chromatograms from reactions containing Bmpr2 exhibited from 105 – 106 relative ion counts for the mass-to-charge and retention time values corresponding to these tyrosine-phosphorylated peptides, no signal was observed from control reactions performed on identical aliquots of peptides but lacking kinase (Figure S10).
Quite surprisingly, we further found that in 61% of motif-containing peptides phosphorylated by Haspin, the modified residue was located two or three residues away from the N-terminus of the peptide (Figure 3A). This is consistent with the localization of the only known endogenous Haspin phosphorylation site: threonine 3 on histone H3 (Dai et al., 2005) and may be a consequence of the unusual structure of the Haspin kinase domain and substrate binding site (Eswaran et al., 2009). In contrast to Haspin, both Bmpr2 and Camkk2b displayed a preference for phosphorylating residues two or two and three amino acids from the C-terminus in 57% and 49% of motif-containing peptides, respectively (Figure 3B), while most other kinases did not display a specific positional preference (Figure S3D-S8D, S9E, S11D-S14D). The observation of a terminal preference for some kinases may explain why some of them, including Haspin, did not yield motifs by other approaches (personal communication, S. Knapp). To further validate these findings, we synthesized optimal motif peptides for Haspin and Bmpr2 with the phosphorylatable residue in the “preferred” position from a terminus. We also synthesized two additional peptides for each kinase with the phosphoacceptor remaining within the same optimal motif elements, but moved consecutively away from the terminus by two positions in each peptide. For the Hasptides, this resulted in a series of peptides with phosphorylatable residues in the third, fifth, and seventh positions from the amino terminus (N3, N5, and N7) of the peptides, while in the Bmpr2tides the phosphorylatable residue was in the second, fourth, and sixth position from the carboxyl terminus (C2, C4, and C6) of the peptides (Figure 3 C and D). Again, activity assays in vitro using these peptides and purified kinase confirmed the preferential activity of Haspin and Bmpr2 for phosphorylatable residues in the N3 and C2 positions, respectively. Initial rates of phosphorylation for Haspin at the N3 position were 6-fold higher than for the N5 position; no activity was detected with the peptide containing the phosphorylatable residue at the N7 position. Similarly, Bmpr2 readily phosphorylated the C2 and C4 position, but displayed no activity towards the peptide substrate with the phosphorylatable residue at the C6 position.
Figure 3. Positional preference of the phosphorylated residue for Haspin, Bmpr2, and Camkk2b kinases.
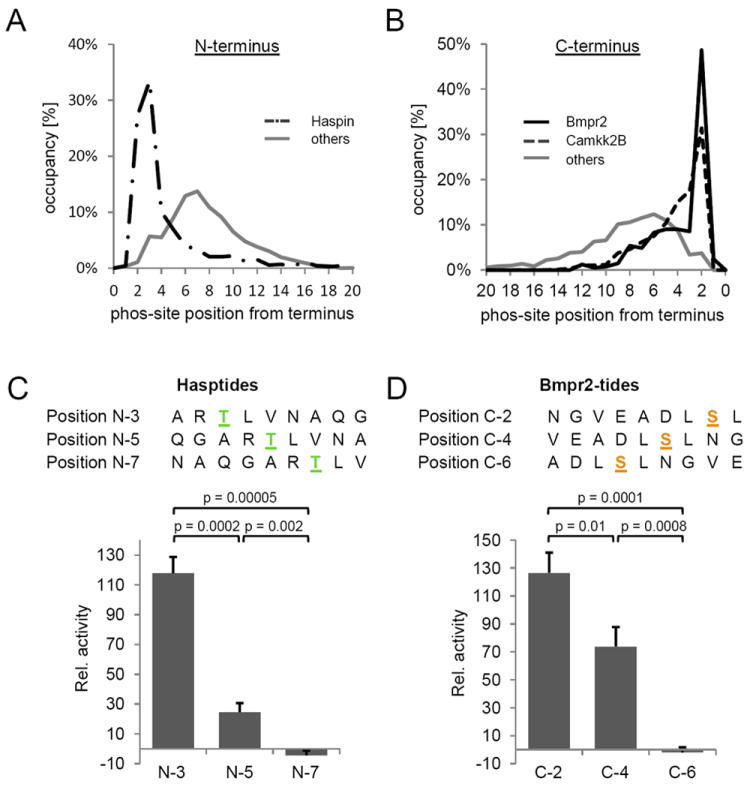
(A) Distribution of phosphorylation site location from the peptide N-terminus for Haspin (dashed-dotted line) and for all other kinases (solid grey line). For 61% of the motif-containing peptides phosphorylated by Haspin, the phosphorylation site was located at the second or third residue from the N-terminus of the peptide. In contrast, phosphorylation sites on motif-containing peptide from all other analyzed kinases were more evenly distributed across the peptide length. (B) Distribution of phosphorylation site location from the peptide C-terminus for Bmpr2 (solid black line), Camkk2b (dashed line), and all other kinases (solid grey line). For 53% of the motif-containing peptides phosphorylated by Bmpr2, the phosphorylation site was located at the second residue away from the C-terminus of the peptide. Similarly, 46% of the motif-containing peptides phosphorylated by Camkk2b exhibited phosphorylated residues two or three positions away from the C-terminus of the peptide. In contrast, phosphorylation sites on motif-containing peptides from all other kinases were more evenly distributed across the length of the peptides. (C) Validation of Haspin positional preference for phosphorylatable residues near the N-terminus of peptides. Three different peptide substrates were synthesized as positional isomers, with the preferred Haspin motif moved sequentially further away from the N-terminus, and were assayed for initial reaction kinetics. (D) Validation of Bmpr2 positional preference for phosphorylatable residues near the C-terminus of peptides. Three different peptide substrates were synthesized as positional isomers, with the preferred Bmpr2 motif moved sequentially further away from the C-terminus, and were assayed for initial reaction kinetics. See also Figures S7 – S9.
It was previously shown that solution-phase methods have several advantages in generating motifs than those using immobilized peptides (Hutti et al., 2004). In some of these approaches, peptide N- and C-termini are often extended with linker sequences, or modified by the addition of biotin, which ablates the peptide terminus and may render them transparent to kinases that are intolerant of chemical constituents at those positions. In addition, most standard peptide library-based approaches utilize a substrate of a specified length, commonly a 13-mer, with the phosphorylatable residue fixed in the center of the peptide sequence. We surmise that the random positioning of the phosphorylatable amino acid and the unmodified nature of the peptides used in our approach overcomes these limitations and reveals kinase preferences beyond the linear substrate motif.
Comparison of Polo-like kinase 1 motifs in vitro and in vivo
Next, we determined the substrate motif for Polo-like kinase 1 (Plk1). In addition to the known motif elements [D/E]Xp[S/T]φ (φ = hydrophobic residue) (Nakajima et al., 2003), we frequently observed asparagine in the -2 position, and a strong preference for leucine in the -3 or another hydrophobic residue in the +2 position (Figure S11 A). This is consistent with a recent report (Alexander et al., 2011) that used an array-based motif approach and found asparagines in addition to glutamic and aspartic acid in the -2 position and a preference for hydrophobic residues in the +1 and +2 position. Interestingly, we found that motif elements that existed either upstream or downstream of the phosphorylatable residue were sufficient to trigger phosphorylation in vitro, a preference that could not have been identified in averaged motifs. For instance, we often observed [D/E/N]Xp[S/T] phosphopeptides that did not contain a hydrophobic amino acid in the +1 position. Similarly, the sub-motif p[S/T]F was frequently found in our dataset in the absence of any acidic residues in the -2 position. We note that the in vivo validated Plk1 sites S676 (EDpSR) on BubR1 (Elowe et al., 2007) and S149 (YSpSF) on Emi1 (Moshe et al., 2004), among others, are consistent with these observations.
Furthermore, we have recently shown that similar motifs were identified in vivo in Taxol-arrested HeLa cells by quantitative chemical phosphoproteomics using the small molecule Polo-like kinase inhibitor BI-2526 (Kettenbach et al., 2011b). We found that the Plk1 motifs identified in vivo represent a subset of the Plk1 motifs identified in vitro and hypothesized that this difference could be dependent on the Taxol-arrest conditions under which the in vivo data was collected. Taxol-treated cells arrest in mitosis in a metaphase-like state due to the stabilization of spindle microtubules. Because Plk1 plays an essential role in mitotic progression from mitotic entry to exit (Barr et al., 2004), we considered that we might identify a different subset of motifs at different phases of mitosis. To test this, we conducted a quantitative phosphoproteomics experiment using BI-2536 in HeLa cells treated with nocodazole, a microtubule depolymerising drug that arrests cells in a prophase-like state. HeLa cells were labelled using either isotopically “heavy” (13C615N2-lysine and 13C615N4-arginine) or natural amino acids in tissue culture (Ong et al., 2002) and arrested in mitosis with nocodazole. Heavy cells were treated with 100nM BI-2526 (dissolved in DMSO), while light cells were control-treated with DMSO (Figure 4A). After treatment, cells were mixed, lysed, trypsin-digested, and peptides were separated by strong-cation exchange (SCX) chromatography. Enrichment of phosphopeptides was performed using titanium dioxide microspheres, peptides were analyzed by LC-MS/MS, and statistically significant motifs were extracted from the inhibited phosphorylation sites. In this analysis, we identified the same motifs as described previously for HeLa cells treated with BI-2536 during Taxol arrest (Figure 4B), suggesting that Plk1 recognizes and phosphorylates sites in vitro and in vivo that are contained within many sub-motifs that do not strictly adhere to the classical [D/E/N]Xp[S/T]φ consensus motif, which previously might not have been predicted based on the classical consensus motif alone. Interestingly, we found that the increase in the number of identified motifs in vitro can be explained in part by the increased number of phosphopeptides identified in vitro in kinase reactions relative to the number identified as down-regulated by more than 2.5-fold after BI-2336 treatment in vivo. Overall, the motifs that are in common between both approaches are the most abundant ones in both datasets (Figure 4C). Motifs unique to the in vitro conditions displayed very similar chemical characteristics as the in vivo motifs (for instance TI, TL, SIL, SLL, SVL, DS, ExxS, and LxxS) and are likely less common variants which only rise to statistical significance when motif detection occurs in larger datasets, such as those from our in vitro kinase reactions, although we cannot rule out the possibility that these additional motifs are due to spurious phosphorylation that might occur under in vitro conditions. Ultimately, as with all large-scale experiments of this nature, any motifs generated in vitro will require careful cellular validation on a case-by-case basis for final confirmation.
Figure 4. In vivo and in vitro identification of Polo-like kinase 1 (Plk1) linear substrate motifs.
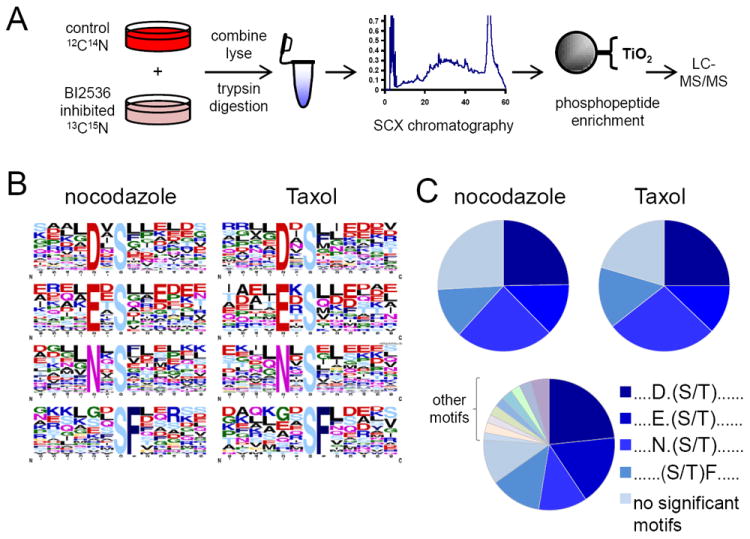
(A) SILAC-based workflow depicting in vivo identification of Plk1 substrates. HeLa cells were labeled with “heavy” and “light” amino acids in tissue culture, arrested in mitosis by nocodazole, differentially treated with a Plk1 inhibitor (BI-2536 dissolved in DMSO, heavy) or DMSO control (light), lysed, and trypsin-digested. Peptides were separated into 24 fractions by strong-cation exchange (SCX) chromatography and phosphopeptides were purified from non-phosphopeptides using titanium dioxide (TiO2) microspheres and analyzed by LC-MS/MS. Statistically significant motifs were extracted from phosphopeptides downregulated by 2.5-fold or more in the BI-2536-treated population by Motif-X. (B) Motif-X output for Plk substrates from nocodazole and Taxol-arrested HeLa cells. (C) Pie charts depicting the relative motif occurrence in HeLa cells arrested with nocodazole and Taxol, and in the in vitro kinase assay using purified Plk1 enzyme. Note that the relative distribution of primary Plk1 motifs observed in vivo with BI-2536 is similar to those with purified Plk1 in vitro. NS, no significant motifs. See also Figure S11 and Table S3.
Unique and common motif elements of polo-like kinase (Plk) family members
An advantage of our approach lies in the extraction of individual, statistically significant motifs for a given kinase, instead of a single, averaged motif as is produced from array-based methods. By way of example, for the closely related family of Polo-like kinases (Plk1-4), we show the value of this feature by identifying common as well as unique substrate sequences that allow for subtle distinctions between these structurally related kinases, which is much more difficult to observe based on single, averaged motifs. In this analysis we found a strong preference for aspartic or glutamic acid downstream and glutamic acid upstream of the phosphorylatable residue for Plk2 and Plk3 (Figure S12A and S13A), and a preference for hydrophobic amino acids (F, I, L, V, Y) in either the +1 or +2 or both for Plk4 (Figure S14A), consistent with a prior analysis using SPOT arrays (Leung et al., 2007). Cluster analysis of the Plk1-4 motifs revealed one motif that is phosphorylated by all four kinases (DXpS) or by three of them (NXpS, EXpS, EXXpS, DpS, DXpT; Figure 5). However, by expanding our motif coverage, we also found a set of motifs that were specifically phosphorylated by Plk2 and/or Plk3 that contained acidic amino acids upstream as well as downstream, or only downstream, of the phosphosite. Similarly, the more complete set of motifs show that Plk1 and Plk4 can recognize hydrophobic residues downstream of the phosphosite, with Plk4 displaying a stronger preference for larger side chain amino acids in +2 position or for hydrophobic amino acids in the +1 as well as +2 position. Most specific to Plk1 was a combination of aspartic or glutamic acid at the -2 and a leucine at the -3 or +2 position.
Figure 5. Hierarchical clustering of in vitro Plk1, Plk2, Plk3, and Plk4 motifs.
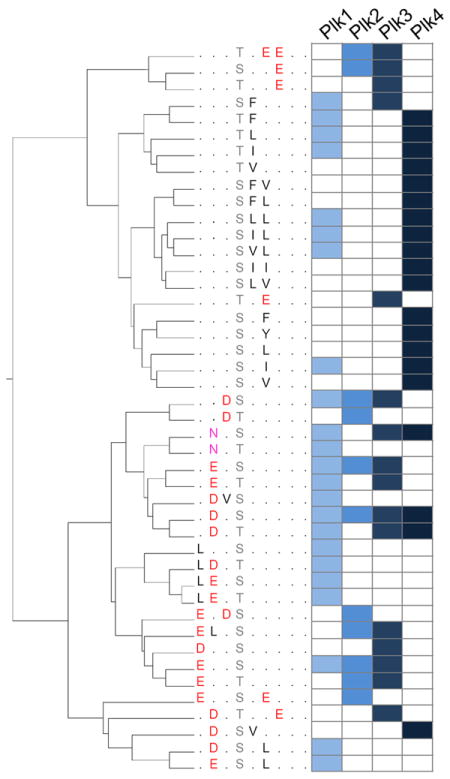
Hierarchical clustering of motifs identified in Plk1, Plk2, Plk3, and Plk4 in vitro kinase assays. Note the close relationship of acidic motifs for Plk2 and Plk3, and of hydrophobic motifs for Plk1 and Plk4. See also Figures S11 – S14.
Significance
Kinase-substrate interactions are based on several mechanisms, one of which involves the recognition of a linear sequence motif in the substrate by the kinase. Many of the 518 kinases encoded in the human genome have at least some of their preferred motif elements in common, represented in part by their broad, general classification as basophilic, acidophilic, hydrophobic, or proline-directed. To develop a more differentiated understanding of kinase specificity, we have developed a mass-spectrometry based approach that identifies a series of linear sequence motifs that are recognized and phosphorylated by a specific kinase. The detection of individual motifs increases sensitivity by recognizing sub-stoichiometric yet highly statistically significant motif elements, thereby elucidating a more complete array of possible phosphorylation targets for a given kinase. Furthermore, using peptide libraries built from the human proteome with phosphorylatable serine, threonine and tyrosine residues at variable positions permits the identification of kinase preferences for these amino acids as well as their position relative to peptide termini in a single analysis.
Detailed knowledge of the full set of sequence specificity determinants, phosphoacceptor preferences, and N- and C-terminal positioning requirements will provide invaluable assets in the quest to predict kinase-substrate interactions robustly and accurately in large scale phosphoproteomics datasets.
Experimental Procedures
Cell culture
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) with 10% fetal bovine serum (Hyclone) and penicillin-streptomycin (100U/mL and 100μg/mL; Invitrogen), at 37°C in a humidified atmosphere with 5% CO2.
Library preparation
HeLa cells were lysed in 8.5M Urea, 50mM Tris pH 8.7, protease inhibitors (Roche), lysate was sonicated using a Branson sonicator equipped with a microtip three times at power level 3 for 15 sec each on ice. Protein concentration of the lysate was determined by BCA protein assay from Pierce (Thermo Fisher Scientific Inc.). Proteins were reduced with 5mM DTT (SIGMA) at 55°C for 30min, cooled to room temperature and alkylated with iodoacetamide (SIGMA) at room temperature for 45 min in the dark. The alkylation reaction was quenched by the addition of another 5mM DTT. After 15 min incubation at room temperature, the lysate was diluted eight-fold in 25mM Tris pH 8.7, 1μg sequencing-grade trypsin (Promega) per 200μg total protein was added, and incubated overnight at 37°C. The same buffer conditions were used for Glu-C (Worthington) and chymotrypsin (Roche) digestion. One μg sequencing-grade Gluc-C per 75μg total protein and one μg sequencing-grade chymotrypsin per 100μg total protein was added, and incubated overnight at 25°C. After digestion, digests were acidified to pH 3 by addition of 20%TFA and incubated for 10 min at room temperature. Precipitates were removed by centrifugation at 3500 rpm for 15 min. The acidified lysates were desalted using a C18 solid-phase extraction (SPE) cartridge (Waters) and the eluate was lyophilized. Lyophilized peptides were dephosphorylated using CIP phosphatase (New England Biolabs) (75 units/mg peptide) in 50mM Tris pH 7.9, 100mM NaCl, 10mM MgCl2, 1mM DTT overnight at 37°C, desalted using a C18 solid-phase extraction (SPE) cartridge and the eluate was lyophilized. Dephosphorylation was repeated with λ-phosphatase (500 units/mg peptide) in 50mM Hepes, pH7.5, 100mM NaCl, 2mM DTT, overnight at 37°C, and desalted using a C18 solid-phase extraction (SPE) cartridge and the eluate was lyophilized. Eight milligrams of lyophilized peptides were separated by strong-cation exchange chromatography as described (Villen et al., 2007). Twelve fractions were collected, lyophilized, and desalted on a 96-well OASIS Elution C18 SPE plate (Waters). Each fraction was separated in 30 aliquots.
Protein expression and purification
The kinase domains of human Haspin (residues 465-798), Bmpr2 (residues 174-519), Dyrk1a (residues 118-476), Pim1 (92-403), Pim3 (1-326), Camkk2b (residues 132-470) and Clk3 (275-632) were cloned into the kanamycin resistant T7 expression vector pNIC28-Bsa4 by ligation-independent cloning. Pim1 (92-403) was cloned into ampicillin resistant pLIC-SGC1. All proteins were expressed as N-terminal His6 fusion with a TEV cleavage site in phage-resistant E. coli expression strain BL21 (DE3)R3-pRARE2 and were purified as described (Bullock et al., 2009; Bullock et al., 2005; Eswaran et al., 2009; Pogacic et al., 2007). All proteins were > 95 % pure as judged by SDS-PAGE and ESI-MS.
Full-length human Plk1, Plk2, Plk3, and Plk4 cDNA was amplified from a sequenced cDNA clone and cloned into a modified version of the pFastBac vector (Invitrogen) containing a 10-His-tag. For bacmid generation, pFastBac constructs were transformed into DH10Bac E. coli (Invitrogen). Recombinant bacmid DNA was purified and recombination was confirmed by PCR. Recombinant bacmid DNA was transfected into Sf9 cells using Cellfectin (Invitrogen) according to the manufacturer’s instructions. Five days after transfection, P1 virus stock was isolated and further amplified. For protein expression, Sf9 cells were infected with amplified virus stocks and 72 hrs after infection cells were harvested. Three hours before harvesting, cells were treated with 100nM okadaic acids (LC Labs) for 3 hrs. Ten-his-tagged proteins were purified using Ni-NTA agarose (Qiagen) according to the manufacturer’s instructions. Purified proteins were dialyzed overnight against 10mM Hepes pH 7.7, 100mM NaCl, 0.1mM EDTA, 1mM DTT, 10% glycerol and stored at -80°C.
In vitro kinase reactions
Pim1, Pim3, Clk3, Bmpr2, and Dyrk1A kinase reactions were performed in 20mM Hepes, pH 7.7 (SIGMA), 10mM MgCl2 (SIGMA), 0.5mM DTT (SIGMA), 5mM β-glycerophosphate (SIGMA), and 100μM ATP (SIGMA). Plk1 kinase reactions were performed in 20mM Hepes, pH 7.7, 20mM MgCl2, 0.5mM DTT, 5mM β-glycerophosphate, and 100μM ATP. Haspin kinase reactions were performed in 20mM Hepes, pH 7.7, 50mM NaCl (SIGMA), 10mM MgCl2, 0.5mM DTT, 5mM β-glycerophosphate, and 100μM ATP. Camkk2b kinase reaction was performed in 20mM Hepes, pH 7.7, 10mM MgCl2, 1mM CaCl2, 0.5mM DTT, 5mM β-glycerophosphate, and 100μM ATP. For each kinase reaction, early and late SCX fractions were incubated with 500ng of kinase in the indicated kinase buffer at 30°C for 3 hrs. For kinases with known basophilic substrate motifs (Pim1, Pim3, and Clk3), kinase reactions were performed on early and late peptide aliquots derived from a Glu-C digestion. Plk1 kinase reactions were performed on peptide aliquots derived from a trypsin digestion. Kinase assays on kinases for which no or only limited knowledge about substrate specificity existed (Bmpr2, Dyrk1a, Haspin, Camkk2b) were performed initially on separate peptide aliquots from both Glu-C and trypsin digestions. After an initial determination of the kinase motif of Bmpr2, additional reactions on chymotryptic peptides were performed. To determine residual phosphorylation sites which were not dephosphorylated in the phosphatase reaction, each peptide pool was incubated with kinase buffer only and analyzed in triplicate by LC-MS/MS; all phosphopeptides identified in these control reaction analyses were summed together and subtracted from each reaction performed with kinase.
Afterwards, the reactions were quenched with 0.1% TFA / 3% methanol and desalted on an OASIS MicroElution C18 SPE plate (Waters), dried by vacuum centrifugation, and phosphopeptides were purified using TiO2 microspheres (GL Sciences).
Phosphopeptide purification
For phosphopeptide purification using titatium dioxide, peptides were dissolved in 50% acetonitrile (Honeywell Burdick & Jackson), 0.1% TFA (Honeywell Burdick & Jackson), 2M lactic acid (SIGMA) and incubated with ~350 μg TiO2 microspheres (Glycan Biosciences) for 45 min with agitation (Kettenbach and Gerber, 2011). After binding, the TiO2 microspheres were washed several times with 50% acetonitrile / 0.1% TFA and phosphopeptides were eluted with 50mM disodium phosphate (SIGMA) pH-adjusted to pH10 with ammonia (SIGMA), dried and desalted. For phosphopeptide purification using immobilized metal (Fe-IMAC), peptides were dissolved in 40% acetonitrile and 25mM formic acid and incubated with 10μl IMAC beads (SIGMA) for 45 min with agitation (Villen and Gygi, 2008). After binding, the IMAC beads were washed several times 40% acetonitrile/ 25mM formic acid and phosphopeptides were eluted with 50mM disodium phosphate (SIGMA) pH-adjusted to pH10 with ammonia (SIGMA), dried and desalted. To increase the number of identification of phosphopeptides derived from Glu-C digest, peptides were further digest with 20ng trypsin in 25mM ammonium bicarbonate for 2hrs at 37°C and dried by vacuum centrifugation. Phosphoepeptide length after double Glu-C/trypsin digestion did not differ from single trypsin digestion (Figure S15).
LC-MS/MS Analysis and database searches
Each phosphopeptide purification was analyzed by nanoscale microcapillary LC-MS/MS essentially as described (Kettenbach et al., 2011b) on a LTQ-Orbitrap (Thermo Electron). Briefly, LC-MS/MS analysis was performed on a LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) equipped with an Agilent 1100 HPLC and LC-Packings FAMOS autosampler. Peptides were re-dissolved in 6% ACN/1% formic acid and loaded onto an in-house pulled and packed fused silica column (18cm length, 100μm inner diameter, ReproSil, C18 AQ 3μm). The peptides were eluted with a 50 min gradient of 0-29% B solvent (Buffer A: 0.0625% FA, 3% ACN, Buffer B: 0.0625% FA, 95% ACN). Peptide precursor ions were measured in the Orbitrap at a resolution of 60,000, and fragmentation spectra were collected in the LTQ. Up to ten of the most intense peptides were selected in each MS scan for MS/MS fragmentation. Raw data were searched using SEQUEST (Eng et al., 1994; Faherty and Gerber, 2010) against a target-decoy human UniProt sequence datasbase (www.uniprot.org; downloaded 09-2010) plus reversed sequences (Elias and Gygi, 2007), requiring for tryptic peptide pools fully tryptic peptides (K, R; not preceding P) with up to two mis-cleavages, for chymotryptic peptide pools, fully chymotryptic peptides (F, Y, W, M, L, I, V; not preceding P) with up to two mis-cleavages, and for Glu-c/tryptic double digest peptide pools, the combination of tryptic plus Glu-C termini (K, R, D, E; not preceding P) plus two mis-cleavages (Gilmore et al., 2012). In addition, carbamidomethylcysteine as fixed modification and oxidized methionine and phosphorylated serine, threonine and tyrosine as variable modifications were included. Searches were conducted at a +/- 1.1 Da precursor tolerance, and results were filtered to be within -/+ 2.5ppm (Haas et al., 2006; Hsieh et al., 2010); typically, XCorr values of > 2 for +2 charge state and > 2.6 for +3 charge state peptides yielded a < 1% FDR at the peptide level. Probability of correct phosphorylation site localization was assessed using PhospoRS (Taus et al., 2011). A localization probability of 0.99 or higher was required for phosphopeptides to be further considered for motif analysis; all other identifications were not utilized for motif generation.
Motif analysis
For motif determination of phosphopeptides identified in in vitro kinase assays the in house developed GrMFPh (Greedy Motif Finder for Phosphopeptides) algorithm was used. GrMFPh follows the basic logic of Motif-X (Schwartz and Gygi, 2005), extended for phosphopeptides from in vitro kinase reactions. Protein-level reduction by CDHIT is performed for both foregrounds and the background (UniProt human FASTA database). For each foreground, the reference protein sequences are collected and input into CDHIT (Li and Godzik, 2006) for clustering with sequence identity threshold at 0.9 and word length at 5. All peptides that are part of any representation of the clusters form the final foreground. The background comprises of all the representative protein sequences from CDHIT’s output of the input UniProtKB human FASTA database. The peptides are aligned at their common phosphorylation site, with pS, pT, and pY peptides handled separately. They remain of variable length, as was the case in the in vitro reactions. A GrMFPh motif then consists of a set of position / amino acid pairs that are statistically enriched in the aligned phosphopeptides, relative to an in silico digest of a background set of proteins (e.g. human proteome database, UniProt). A set of motifs is found, supported by disjoint sets of phosphopeptides. GrMFPh proceeds as follows. (1) Calculate the binomial probability of each amino acid a at each upstream and downstream position j. Since both foreground and background peptides are of variable length, only those that extend to the position in question are used in that calculation. The binomial probability of finding cja or more instances of amino acid a at position j in the n sufficiently-long foreground sequences, with respect to background probability pja, is:
The background probabilities are estimated in a preprocessing step, based on occurrence counts in the background peptides (considering each S, T, and Y separately). (2) Find the lowest-probability position j / amino acid a pair, such that the binomial probability P(X ≥ cja) is below a user-specified threshold (10-6 for the presented results) and the occurrence count cja is above another user-specified threshold (20 for the presented results). (3) If no such pair exists, the motif is complete; its score is the sum of the negative log binomial probabilities for its position / amino acid pairs (larger is better). The algorithm attempts to find additional new motifs by returning to step (1) with the remaining foreground peptides. (4) Otherwise, continue extending the current motif by returning to step (1) after restricting the foreground to the supporting peptides in which the motif occurs.
Phosphopeptides derived from Glu-C digestion were digested by trypsin before LC-MS/MS analysis. Thus before performing motif analysis, these peptides are in silico reconstituted to Glu-C peptides likely seen by the kinases, by finding in the UniProt database the protein containing the peptide, and extending from the tryptic cleavage sites out to the first Glu-C ones. Non-unique protein mappings from tryptic to Glu-C termini in the human proteome database were discarded (only unique sequence matches were retained). The number of occurrences was adjusted depending on the size of the data set (15 for 0-499; 20 for 500-999; 25 for 1000-1499; 30 for 1500-1999; and so on). For each motif, a sequence logo is generated by WebLogo (Crooks et al., 2004)
A GrMFPh webserver is available at http://www.cs.dartmouth.edu/~cbk/grmfph/ and the platform-independent Java code is freely available for academic use by contacting the authors.
Quantitative phosphoproteomics and inhibitor treatment
HeLa cells were grown in arginine- and lysine-free DMEM with 10% dialyzed fetal bovine serum supplemented with either 100mg/L 13C615N2-lysine and 100mg/L 13C615N4-arginine (Cambridge Isotope Laboratories) (“Heavy”) or identical concentrations of isotopically-normal lysine and arginine (“Light”) for at least six cell doublings. HeLa cells were synchronized in G1/S by a double thymidine block (1mM, SIGMA) for 16 hrs, with an 8 hour release between each cycle. To arrest cells in mitosis, nocodazole (100ng/mL, SIGMA) was added to the media 3hrs after the washout of the second thymidine block. Heavy labeled HeLa cells were incubated with 100nM BI2536 (synthesized in house, (Kettenbach et al., 2011b) along with MG132 (1μM, SIGMA) for 45min, while light cells were incubated with 1μM MG132 and DMSO. Heavy and light HeLa cells were counted and equal numbers of cells were mixed, washed twice in PBS, and lysed. Lysis and strong-cation exchange chromatography were carried out as previously described (Villen et al., 2007). Twenty-four fractions were collected, lyophilized, and desalted on a 96-well OASIS C18 SPE plate (Waters).
Data analysis for in vivo motif determination
The collected tandem mass spectra were data-searched using the SEQUEST algorithm (Link et al., 1999), filtered a 0.2% false discovery rate using the target-decoy strategy (Elias and Gygi, 2007) and reported. SILAC quantification was performed using a highly in-house-modified version of the XPRESS algorithm (http://tools.proteomecenter.org, Han et al., 2001). All H/L ratios were log2 transformed and fit to a Gaussian distribution using Sigma Plot software (San Jose, CA). Log2 transformed ratios were adjusted to the calculated, experiment-specific distribution offset. Motif analysis was performed on in vivo identified singly phosphopeptides with a log2 ratio of -1.4 or less using Motif-X (Schwartz and Gygi, 2005).
Positional preference
To determine positional preferences, the location of the phosphorylation site relative to the C- and N-terminus of motif-containing peptides was calculated. The distribution of phosphorylation sites was plotted in Excel and displayed for the first 20 amino acids from the C- and N-terminus. For Bmpr2 and Camkk2b, motif analysis led to the identification of motifs containing a protease cleavage site (arginine and lysine, trypsin, Supp. Table 2). Closer inspection of the peptides in these motifs showed that these residues were C-terminal cleavage sites of the respective peptide. To determine if these Arg and Lys residues were in fact part of the kinase substrate motifs, or just statistically enriched because of their location in the peptide, we investigated motifs derived from kinase assays using Glu-C peptides for Camkk2b and chymotryptic peptides for Bmpr2. For Bmpr2, we found the same upstream motifs as identified with tryptic peptides; however, instead of arginine and lysine residues, chymotryptic cleavage sites (leucine) occurred in a subset of downstream motifs (Supp. Table 2). Similarly, for Camkk2b we identified the same upstream motifs with Glu-C and trypsin proteases; however, glutamic acid, the Glu-C cleavage site, was observed downstream of the phosphorylation site instead of arginine and lysine residues (Supp. Table 2). Analysis of positional preference for both kinases show that 53% of motif-containing sites for Bmpr2 and 46% for Camkk2b were located two or two and three amino acids from the C-terminus, respectively. We concluded that for these kinases, the arginine and lysine residues in the motifs from tryptic digests, leucine from chymotryptic digests, and glutamic acid from Glu-C digests were indeed not part of kinase substrate motif, but rather a reflection of the preference for these kinases to phosphorylate C-termini. For comparison of positional preference, an equal number of motif-containing phosphopeptides from each of the other kinases were combined, and the distribution of locations of the phosphorylated residues relative to the C- and N-terminus of the respective peptide was determined.
Kinase-substrate kinetic assays
Variable sequence substrates were purchased as >95% HPLC-pure synthetic peptides from New England Peptide, LLC (Gardner, MA) and verified by LC-MS (Hasptides: ARSLVNAQG, ARTLVNAQG, ARYLVNAQG, QGARTLVNA, NAQGARTLV; Bmpr2tides: NGVEADLSL, NGVEADLTL, NGVEADLYL, VEADLSLNG, ADLSLNGVE). 2 nmol (~3 μg) peptide substrate was placed in separate PCR tubes, dried by vacuum centrifugation, and resuspended in 20 μl kinase buffer (Hapsin: 20mM HEPES pH 7.5, 125mM NaCl, 15mM MgCl2, 2mM MnCl2, 1mM DTT, 100μM ATP; Bmpr2: as for Haspin but without MnCl2) to produce a 100μM substrate concentration. Control reactions containing kinase buffer but no peptide substrate were included to correct for kinase autophosphorylation activities. Reactions were initiated by addition of 0.5pmol kinase, and allowed to proceed for 30 minutes at 28°C in a dry incubator. Kinase reactions were then processed using the ADP-Glo assay kit per manufacturer’s instructions (Promega, Madison, WI) and analyzed on an LMax II plate luminometer (Molecular Devices, Sunnyvale, CA). All reactions were analyzed in triplicate, and the average luminescence from each kinase reaction was background-corrected with the average control reaction values for that kinase. Data were reported as arbitrary luminescence units.
Selected-ion monitoring
Phosphotyrosine-containing peptides identified in Bmpr2 kinase reactions were used to create selected-ion monitoring (SIM) methods for two different trypsin-SCX fractions (fractions 5 and 6) essentially as described (Kettenbach et al., 2011a). 10 m/z-wide SIM windows were used to profile nine different pTyr peptides (SCX Fraction 5: VDDDFTAQDpYR @ 712.7740, GQLCELSCSTDpYR @ 834.8248, TIAQDpYGVLK @ 594.2911, GDpYPLEAVR @ 550.2467, SITADPLDpYR @ 615.7759; SCX fraction 6: LSEDpYGVLK @ 552.2568, EIMDAAEDpYAK @668.2645, DIYETDpYYR @ 659.2575, LDVTSVEDpYK @ 624.7755) in scheduled SIMs across relevant elution profile time windows, with a maximum of 3 SIMs in any one scan segment. SIMs were performed at R = 30,000 in an LTQ-Orbitrap mass spectrometer, and the resulting ion chromatograms were ion extracted for the observed mass of the precursor at maximum intensity +/- 2.5 ppm and plotted for reactions with and without Bmpr2 kinase (Figure S10).
Motif clustering
Plk1, Plk2, Plk3 and Plk4 motifs were clustered by average linkage hierarchical clustering, with scores computed for each pair of motifs according to a Needleman-Wunsch global alignment (Needleman and Wunsch, 1970). In constructing a motif alignment, the phosphorylation site was not scored (so that pS and pT were not distinguished), each pair of fixed amino acids was scored according to the Blosum-62 substitution matrix, each pair of variable amino acids (‘X’) contributed a score of 0, and each pair with one fixed and one variable contributed -4. The gap penalty was -4 for each gap character inserted.
Supplementary Material
Highlights.
Mass spectrometry-based kinase motif assay
Generates multiple independent, statistically significant linear motifs
Utilizes naturally occurring peptides as substrates, facilitating identification of substrates
Allows low-frequency but significant motif residues to be well represented
Acknowledgments
We thank the Gerber lab for helpful discussions and comments on the manuscript. We thank J.M. Higgins for helpful discussions. We thank Karl Mechtler for providing a standalone version of PhosphoRS. This work was supported by the National Institutes of Health grant P20-RR018787 from the IDeA Program of the National Center for Research Resources (to S.A.G.). T.W. is funded by National Science Foundation grant IIS-0905206. S.K. is an investigator of the Structural Genomics Consortium (SGC), a registered charity (no. 1097737) funded by the Wellcome Trust, GlaxoSmithKline, Genome Canada, the Canadian Institutes of Health Research, the Ontario Innovation Trust, the Ontario Research and Development Challenge Fund, the Canadian Foundation for Innovation, VINNOVA, The Knut and Alice Wallenberg Foundation, The Swedish Foundation for Strategic Research, and Karolinska Institutet. We would like to thanks Drs J. Elkins, A. Bullock, J. Eswaran and M Soundararajan at the SGC for making recombinant kinases available.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander J, Lim D, Joughin BA, Hegemann B, Hutchins JR, Ehrenberger T, Ivins F, Sessa F, Hudecz O, Nigg EA, et al. Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci Signal. 2011;4:ra42. doi: 10.1126/scisignal.2001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nature Reviews Molecular Cell Biology. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- Bullock AN, Das S, Debreczeni JE, Rellos P, Fedorov O, Niesen FH, Guo K, Papagrigoriou E, Amos AL, Cho S, et al. Kinase domain insertions define distinct roles of CLK kinases in SR protein phosphorylation. Structure. 2009;17:352–362. doi: 10.1016/j.str.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock AN, Debreczeni J, Amos AL, Knapp S, Turk BE. Structure and substrate specificity of the Pim-1 kinase. J Biol Chem. 2005;280:41675–41682. doi: 10.1074/jbc.M510711200. [DOI] [PubMed] [Google Scholar]
- Campbell LE, Proud CG. Differing substrate specificities of members of the DYRK family of arginine-directed protein kinases. FEBS Lett. 2002;510:31–36. doi: 10.1016/s0014-5793(01)03221-5. [DOI] [PubMed] [Google Scholar]
- Cohen P. The regulation of protein function by multisite phosphorylation--a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Sultan S, Taylor SS, Higgins JM. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005;19:472–488. doi: 10.1101/gad.1267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols N, Harrison P, Balasubramanian S, Luscombe NM, Bertone P, Zhang Z, Gerstein M. Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes. Nucleic Acids Res. 2002;30:2515–2523. doi: 10.1093/nar/30.11.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Elowe S, Hummer S, Uldschmid A, Li X, Nigg EA. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 2007;21:2205–2219. doi: 10.1101/gad.436007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR., Iii An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Eswaran J, Patnaik D, Filippakopoulos P, Wang F, Stein RL, Murray JW, Higgins JM, Knapp S. Structure and functional characterization of the atypical human kinase haspin. Proc Natl Acad Sci U S A. 2009;106:20198–20203. doi: 10.1073/pnas.0901989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faherty BK, Gerber SA. MacroSEQUEST: efficient candidate-centric searching and high-resolution correlation analysis for large-scale proteomics data sets. Anal Chem. 2010;82:6821–6829. doi: 10.1021/ac100783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore JM, Kettenbach AN, Gerber SA. Increasing phosphoproteomic coverage through sequential digestion by complementary proteases. Anal Bioanal Chem. 2012;402:711–720. doi: 10.1007/s00216-011-5466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, Li X, Villen J, Gygi SP. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics. 2006;5:1326–1337. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himpel S, Tegge W, Frank R, Leder S, Joost HG, Becker W. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J Biol Chem. 2000;275:2431–2438. doi: 10.1074/jbc.275.4.2431. [DOI] [PubMed] [Google Scholar]
- Hsieh EJ, Hoopmann MR, Maclean B, Maccoss MJ. Comparison of Database Search Strategies for High Precursor Mass Accuracy MS/MS Data. J Proteome Res. 2010;9:1138–1143. doi: 10.1021/pr900816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE. A rapid method for determining protein kinase phosphorylation specificity. Nat Methods. 2004;1:27–29. doi: 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- Kettenbach AN, Gerber SA. Rapid and reproducible single-stage phosphopeptide enrichment of complex peptide mixtures: Application to general and phosphotyrosine-specific phosphoproteomics experiments. Anal Chem. 2011;83:7635–7644. doi: 10.1021/ac201894j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach AN, Rush J, Gerber SA. Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat Protoc. 2011a;6:175–186. doi: 10.1038/nprot.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of aurora and polo-like kinase activities in mitotic cells. Sci Signal. 2011b;4:rs5. doi: 10.1126/scisignal.2001497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung GC, Ho CS, Blasutig IM, Murphy JM, Sicheri F. Determination of the Plk4/Sak consensus phosphorylation motif using peptide spots arrays. FEBS Lett. 2007;581:77–83. doi: 10.1016/j.febslet.2006.11.080. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Linding R, Jensen LJ, Pasculescu A, Olhovsky M, Colwill K, Bork P, Yaffe MB, Pawson T. NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 2008;36:D695–699. doi: 10.1093/nar/gkm902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., 3rd Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Miller ML, Jensen LJ, Diella F, Jorgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, et al. Linear motif atlas for phosphorylation-dependent signaling. Sci Signal. 2008;1:ra2. doi: 10.1126/scisignal.1159433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, et al. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal. 2010;3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshe Y, Boulaire J, Pagano M, Hershko A. Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc Natl Acad Sci U S A. 2004;101:7937–7942. doi: 10.1073/pnas.0402442101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem. 2003;278:25277–25280. doi: 10.1074/jbc.C300126200. [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Pinkse MWH, Uitto PM, Hilhorst MJ, Ooms B, Heck AJR. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-nanoLC-ESI-MS/MS and titanium oxide precolumns. Analytical Chemistry. 2004;76:3935–3943. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]
- Pogacic V, Bullock AN, Fedorov O, Filippakopoulos P, Gasser C, Biondi A, Meyer-Monard S, Knapp S, Schwaller J. Structural analysis identifies imidazo[1,2-b]pyridazines as PIM kinase inhibitors with in vitro antileukemic activity. Cancer Res. 2007;67:6916–6924. doi: 10.1158/0008-5472.CAN-07-0320. [DOI] [PubMed] [Google Scholar]
- Schilling O, Overall CM. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnol. 2008;26:685–694. doi: 10.1038/nbt1408. [DOI] [PubMed] [Google Scholar]
- Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005;23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr Biol. 1994;4:973–982. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Sugiyama N, Masuda T, Shinoda K, Nakamura A, Tomita M, Ishihama Y. Phosphopeptide enrichment by aliphatic hydroxy acid-modified metal oxide chromatography for nano-LC-MS/MS in proteomics applications. Mol Cell Proteomics. 2007;6:1103–1109. doi: 10.1074/mcp.T600060-MCP200. [DOI] [PubMed] [Google Scholar]
- Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res. 2011;10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci U S A. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc. 2008;3:1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB. Novel at the library. Nat Methods. 2004;1:13–14. doi: 10.1038/nmeth1004-13. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol. 2001;19:348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.