

Abstract
Rationale and Objective
Hemizygous deficiency of the transcription factor Krüppel-like factor 2 (KLF2) has been shown previously to augment atherosclerosis in hypercholesterolemic mice. However, the cell type responsible for the increased atherosclerosis due to KLF2 deficiency has not been identified. This study examined the consequence of myeloid cell-specific KLF2 inactivation in atherosclerosis.
Methods and Results
Cell-specific knockout mice were generated by Cre/loxP recombination. Macrophages isolated from myeloid-specific Klf2 knockout (myeKlf2-/-) mice were similar to myeKlf2+/+ macrophages in response to activation, polarization, and lipid accumulation. However, in comparison to myeKlf2+/+ macrophages, myeKlf2-/- macrophages adhered more robustly to endothelial cells. Neutrophils from myeKlf2-/- mice also adhered more robustly to endothelial cells, and less myeKlf2-/- neutrophils survived in culture over a 24 hr period in comparison with myeKlf2+/+ neutrophils. When myeKlf2-/- mice were mated to Ldlr-/- mice and then fed a high fat and high cholesterol diet, significant increase in atherosclerosis was observed in the myeKlf2-/-Ldlr-/- mice compared to myeKlf2+/+Ldlr-/- littermates. The increased atherosclerosis in myeKlf2-/-Ldlr-/- mice was associated with elevated presence of neutrophils and macrophages, with corresponding increase of myeloperoxidase as well as chlorinated- and nitrosylated-tyrosine epitopes in their lesion areas compared to myeKlf2+/+Ldlr-/- mice.
Conclusions
This study documents a role for myeloid KLF2 expression in modulating atherosclerosis. The increased neutrophil accumulation and atherosclerosis progression with myeloid-specific KLF2 deficiency also underscores the importance of neutrophils in promoting vascular oxidative stress and atherosclerosis. Collectively, these results suggest that elevating KLF2 expression may be a novel strategy for prevention and treatment of atherosclerosis.
Keywords: Krüppel-like factor, neutrophils, oxidative stress, atherosclerosis
The zinc finger transcription factor, Krüppel-like factor 2 (KLF2), is required for vascular integrity. Mice lacking KLF2 fail to develop beyond embryonic day 12.5 - 14.51, 2 and animals exhibit severe hemorrhaging, endothelial-mediated defects in blood vessel wall integrity, and abnormalities in the tunica media with decreased deposition of extracellular matrix surrounding the vessel.1 The embryonic lethality observed in whole animal knockouts of KLF2 occurs with endothelial specific knockouts of KLF2 but not with smooth muscle loss of KLF2, indicating that endothelial expression of KLF2 is necessary for vascular function.3 Mice lacking endothelial KLF2 die in utero due to high cardiac output resulting from a loss of peripheral vascular resistance.3
The importance of KLF2 in vascular development has led to the hypothesis that KLF2 may also have a protective role in the vascular system under pathological conditions. This hypothesis is supported by the finding that mice lacking one copy of the KLF2 gene exhibit increased atherosclerosis when mated with apoE-deficient mice and fed a high fat and cholesterol diet.4 However, as these mice have reduced KLF2 levels in all cells, and the pathogenesis of atherosclerotic lesions includes vascular endothelial and smooth muscle cell abnormalities as well as infiltration and pro-inflammatory activation of circulating leukocytes, the importance of KLF2 expression in each of these cell types toward atheroprotection has not been completely elucidated.
In the vascular endothelium, KLF2 is expressed in areas of high laminar flow, but at low levels where little shear stress occurs such as at bifurcations and bends.5 The shear stress element of the KLF2 gene has been identified 6 and shown to bind several transcription factors 7-9 with MEF2 acting as one of the factors that links shear stress with KLF2 expression.9 Under laminar flow conditions, KLF2 expression is induced,10, 11 leading to the down regulation of inflammatory genes such as vascular cell adhesion molecule and E-selectin, and the up regulation of vascular protective genes such as endothelial nitric oxide synthase.12-14 Several anti-thrombotic genes are also up regulated including thrombomodulin and tissue plasminogen activator, while plasminogen activator inhibitor-1, protease activated receptor 1, and tissue factor are down regulated.4, 6, 10-14 Two of these genes, endothelial nitric oxide synthase and thrombomodulin, have been shown to be KLF2 regulated and the promoter regions which bind KLF2 have been identified.12-14 These results indicate that KLF2 expression in endothelial cells protects against endothelial activation by modulating the expression of these genes.
In the peripheral circulation, KLF2 is expressed in mature thymocytes, naïve T cells, and memory T cells as well as in cells from the myeloid lineage. In the hematopoietic T cells, KLF2 binds to and trans-activates the promoter for sphingosine-1-phosphate receptor-1 to promote thymocyte emigration and recirculation through peripheral lymphoid organs.15 KLF2 has also been shown to be involved in monocyte differentiation, with diminished expression upon monocyte differentiation into macrophages.16 The expression of KLF2 is required to maintain immune cell quiescence, but its expression in both T cells and monocytes is suppressed during cell activation and inflammation.16-18 Over-expression of KLF2 inhibits immune cell activation and the expression of inflammatory genes is repressed.16 Interestingly, KLF2 expression can be induced by statin treatment,18 suggesting that the lipid-independent atheroprotective effects of statin therapy may be due to induction of KLF2 gene expression.
Another related transcription factor KLF4 has also been shown to play a role in the proinflammatory response of macrophages.19 Interestingly, KLF4-deficient progenitor cells of the myeloid lineage differentiate preferentially into granulocytes at the expense of monocytes, whereas over-expression of KLF4 results in almost exclusive differentiation of these progenitor cells into monocytes.20 Since neutrophil infiltration is an early event of atherogenesis and the depletion of neutrophils reduces atherosclerotic lesion size,21 expression of these kruppel-like factors in myeloid cells may significantly impact on atherosclerosis development and progression. To explore the role of myeloid cell KLF2 in lesion formation, mice were developed where KLF2 was deleted in myeloid cells. The myeloid KLF2 deficient mice were then placed on an Ldlr-/- background and fed a high fat diet. The resulting animals exhibited an increase in vascular lesion formation emphasizing the importance in myeloid KLF2 in this process.
METHODS
The Klf2fl/lfl mice in C57BL/6 background were generated by homologous recombination using the strategy described in Online Supplemental Methods and shown schematically in Figure 1A. The Klf2fl/fl mice were mated with Ldlr-/- mice (Jackson Laboratories) and then crossed with transgenic mice expressing Cre recombinase under the control of the LysM promoter (LysM-cre, Jackson Laboratories) to generate Ldlr-/- mice with or without KLF2 expression in myeloid cells. The Klf2fl/fl mice without the Cre recombinase transgene were used as controls throughout the study. Detailed experimental protocols, including animal maintenance and dietary conditions, blood cell count, macrophage and neutrophil functional studies, immunoblot analysis, and atherosclerosis lesion analysis and characterization, are also described in the Online Supplemental Methods. All polymerase chain reaction primers used are in Online supplemental Table I. All animal care and experimental procedures were reviewed and approved by the University of Cincinnati Institutional Animal Care and Use Committee.
Figure 1.
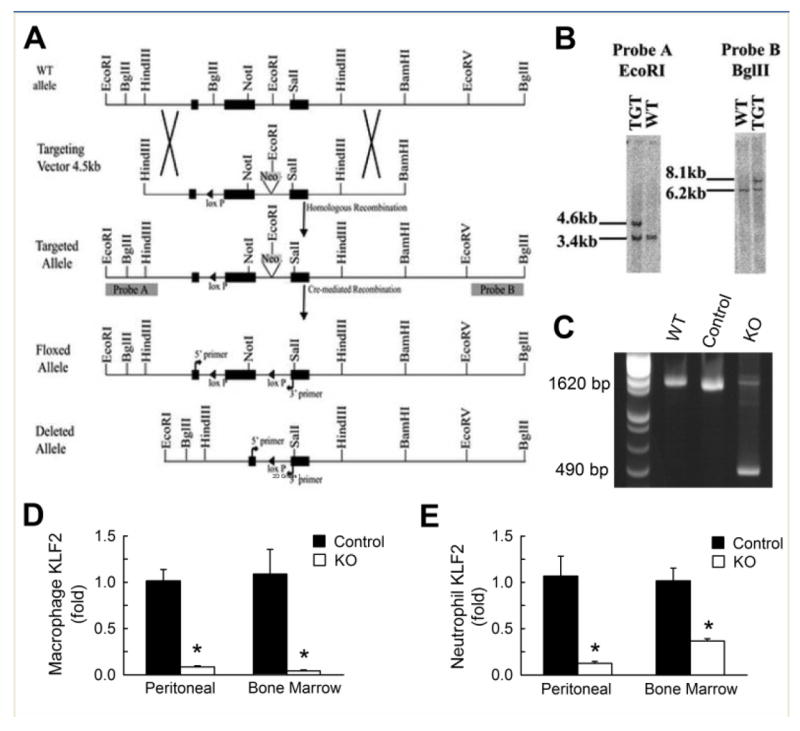
Generation of myeloid-specific Klf2 knockout mice. (A) Schematic diagram illustrating strategy used to insert LoxP site into the mouse Klf2 gene. The positions of 5’ and 3’ probes (probes A and B, respectively) for Southern screening of clones are shown. (B) Diagnostics illustrating the different sized bands recognized by probes after restriction digestion and Southern hybridization of DNA isolated from wild type (WT) and targeted (TGT) embryonic stem cells. (C) PCR analysis of DNA isolated from bone marrow macrophages from WT, cre-Klf2fl/fl (Control) and cre+Klf2fl/fl (KO) mice confirming DNA recombination as depicted in the 1620bp wild type allele and the 490bp recombined allele. Real-time quantitative PCR analysis of KLF2 mRNA abundance in (D) elicited peritoneal macrophages and bone marrow (BM) macrophages as well as (E) elicited peritoneal neutrophils and bone marrow neutrophils. *P< 0.01 compared to control as analyzed by Student’s t test.
RESULTS
Development of Myeloid-specific KLF2-Deficient Mice
The approach for determining whether KLF2 in myeloid cells alters the course of vascular lesion formation and atherosclerosis was to develop mice which lack this transcription factor in the myeloid cell lineage. This was accomplished by mating mice carrying conditional Klf2 allele in which a portion of the Klf2 gene was flanked by LoxP sites with transgenic mice with LysM promoter driven Cre recombinase expression (Figure 1A, B). Analysis of genomic DNA from bone marrow cells of the progeny mice by PCR amplification showed the replacement of a DNA fragment in the Klf2fl/fl gene with a smaller DNA fragment expected from cre-mediated recombination indicative of successful Klf2 gene knockout in bone marrow cells (Figure 1-C). Real-time PCR quantification of RNA from bone marrow-derived and peritoneal-elicited macrophages and CD11b+Ly6G+Ly6C+ neutrophils confirmed the absence of Klf2-specific reaction products in myeloid cells of the LysM-Cre, Klf2flox/flox mice (Figures 1D, E). Therefore, these mice can be referred to as myeloid-specific Klf2 knockout (myeKlf2-/-) mice. The Klf2fl/fl mice without cre recombinase expression are referred to as myeKlf2+/+ mice. The inactivation of KLF2 in myeloid cells did not influence KLF4 gene expression in macrophages of myeKlf2+/+ and myeKlf2-/- mice (Online supplemental Figure I). Additionally, KLF4 expression was not detectable in neutrophils of either myeKlf2+/+ and myeKlf2-/- mice.
Myeloid KLF2 Inactivation Reduces Lymphocyte Cell Count and Alters Neutrophil-Lymphocyte Ratio
The influence of myeloid KLF2 expression on leucopoiesis and cell differentiation was examined by comparing total leukocyte cell counts between myeKlf2+/+ and myeKlf2-/- mice. Interestingly, total number of monocytes and granulocytes, including neutrophils, eosinophils, and basophils, was not affected by KLF2 expression. In contrast, total lymphocyte cell count was significantly reduced in myeKlf2-/- mice compared to myeKlf2+/+ mice (Online supplemental Figure IIA). The reduced lymphocyte cell count resulted in alteration in leukocyte cell population with a trend toward higher monocyte:lymphocyte and neutrophil:lymphocyte ratios in myeKlf2-/- mice that did not reach statistical significance (Online supplemental Figure IIB, C).
Myeloid KLF2 Inactivation Increases Atherosclerosis in Hypercholesterolemic Mice
Elevation of neutrophil-lymphocyte ratio is an indicator of inflammation and is associated with poor cardiovascular outcome.22, 23 Although this relationship is generally attributed to inflammation associated with atherosclerosis 24 and inflammation-induced granulopoiesis at the expense of lymphopoiesis,25-27 potential direct contribution of elevated neutrophil-lymphocyte ratio toward atherosclerosis has not been explored. Therefore, myeKlf2-/- mice were mated to Ldlr-/- mice to generate myeKlf2-/-Ldlr-/- mice. The Klf2flox/floxLdlr-/- mice without the LysM-cre transgene (myeKlf2+/+Ldlr-/-) retained normal myeloid Klf2 expression in Ldlr-/- background and therefore were used as control. The LysM-cre transgenic mice with normal Klf2 gene expression were not included as controls because numerous studies from several laboratories have reported that myeloid cre recombinase expression in this mouse line has no effect on atherosclerosis lesion area in the aortic roots.28-30 The mice were fed a high fat and high cholesterol Western type diet for 8 weeks beginning at 6 weeks of age. Plasma cholesterol and triglyceride levels were similar between myeKlf2+/+Ldlr-/- and myeKlf2-/-Ldlr-/- mice under both basal and Western diet feeding conditions (Figure 2A). However, significant increase in atherosclerosis was observed in the aortic roots in mice with myeloid KLF2 deficiency (Figure 2B). The difference in atherosclerotic lesion area was evident when data from both male and female mice were analyzed together (Figure 2C). While lesion areas in male myeKlf2+/+Ldlr-/- and myeKlf2-/-Ldlr-/- mice did not reach statistically significant difference, the difference in lesion area was striking between female myeKlf2+/+Ldlr-/- and myeKlf2-/-Ldlr-/- mice (Figure 2D). The gender difference is not immediately obvious, but may be related to the generally more robust atherosclerotic response of female mice compared to male mice,31, 32 and the effect of KLF2 expression was less evident in mice with less severe atherosclerosis.
Figure 2.
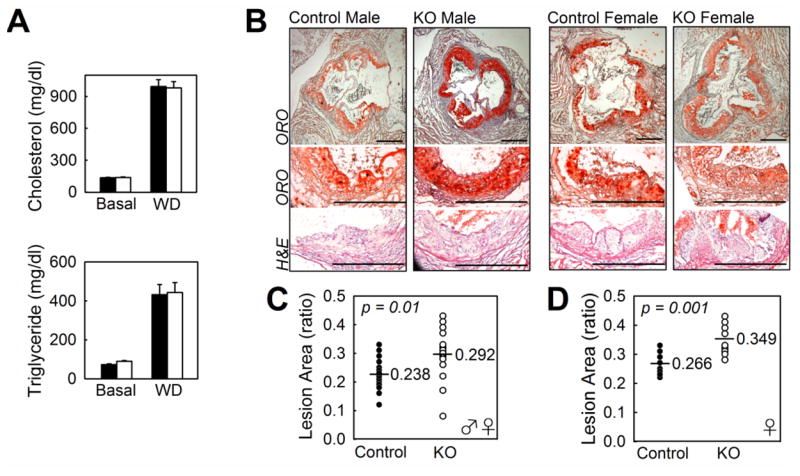
Plasma lipid levels and atherosclerotic lesion development in the aortic root of Ldlr-/- mice with or without myeloid-specific KLF2 expression. Male and female myeKlf2+/+Ldlr-/- (control) and myeKlf2-/-Ldlr-/- (KO) mice were fed a high-fat, high-cholesterol western-type diet for 8 weeks. (A) Plasma cholesterol and triglyceride levels in myeKlf2+/+Ldlr-/- (filled bars) and myeKlf2-/-Ldlr-/- (open bars) mice at basal level and after western diet feeding (WD). (B) Representative photomicrographs of aortic root lesions stained with oil-red-O (bar represents 500 μm) or hematoxylin and eosin from Control and KO male and female mice. (C) Morphometric analysis of lesion areas expressed as ratios of lesion area to total valve area from 22 control (●) and 21 KO (○) male and female mice or (D) female mice alone, with the geometric mean indicated with a line. P values for significance were indicated with each chart.
KLF2 Deficiency Increases Macrophage Adhesion to Endothelial Cells
The increase in atherosclerosis observed in myeloid KLF2 deficient hypercholesterolemic mice may be due to elevated macrophage inflammation and/or propensity for lipid accumulation in response to atherogenic lipoprotein stimulation. To test these possibilities, bone marrow-derived macrophages from myeKlf2+/+ and myeKlf2-/- mice were incubated with or without LPS and IL-4. Both myeKlf2+/+ and myeKlf2-/- macrophages responded to LPS-induced M1 polarization with elevated expression of IL-6, TNF-α, and iNOS (Nos2) (Figure 3A). Macrophages from myeKlf2+/+ and myeKlf2-/- mice also responded to IL-4-induced M2 polarization with elevated expression of Arg-1, Ym1, and Fizz1 (Figure 3B). Interestingly, both LPS and IL-4 also increased KLF2 expression in myeKlf2+/+ macrophages, which may explain the lower LPS-induction of iNOS and IL-6 and IL-4-induction of Fizz1 observed in myeKlf2-/- cells. When the thioglycolate-elicited peritoneal macrophages were added to endothelial monolayer, more myeKlf2-/- macrophages were found to adhere to endothelial cells in comparison with that observed with myeKlf2+/+ macrophages (Figure 4A). Additional experiments revealed no statistically significant difference in neutral lipid accumulation between peritoneal macrophages isolated from myeKlf2+/+ and myeKlf2-/- mice (Figure 4B). Taken together, these data indicate that KLF2 deficiency does not alter lipid accumulation in macrophages, but may promote atherosclerotic lesion progression via increased adhesion to endothelial cells.
Figure 3.
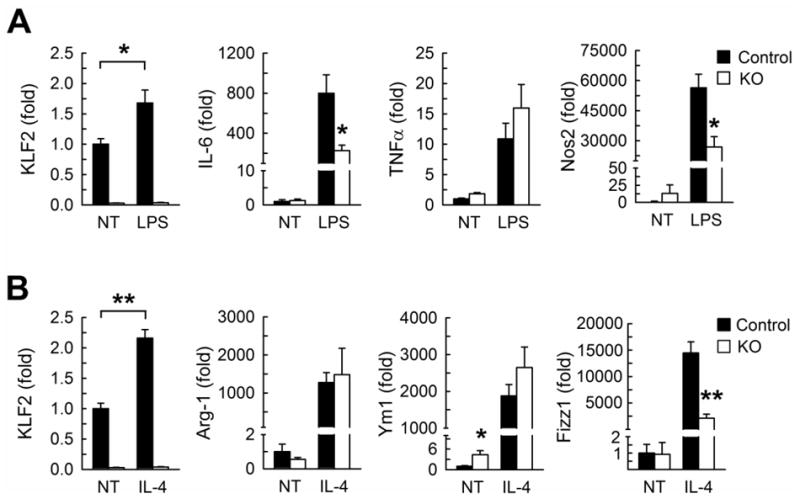
Lipopolysaccharide (LPS) or interleukin-4 (IL-4) mediated polarization of macrophages from male and myeKlf2+/+ and myeKlf2-/- mice. (A) Bone marrow-derived macrophages from myeKlf2+/+ (Control, filled bars) and myeKlf2-/- mice (KO, open bars) were treated with LPS for 24 hr (pooled from 3 mice per genotype, three wells per treatment). Expression levels of KLF2 and M1 macrophage markers were measured by qRT-PCR and normalized to expression levels observed without treatment (NT). (B) M2 marker gene expression in bone marrow-derived macrophages of control and KO mice after 24 hr treatment with IL-4. *P < 0.05, **P < 0.01.
Figure 4.
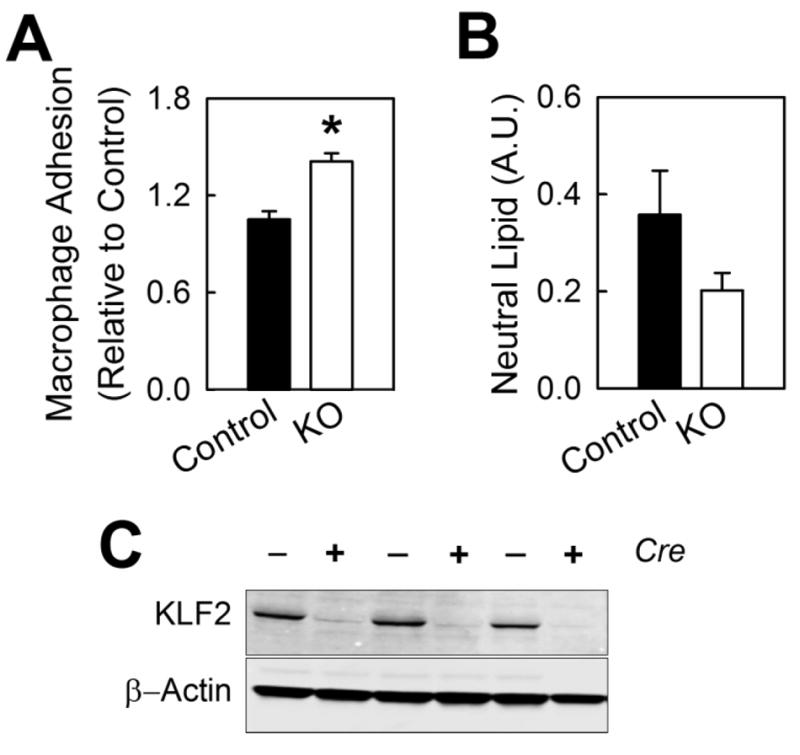
Functional characteristics of peritoneal macrophages from female myeKlf2+/+ and myeKlf2-/- mice. (A) Thioglycolate-elicited peritoneal macrophages from myeKlf2-/- (KO) and cre-negative littermates (Control) were added to endothelial monolayer in culture. The number of adherent cells is expressed relative to macrophages from control mice. (B) Control and KO macrophages were incubated with acLDL. Neutral lipid accumulation was quantified after Oil Red O incubation. (C) Representative KLF2 immunoblot of cellular proteins collected from peritoneal macrophages demonstrating deletion of the klf2 gene in the presence of cre recombinase. *P < 0.05.
KLF2 Deficiency Increases Neutrophil Adhesion and Cell Death
Neutrophils from myeKlf2-/- mice were also defective in KLF2 expression compared to neutrophils from myeKlf2+/+ mice (Figure 1E). Neutrophils from myeKlf2-/- mice showed elevated CD11b expression during fasting as well as after feeding a lipid-rich meal (Figure 5A). Additionally, more myeKlf2-/- neutrophils than myeKlf2+/+ neutrophils were found to adhere to activated endothelial cells (Figure 5B), which is likely due to the elevated expression of CD11b in myeKlf2-/- neutrophils.33 Expression of CD11b in polymorphonuclear leukocytes has also been shown to promote apoptosis.34, 35 Indeed, fewer myeKlf2-/- neutrophils were found to survive after 24 hr in culture compared to myeKlf2+/+ neutrophils (Figure 5C).
Figure 5.
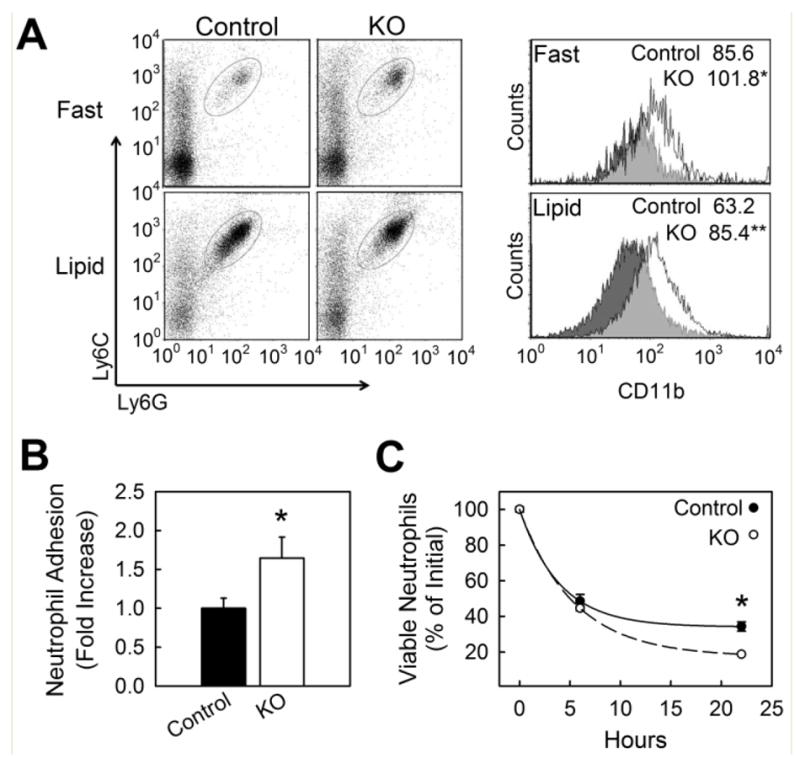
Neutrophil adhesion and survival in KLF2-deficient mice. (A) Whole blood neutrophils pooled from male and female mice were analyzed for cell surface expression of integrin receptor CDllb in fasted and hypertriglyceridemic state; n = 10 per group. Cells were gated to identify the neutrophil population (Ly6Gpos:Ly6Cpos) and analyzed for surface expression of CD11b during fasting conditions or two hours after an oral lipid gavage. Neutrophil expression of CD11b is shown in representative histogram plots with the mean ±SEM of the median fluorescence intensity (MFI) displayed (inset). (B) In vitro adhesion of peritoneal neutrophils to TNFα-activated bEND.3 endothelial cells. (C) Viability of peritoneal neutrophils cultured in IMBM containing 10% serum (percent annexin V and propidium iodide negative cells). *P < 0.05, **P < 0.01.
Myeloid KLF2 Deficiency Promotes Macrophage and Neutrophil Accumulation and Activity in Atherosclerotic Lesions of Hypercholesterolemic Mice
The increased propensity of myeKlf2-/- macrophage and neutrophil adherence to endothelial cells observed in vitro suggested that increased atherosclerosis observed in myeKlf2-/-Ldlr-/- mice may be due to elevated infiltration of these inflammatory cells to the lesion area. Analysis of atherosclerotic lesions in female myeKlf2+/+Ldlr-/- and myeKlf2-/-Ldlr-/- mice did reveal statistically significant increase in both macrophage (Figure 6A) and neutrophil (Figure 6B) numbers in the lesions of myeKlf2-/-Ldlr-/- mice. The higher number of neutrophils present in the lesion areas of myeKlf2-/-Ldlr-/ mice coincided with increased accumulation of myeloperoxidase (Figure 6C), an enzyme that directly contributes to the oxidative stress and endothelial dysfunction characteristics of atherosclerosis.36 Indeed, increased chlorinated tyrosine (Figure 6D) and nitrosylated tyrosine (Figure 6E) immunoreactivities, indicative of myeloperoxidase- and oxidative stress-induced reactive oxygen and reactive nitrogen species, respectively, were observed in the lesion areas of myeKlf2-/-Ldlr-/- mice.
Figure 6.
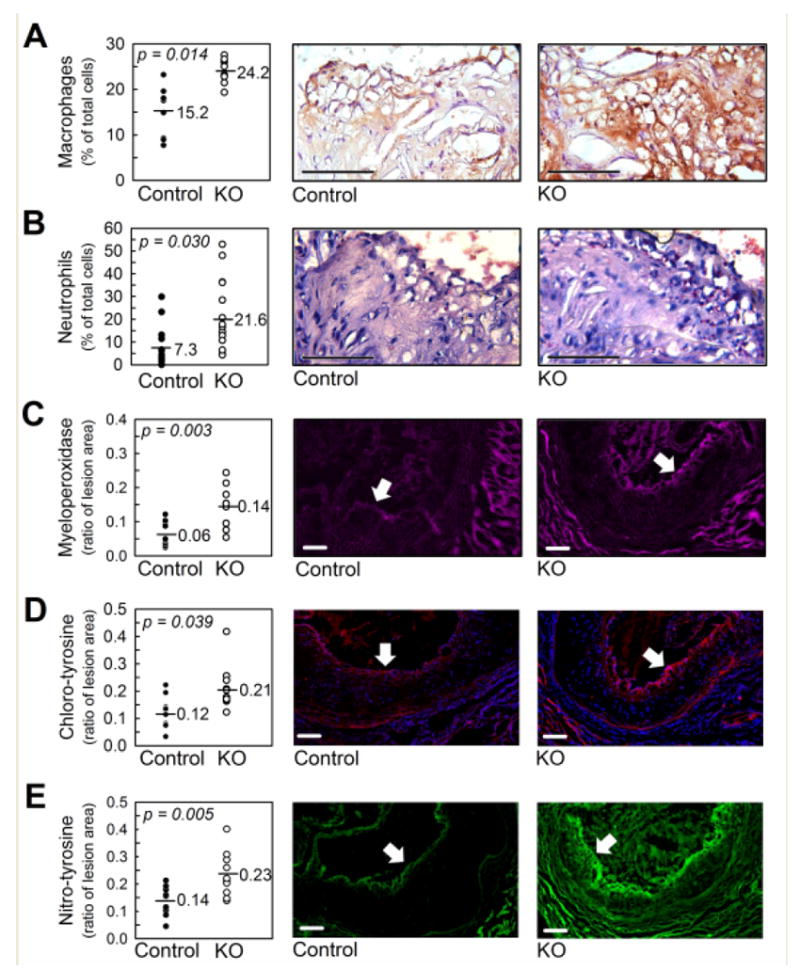
Aortic root atherosclerotic lesions from female myeKlf2+/+Ldlr-/- (Control) and myeKlf2-/-Ldlr-/-(KO) mice. (A) Macrophages identified by anti-CD68 immune-staining with hematoxylin counterstain. Negative control IgG staining is included in the supplemental section as Figure IIIA. (B) Neutrophils were identified by Leder staining for neutrophil esterases. A magnified image of positive Leder staining can be found in the supplement as Figure IIIC. (C) Presence of myeloperoxidase identified using anti-MPO antibody (pink). Accumulation of (D) chlorinated-tyrosine or (E) nitrosylated tyrosine specific protein adducts were identified using respective antibodies. Donkey IgG control antibody staining for all immunofluorescence experiments can be found in the supplement section as Figure IIIB. The panels on the left indicate morphometric analysis of the corresponding images from at least 10 mice in each group, with the horizontal line indicating the geometric mean. The arrows within each representative photomicrograph mark the lumenal margin of the atherosclerotic lesion. Scale bar, 100 μm. P values were determined by Student’s t test.
DISCUSSION
The importance of KLF2 in cell quiescence, trafficking, and differentiation has been demonstrated in several cell types, including T lymphocytes, adipocytes, monocytes, and erythroid cells.15, 37-46 The role of KLF2 in the vascular system was documented by observations that mice lacking the KLF2 gene die in utero due to vascular abnormalities.1, 2 Endothelial specific KLF2 knockout mice are also embryonically lethal, thus establishing the importance of endothelial KLF2 expression in vascular functions.3 Mice with hemizygous deficiency of KLF2 have been shown to augment atherosclerosis in apoE-deficient mice.4 The latter study showed that increased atherosclerosis due to heterozygous KLF2 deficiency was due to elevated macrophage infiltration into the atherosclerotic lesion areas as well as increased oxidized LDL accumulation in the macrophages of the Klf2+/- mice.4 Since KLF2 expression in the hemizygous mice is reduced in all tissues, including endothelial cells of the vessel wall and leukocytes in circulation, the cell type responsible for the accelerated atherosclerosis in the Klf2+/- mice has not been identified. The current study used conditional gene knockout approach to establish the importance of KLF2 expression in myeloid cell lineage in atheroprotection. The data revealed myeloid-specific KLF2 inactivation increases atherosclerosis in hypercholesterolemic Ldlr-/- mice. The mechanism appears to be related to increased myeloid cell adhesion to endothelial cells, with the consequence of elevated macrophage and neutrophil accumulation in atherosclerotic lesion areas to promote oxidative stress.
Although our results of increased atherosclerosis with myeloid-specific KLF2 inactivation are consistent with results reported earlier with hemizygous Klf2+/- mice, the two studies differed with respect to macrophage lipid accumulation in vitro. Atkins et al showed that peritoneal macrophages isolated from hemizygous Klf2+/- mice accumulated more neutral lipids accompanied by elevated fatty acid binding protein 4 expression in response to oxLDL.4 In contrast, our results showed that peritoneal macrophages with total KLF2 deficiency were similar to control macrophage in lipid accumulation in response to acLDL stimulation. Difference between these two studies are not immediately evident, but may be related to KLF2 expression levels in the macrophages or due to different forms of modified lipoproteins used in the in vitro experiments. Additionally, while both our study with conditional myeloid-specific KLF2 knockout mice and the Atkins study4 with global hemizygous Klf2+/- mice showed increased atherosclerosis with elevated macrophage infiltration into the lesion areas, our data additionally indicated that KLF2 expression in macrophages is not the only contributing factor, with KLF2 expression in neutrophils also playing prominent role in accounting for the difference in atherosclerosis between control and KLF2-deficient mice.
It is interesting to note that, in comparison to control macrophages, KLF2-deficient macrophages expressed less IL-6 and iNOS in response to LPS-induced M1 polarization, yet more atherosclerosis was observed in myeKlf2-/-Ldlr-/- mice compared to myeKlf2+/+Ldlr-/- mice. Although lower levels of inflammatory M1 markers are typically associated with less atherosclerosis,47 the robust atherosclerosis observed in myeKlf2-/-Ldlr-/- mice may be due to more myeKlf2-/- macrophages recruited to the atherosclerotic lesion areas as a consequence of increased CD11b expression and their adhesion to endothelial cells. Alternatively, the myeKlf2-/- macrophages were also less polarized to M2 macrophages with reduced Fizz1 expression in response to IL-4, suggesting their reduced ability to resolve inflammation at the lesion site.48 Our results showing persistent presence of significantly more neutrophils in the lesion areas of myeKlf2-/- mice compared to myeKlf2+/+ mice is consistent with the latter possibility. Typically, neutrophils are prominent in atherosclerotic lesions only during the early phase of atherogenesis preceding the recruitment of macrophages and the establishment of mature plaques.49 However, significant presence of neutrophils was observed in hypercholesterolemic myeKlf2-/- mice even after 8 weeks of feeding the high fat and high cholesterol diet when mature lesions were well established. The high levels of neutrophils in the atherosclerotic plaques of myeKlf2-/- mice may also reflect increased CD11b expression in myeKlf2-/- neutrophils, leading to their sustained infiltration into atherosclerotic lesion areas throughout the atherosclerosis process.
Results of the current study also showed that myeKlf2-/- mice have reduced lymphocyte cell count in the blood. Since the LysM promoter sequence used in the current study to drive cre expression and conditional Klf2 knockout is specific for myeloid cells without direct influence on gene expression in lymphoid cells,50 the reduced number of lymphocytes in myeKlf2-/- mice is probably an indirect effect reflecting myeloid cell regulation of lymphopoiesis.27, 51 However, despite the contributing role of lymphocytes in vascular inflammation and atherosclerosis,52 the reduction in the number of lymphocytes in circulation of myeKlf2-/- mice did not reduce atherosclerosis in hypercholesterolemic Ldlr-/- mice. In contrast, atherosclerosis was significantly increased in myeKlf2-/-Ldlr-/- mice compared to myeKlf2+/+Ldlr-/- mice. The difference is likely due to elevated macrophage and neutrophil adhesion to activated endothelial cells and their infiltration into the atherosclerotic lesion areas. The contributory role of neutrophils toward atherosclerosis progression is recently highlighted by data showing neutrophilia promotes atherosclerosis whereas neutropenia reduces atherosclerosis.53 Neutrophil accumulation in the vessel wall may increase atherosclerosis through its abundant expression of myeloperoxidase, which promotes endothelial dysfunction and elevates oxidative stress.36, 54 Increases in myeloperoxidase as well as reactive oxygen and nitrogen species were indeed observed in the lesion areas of myeKlf2-/-Ldlr-/- mice compared to myeKlf2+/+Ldlr-/- mice. In view of reports showing statin induces KLF2 expression in several cell types,55-57 including its reduction of CD11b expression58, 59 and transendothelial migration of leukocytes in the vessel wall, 60-63 the cholesterol-independent properties of statin in atheroprotection may in part be related to induction of KLF2 expression in macrophages and neutrophils, leading to their reduced adherence to activated endothelial cells.
Supplementary Material
Novelty and Significance
What is known?
The transcription factor Krüppel-like factor 2 (KLF2) plays a protective role in the vascular system and suppresses atherosclerotic lesion formation in hypercholesterolemic mice.
KLF2 expression in vascular endothelial cells protects against endothelial activation by down-regulating endothelial adhesion molecules and increasing the expression of vascular protective genes.
KLF2 is also expressed in myeloid cells and participates in inflammatory responses.
What new information does this article contribute?
Myeloid cell-specific inactivation of KLF2 increases atherosclerosis in hypercholesterolemic mice.
KLF2 inactivation elevated cell surface expression of CD11b in neutrophils and macrophages, leading to their increased adhesion to endothelial cells and recruitment to atherosclerotic lesions.
The increased presence of neutrophils and macrophages in atherosclerotic lesion areas of myeloid-specific KLF2-deficient mice exacerbates vascular oxidative stress to further promote atherosclerosis.
The transcription factor KLF2 plays a protective role in endothelial cells by repressing pro-inflammatory genes and inducing those that are anti-inflammatory. Nevertheless, the role of KLF2 in myeloid cells in atherosclerosis is currently unknown. Therefore we examined the effects of deleting KLF2 in neutrophils and macrophages on atherosclerotic lesion formation in LDL receptor-deficient mice fed with a high- fat, high-cholesterol diet. We found that mice lacking KLF2 in myeloid cells (myeKlf2-/-) displayed increased atherosclerosis, with elevated neutrophil and macrophage infiltration due to their increased adhesion to endothelial cells. Increased presence of neutrophils and macrophages in the lesions of myeKlf2-/- mice was associated with increasedoxidative stress, myeloperoxidase, and chlorinated and nitrosylated tyrosine residues. These findings suggest that myeloid cell KLF2 plays a critical role in protecting against atherosclerotic lesion formation by limiting neutrophil and macrophage recruitment in to the lesions and reducing oxidative stress in the vasculature in response to hypercholesterolemia. An important implication of this study is that induction of KLF2 expression in myeloid cells may confer atheroprotectiion. The atheroprotective effects of statins, independent of cholesterol lowering may also be related, in part, to KLF2 induction in myeloid cells.
Acknowledgments
We thank Dr. Marie-Dominique Filippi for her expert advice in neutrophil biology and Dr. Junqi Yang for assistance with cell sorting.
Sources of Funding
This study was supported by NIH grants HL78806 (to J.B.L) and DK74932 (to D.Y.H.).
Non-Standard Abbreviations
- acLDL
Acetylated LDL
- DAPI
4’,6-diamidino-2-phenylindole
- FBS
Fetal bovine serum
- IFNγ
Interferon-γ
- IL-4
Interleukin-4
- iNOS
Inducible nitric oxide synthase
- KLF
Krüppel-like factor
- LPS
Lipopolysaccharide
Footnotes
Disclosures None
References
- 1.Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 1997;11:2996–3006. doi: 10.1101/gad.11.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wani MA, Means RT, Jr, Lingrel JB. Loss of LKLF function results in embryonic lethality in mice. Transgenic Res. 1998;7:229–238. doi: 10.1023/a:1008809809843. [DOI] [PubMed] [Google Scholar]
- 3.Lee JS, Yu Q, Shin JT, Sebzda E, Bertozzi C, Chen M, Mericko P, Stadtfeld M, Zhou D, Cheng L, Graf T, MacRae CA, Lepore JJ, Lo CW, Kahn ML. KLF2 is an essential regulator of vascular hemodynamic forces in vivo. Dev Cell. 2006;11:845–857. doi: 10.1016/j.devcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Atkins GB, Wang Y, Mahabeleshwar GH, Shi H, Gao H, Kawanami D, Natesan V, Lin Z, Simon DI, Jain MK. Hemizygous deficiency of kruppel-like factor 2 augments experimental atherosclerosis. Circ Res. 2008;103:690–693. doi: 10.1161/CIRCRESAHA.108.184663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, de Vries CJ, Biessen EA, van Berkel TJ, Pannekoek H, Horrevoets AJ. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol. 2005;167:609–618. doi: 10.1016/S0002-9440(10)63002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huddleson JP, Srinivasan S, Ahmad N, Lingrel JB. Fluid shear stress induces endothelial KLF2 gene expression through a defined promoter region. J Biol Chem. 2004;385:723–729. doi: 10.1515/BC.2004.088. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad N, Lingrel JB. Kruppel-like factor 2 transcriptional regulation involves heterogeneous nuclear ribonucleoproteins and acetyltransferases. Biochemistry. 2005;44:6276–6285. doi: 10.1021/bi050018s. [DOI] [PubMed] [Google Scholar]
- 8.Huddleson JP, Ahmad N, Lingrel JB. Up-regulation of the KLF2 transcription factor by fluid shear stress requires nucleolin. J Biol Chem. 2006;281:15121–15128. doi: 10.1074/jbc.M513406200. [DOI] [PubMed] [Google Scholar]
- 9.Kumar A, Lin Z, SenBanerjee S, Jain MK. Tumor necrosis factor-α alpha-mediated reduction of KLF2 is due to inhibition of MEF2 by NF-κB and histone deacetylases. Mol Cell Biol. 2005;25:5893–5903. doi: 10.1128/MCB.25.14.5893-5903.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJ. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung kruppel-like factor (KLF2) Blood. 2002;100:1689–1698. doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- 11.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr, Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Jr, Balasubramanian V, Garcia-Cardena G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48–57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- 13.SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA, Jr, Garcia-Cardena G, Jain MK. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Z, Hamik A, Jain R, Kumar A, Jain MK. Kruppel-like factor 2 inhibits protease activated receptor-1 expression and thrombin-mediated endothelial activation. Arterioscler Thromb Vasc Biol. 2006;26:1185–1189. doi: 10.1161/01.ATV.0000215638.53414.99. [DOI] [PubMed] [Google Scholar]
- 15.Carlson CM, Endrizzi bT, Wu J, Ding X, Weinreich MA, Walsh ER, Wani MA, Lingrel JB, Hogquist KA, Jameson SC. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442:299–302. doi: 10.1038/nature04882. [DOI] [PubMed] [Google Scholar]
- 16.Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW, Majumder PK, Jain MK. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci U S A. 2006;103:6653–6658. doi: 10.1073/pnas.0508235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao Z, Sun X, Icli B, Wara AK, Feinberg MW. Role of kruppel-like factors in leukocyte development, function, and disease. Blood. 2010;116:4404–4414. doi: 10.1182/blood-2010-05-285353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bu DX, Tarrio M, Grabie N, Zhang Y, Yamazaki H, Stavrakis G, Maganto-Garcia E, Pepper-Cunningham Z, Jarolim P, Aikawa M, Garcia-Cardena G, Lichtman AH. Statin-induced kruppel-like factor 2 expression in human and mouse t cells reduces inflammatory and pathogenic responses. J Clin Invest. 2010;120:1961–1970. doi: 10.1172/JCI41384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feinberg MW, Cao Z, Wara AK, Lebedeva MA, Senbanerjee S, Jain MK. Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J Biol Chem. 2005;280:38247–38258. doi: 10.1074/jbc.M509378200. [DOI] [PubMed] [Google Scholar]
- 20.Feinberg MW, Wara AK, Cao Z, Lebedeva MA, Rosenbauer F, Iwasaki H, Hirai H, Katz JP, Haspel RL, Gray S, Akashi K, Segre J, Kaestner KH, Tenen DG, Jain MK. The kruppel-like factor klf4 is a critical regulator of monocyte differentiation. EMBO J. 2007;26:4138–4148. doi: 10.1038/sj.emboj.7601824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, Meiler S, Krohn R, Schober A, Sperandio M, Soehnlein O, Bornemann J, Tacke F, Biessen EA, Weber C. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–217. doi: 10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 22.Zahorec R. Ratio of neutrophil to lymphocyte counts--rapid and simple parameter of systemic inflammation and stress in critically ill. Bratisl Lek Listy. 2001;102:5–14. [PubMed] [Google Scholar]
- 23.Duffy BK, Gurm HS, Rajagopal V, Gupta R, Ellis SG, Bhatt DL. Usefulness of an elevated neutrophil to lymphocyte ratio in predicting long-term mortality after percutaneous coronary intervention. Am J Cardiol. 2006;97:993–996. doi: 10.1016/j.amjcard.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 24.Hansson GK, Robertson A-KL, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol Mech Dis. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 25.Terashima T, English D, Hogg JC, van Eeden SF. Release of polymorphonuclear leukocytes from the bone marrow by interleukin-8. Blood. 1998;92:1062–1069. [PubMed] [Google Scholar]
- 26.Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med. 2004;199:47–58. doi: 10.1084/jem.20031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uedo Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu X, Lee J-Y, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS. Increased cellular free cholesterol in macrophage-specific ABCA1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA. Macrophage deficiency of p38α MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest. 2009;119:886–898. doi: 10.1172/JCI37262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kardakaris R, Gareus R, Xanthoulea S, Pasparakis M. Endothelial and macrophage-specific deficiency of p38α mapk does not affect the pathogenesis of atherosclerosis in apoe-/- mice. PLoS One. 2011;6:e21055. doi: 10.1371/journal.pone.0021055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 32.Tangirala R, Rubin E, Palinski W. Quantitation of atherosclerosis in murine models: Correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J Lipid Res. 1995;36:2320–2328. [PubMed] [Google Scholar]
- 33.Lo S, Van Seventer G, Levin S, Wright S. Two leukocyte receptors (CD11a/CD18 and CD11b/CD18) mediate transient adhesion to endothelium by binding to different ligands. The Journal of Immunology. 1989;143:3325–3329. [PubMed] [Google Scholar]
- 34.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 35.Walzog B, Jerblonski F, Zakzewicz A, Gaehtgens P. β2 integrins (CD11/CD18) promote apoptosis of human neutrophils. FASEB J. 1997;11:1177–1186. doi: 10.1096/fasebj.11.13.9367353. [DOI] [PubMed] [Google Scholar]
- 36.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 37.Kuo CT, Leiden JM. Transcriptional regulation of T lymphocyte development and function. Annu Rev Immunol. 1999;17:149–187. doi: 10.1146/annurev.immunol.17.1.149. [DOI] [PubMed] [Google Scholar]
- 38.Kuo CT, Veselits ML, Leiden JM. Lklf: A transcriptional regulator of single-positive T cell quiescence and survival. Science. 1997;277:1986–1990. doi: 10.1126/science.277.5334.1986. [DOI] [PubMed] [Google Scholar]
- 39.Buckley AF, Kuo CT, Leiden JM. Transcription factor LKLF is sufficient to program T cell quiescence via a c-myc--dependent pathway. Nat Immunol. 2001;2:698–704. doi: 10.1038/90633. [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Lingrel JB. KLF2 inhibits jurkat T leukemia cell growth via upregulation of cyclin-dependent kinase inhibitor p21WAF1/CIP1. Oncogene. 2004;23:8088–8096. doi: 10.1038/sj.onc.1207996. [DOI] [PubMed] [Google Scholar]
- 41.Haaland RE, Yu W, Rice AP. Identification of LKLF-regulated genes in quiescent CD4+ T lymphocytes. Mol Immunol. 2005;42:627–641. doi: 10.1016/j.molimm.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 42.Bai A, Hu H, Yeung M, Chen J. Kruppel-like factor 2 controls T cell trafficking by activating l-selectin (CD62l) and sphingosine-1-phosphate receptor 1 transcription. J Immunol. 2007;178:7632–7639. doi: 10.4049/jimmunol.178.12.7632. [DOI] [PubMed] [Google Scholar]
- 43.Sebzda E, Zou Z, Lee JS, Wang T, Kahn ML. Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nature Immunology. 2008;9:292–300. doi: 10.1038/ni1565. [DOI] [PubMed] [Google Scholar]
- 44.Banerjee SS, Feinberg MW, Watanabe M, Gray S, Haspel RL, Denkinger DJ, Kawahara R, Hauner H, Jain MK. The kruppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-γ expression and adipogenesis. J Biol Chem. 2003;278:2581–2584. doi: 10.1074/jbc.M210859200. [DOI] [PubMed] [Google Scholar]
- 45.Basu P, Morris PE, Haar JL, Wani MA, Lingrel JB, Gaensler KM, Lloyd JA. KLF2 is essential for primitive erythropoiesis and regulates the human and murine embryonic β-like globin genes in vivo. Blood. 2005;106:2566–2571. doi: 10.1182/blood-2005-02-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basu P, Lung TK, Lemsaddek W, Sargent TG, Williams DC, Jr, Basu M, Redmond LC, Lingrel JB, Haar JL, Lloyd JA. EKLF and KLF2 have compensatory roles in embryonic β-globin gene expression and primitive erythropoiesis. Blood. 2007;110:3417–3425. doi: 10.1182/blood-2006-11-057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 48.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 49.Baetta R, Corsini A. Role of polymorphonuclear neutrophils in atherosclerosis: Current state and future perspectives. Atherosclerosis. 2010;210:1–13. doi: 10.1016/j.atherosclerosis.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 50.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using lysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 51.Li CY, Zhan YQ, Li W, Xu CW, Xu WX, Yu DH, Peng RY, Cui YF, Yang X, Hou N, Li YH, Dong B, Sun HB, Yang XM. Overexpression of a hematopoietic transcriptional regulator EDAG induces myelopoiesis and suppresses lymphopoiesis in transgenic mice. Leukemia. 2007;21:2277–2286. doi: 10.1038/sj.leu.2404901. [DOI] [PubMed] [Google Scholar]
- 52.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis. J Am Coll Cardiol. 2009;54:2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Drechsler M, Megens RTA, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837–1845. doi: 10.1161/CIRCULATIONAHA.110.961714. [DOI] [PubMed] [Google Scholar]
- 54.Soehnlein O, Weber C. Myeloid cells in atherosclerosis: Initiators and decision shapers. Semin Immunopathol. 2009;31:35–47. doi: 10.1007/s00281-009-0141-z. [DOI] [PubMed] [Google Scholar]
- 55.Ali F, Hamdulay SS, Kinderlerer AR, Boyle JJ, Lidington EA, Yamaguchi T, Soares MP, Haskard DO, Randi AM, Mason JC. Statin-mediated cytoprotection of human vascular endothelial cells: A role for kruppel-like factor 2-dependent induction of heme oxygenase-1. J Thromb Haemost. 2007;5:2537–2546. doi: 10.1111/j.1538-7836.2007.02787.x. [DOI] [PubMed] [Google Scholar]
- 56.Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, Jr, Garcia-Cardena G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714–26719. doi: 10.1074/jbc.C500144200. [DOI] [PubMed] [Google Scholar]
- 57.Tuomisto TT, Lumivuori H, Kansanen E, Hakkinen S-K, Turunen MP, van Thienen JV, Horrevoets AJ, Levonen A-L, Yla-Herttuala S. Simvastatin has an anti-inflammatory effect on macrophages via upregulation of an atheroprotective transcription factor, kruppel-like factor 2. Cardiovasc Res. 2008;78:175–184. doi: 10.1093/cvr/cvn007. [DOI] [PubMed] [Google Scholar]
- 58.Van Oostrom AJHHM, Plokker HWM, Van Asbeck BS, Rabelink TJ, van Kessel KPM, Jansen EHJM, Stehouwer CDA, Cabezas MC. Effects of rosuvastatin on postprandial leukocytes in mildly hyperlipidemic patients with premature coronary sclerosis. Atherosclerosis. 2006;185:331–339. doi: 10.1016/j.atherosclerosis.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 59.Walter T, Suselbeck T, Borggrefe M, Swoboda S, Hoffmeister HM, Dempfle CE. Effect of atovastatin on cellular adhesion molecules on leukocytes in patients with normocholesterolemic coronary artery disease. In vivo. 2010;24:189–193. [PubMed] [Google Scholar]
- 60.Sugano R, Matsuoka H, Haramaki N, Umei H, Murase E, Fukami K, Iida S, Ikeda H, Imaizumi T. Polymorphonuclear leukocytes may impair endothelial function: Results of crossover randomized study of lipid-lowering therapies. Arterioscler Thromb Vasc Biol. 2005;25:1262–1267. doi: 10.1161/01.ATV.0000163842.91226.ba. [DOI] [PubMed] [Google Scholar]
- 61.Guasti L, Marino F, Cosentino M, Cimpanelli M, Maio RC, Klersy C, Crespi C, Restelli D, Simoni C, Franzetti I, Gaudio G, Marnini P, Grandi AM, Lecchini S, Venco A. Simvastatin treatment modifies polymorphonuclear leukocyte function in high-risk individuals: A longitudinal study. J Hypertens. 2006;24:2423–2430. doi: 10.1097/01.hjh.0000251903.62804.77. [DOI] [PubMed] [Google Scholar]
- 62.Guasti L, Marino F, Cosentino M, Maio RC, Rasini E, Ferrari M, Castiglioni L, Klersy C, Gaudio G, Grandi AM, Lecchini S, Venco A. Prolonged statin-associated reduction in neutrophil reactive oxygen species and angiotensin II type 1 receptor expression: 1-year follow-up. Eur Heart J. 2008;29:1118–1126. doi: 10.1093/eurheartj/ehn138. [DOI] [PubMed] [Google Scholar]
- 63.Maher BM, Dhonnchu TN, Burke JP, Soo A, Wood AE, Watson RW. Statins alter neutrophil migration by modulating cellular rho activity--a potential mechanism for statins-mediated pleotropic effects? J Leukoc Biol. 2009;85:186–193. doi: 10.1189/jlb.0608382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.