

Abstract
The class of chemicals known as the “organophosphates” (OPs) comprises many of the most common agricultural and commercial pesticides that are used worldwide as well as the highly toxic chemical warfare agents. The mechanism of the acute toxicity of OPs in both target and non-target organisms is primarily attributed to inhibitory actions on various forms of cholinesterase leading to excessive peripheral and central cholinergic activity. However, there is now substantial evidence that this canonical (cholinesterase-based) mechanism cannot alone account for the wide-variety of adverse consequences of OP exposure that have been described, especially those associated with repeated exposures to levels that produce no overt signs of acute toxicity. This type of exposure has been associated with prolonged impairments in attention, memory, and other domains of cognition, as well as chronic illnesses where these symptoms are manifested (e.g., Gulf War Illness, Alzheimer’s disease). Due to their highly reactive nature, it is not surprising that OPs might alter the function of a number of enzymes and proteins (in addition to cholinesterase). However, the wide variety of long-term neuropsychiatric symptoms that have been associated with OPs suggests that some basic or fundamental neuronal process was adversely affected during the exposure period. The purpose of this review is to discuss several non-cholinesterase targets of OPs that might affect such fundamental processes and includes cytoskeletal and motor proteins involved in axonal transport, neurotrophins and their receptors, and mitochondria (especially their morphology and movement in axons). Potential therapeutic implications of these OP interactions are also discussed.
Keywords: Pesticide, Cholinesterase inhibitor, Chronic, Memory, Cognition
1. Introduction
1.1 Purpose of this review
The purpose of this review is to: 1) describe the variety of means by which individuals come in contact with organophosphates (OPs), 2) provide an overview of the various toxicological symptoms, particularly the neurobehavioral symptoms associated with repeated exposures to OPs and 3) discuss recent evidence to support the argument that the canonical (cholinesterase-based) mechanism of OP toxicity cannot alone explain the wide-variety of adverse consequences of OP exposure that have been described. For the second and third objectives, acute OP toxicity will be briefly discussed; however, the neurobehavioral symptoms emphasized in this review (and the proposed mechanisms thereof) will primarily apply to those observed in the absence of overt signs of acute toxicity. Diverse semantics have been used for this form of toxicity (subacute, subtoxic, subclinical, subthreshold, etc.), thus it is important to reiterate that in this review, protracted neurobehavioral symptoms occurring in the absence of antecedent signs of acute toxicity, but not necessarily in the absence of cholinesterase inhibition, will be emphasized. It is also important to note that cholinesterase inhibition can occur in the absence of acutely toxic effects, but not vice versa; thus these two descriptors may represent a gradation of direct effects. The non-cholinesterase targets discussed in this review could be affected in both the case of acute-high level exposure as well as repeated lower level exposure (however, in the acute setting the non-cholinesterase related physiologic effects might be difficult to distinguish). Moreover, while several non-cholinesterase-based mechanisms of OP toxicity will be briefly discussed (e.g., OP target proteins, oxidative stress, neuroinflammation), the primary focus here will be on more recently introduced (potential) targets of OPs (e.g., axonal transport, neurotrophins and mitochondrial dynamics).
1.2 OPs-General Background
The generic term “organophosphate” or “OP” is used for a wide variety of chemicals that are derived from phosphoric, phosphonic and phosphinic acids (see Fig 1). The French chemists, Jean Louis Lassaigne and Philip de Clermount are credited with the synthesis of the first OPs in the nineteenth century, while the initial development of OPs as insecticides and chemical warfare agents early in the twentieth century is primarily attributed to the German chemist Gerhard Schrader (Gallo and Lawryk, 1991; Tucker, 2006). Since these early years literally hundreds of OP-based compounds have been synthesized and they are found in insecticides (e.g., malathion, parathion, diazinon, chlorpyrifos), chemical warfare (“nerve”) agents (e.g., soman, sarin, tabun, VX), some ophthalmic agents (e.g., echothiophate, isoflurophate), antihelmintics (e.g., trichlorfon), herbicides (e.g., tribufos, merphos), as well as solvents, plasticizers, and extreme pressure additives for lubricants (Katz and Brooks, 2010). The widespread use of OPs (especially as insecticides, see below) has been an environmental health concern for many years and there are a number of reports suggesting that OPs might be associated with an increased risk of a variety of chronic illnesses including respiratory (e.g., chronic obstructive respiratory disease), metabolic (e.g., obesity, diabetes) and neurologic (e.g., Alzheimer’s Disease Parkinson’s disease) disorders (Hancock et al., 2008; Chakraborty et al., 2009; Hayden et al., 2010; Slotkin 2011).
Fig. 1.
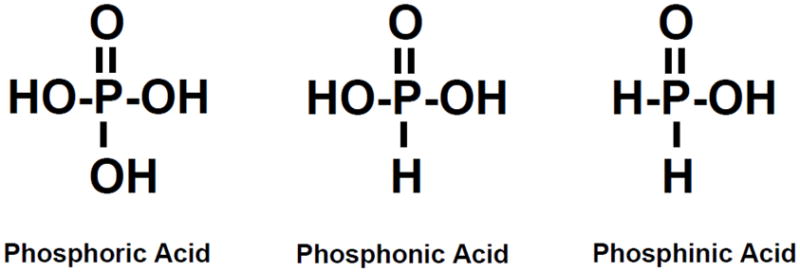
Chemical structures of phosphoric, phosphonic, and phosphinic acid.
1.3 OP-Pesticides
Pesticides (including OPs) have multiple applications in agricultural, industrial, and household settings and as a result they are used extensively worldwide. Their value in optimizing agricultural productivity, the control of deadly vector-borne illnesses (e.g., malaria, yellow fever, viral encephalitis, typhus, etc.), and “nuisance” pests (e.g., flies, roaches, ants, mosquitoes, etc.) is clear (reviewed, Cooper and Dobson, 2007). As consequence of their widespread use, however, pesticides (and their residues) are now among the most ubiquitous synthetic chemicals in our environment. Accordingly, inherent dangers to the public health exist since no pesticide is innocuous and all carry significant toxicological risks. The use of OP-based pesticides (specifically) is now considered a worldwide health problem since they are the most commonly used and the most often associated with toxicity to humans (see reviews, Buckley et al., 2004; Rohlman et al., 2011). In the United States (US) alone, the most recent estimates available (from 2007) indicate that approximately 33 million pounds of OP-based pesticides are applied annually (US EPA, 2011). Internationally, OP-pesticide poisonings are among the most common modes of poisoning-related fatalities (i.e., both intentional and unintentional), a phenomenon that has been attributed to ease of access to OPs and the relatively low level of regulations governing their use especially in developing countries (see Buckley et al., 2004; Dharmani and Jaga 2005 for reviews). In the US there are multiple regulations and safety training requirements under the purview of the Environmental Protection Agency (EPA) for the handling and transport of OPs. However, recent studies suggest that acute pesticide poisoning particularly in the agricultural industry (including poisonings with OP-based pesticides) continues to be a significant problem (Calvert et al., 2008) and moreover, pesticide poisonings are most likely underreported. It has been suggested that disproportionate numbers of agricultural workers are deterred from seeking health care in the US due to a number of factors including concerns related to immigration status, the lack of health insurance, unfamiliarity with (or the inability to qualify for) workers’ compensation benefits, and the fear of job loss if they miss time from work to seek health care. In addition, other sources of the underreporting of pesticide poisonings include misdiagnosis by health care workers as well as their lack of awareness that they are required to report such incidents (i.e., when they are properly diagnosed) to public health officials (see Calvert et al., 2008).
1.4 OP Nerve Agents as Chemical Weapons
While the risk of OP exposure as a result of extensive pesticide use is considerably higher for most people, the threat from intentional poisonings by rogue governments and terrorist organizations is an ongoing concern. It is now relatively well documented that in the 1980s the Iraqi military attacked Iranian military soldiers (Majnoon Island) and Kurdish civilians (Halabja) with OP-based nerve agents producing casualties estimated to be as high as “tens of thousands” (Barnaby, 1988; Macilwain, 1993; O’Leary, 2002; Hawrami et al., 2004). Moreover, international news reports provide an almost daily reminder of the escalating terrorist activities throughout the world and it is clear that the use of toxic chemicals is a major goal of such groups. The Tokyo Sarin attack in March of 1995 revealed the danger of even a limited chemical attack given that 12 people were killed and over 5,000 others required emergency medical evaluation and/or treatment (Suzuki et al., 1995; Nagao et al., 1997). This incident also clearly indicated that terrorist groups have the desire to use nerve agents on civilian populations and that they are capable of both acquiring and deploying them. Since the Tokyo attacks an increasing terrorist threat level can be surmised from a number of factors such as: the growth of militant religious groups with increasingly sophisticated and international capabilities, the increasing global availability of highly technical information regarding chemical (and biological) weapons on the internet, and the clear evidence of terrorist’s interest in such weapons (Cronin, 2003). Based on several factors, the odds for a chemical attack by terrorists may be actually higher than biological or nuclear attacks due to the more widespread availability of raw materials for making chemical weapons. These materials include large stockpiles of military-grade chemical weapons that remain undestroyed or unaccounted for in multiple countries around the world. The Organization for the Prohibition of Chemical Weapons (OPCW) estimates that (as of September 30, 2010) 44,131, or 61.99%, of the world’s declared stockpile of 71,194 metric tons of chemical agent have been verifiably destroyed (OPCW, 2011). However, this leaves nearly 30,000 metric tons undestroyed, and these numbers do not include the stockpiles of non-member states (e.g., Syria, North Korea) that have neither signed nor acceded to the Chemical Weapons Convention.
1.5 OP Pesticides as Chemical Weapons
As opposed to the use of nerve agents, an equally significant and perhaps more likely domestic terrorist scenario would be the use of industrial or agricultural chemicals as weapons (Burklow et al., 2003). Industrial chemicals have (in fact) been used by terrorists as improvised explosives, incendiaries, and poisons in several incidents (Hughart, 1999). Notwithstanding the potential access of terrorists to nerve agent stockpiles in foreign countries (discussed above), in the US, the use of these weapons is limited by the security surrounding government chemical agent stockpiles and binary chemical agent storage, as well as the controlled access to precursor chemicals. While improvised chemical agents may be less toxic than weaponized (military) agents, they have rapid, highly visible impacts on human health and they can be dispersed by smoke, gas clouds, or food and medicine distribution networks (Hughart, 1999). Of the variety of chemicals that could be used as domestic chemical weapons, OPs certainly must rank near the top given their wide availability on hundreds of insecticide products.
2. Neurobehavioral Effects of OP Exposure
2.1 Acute and Chronic Behavioral Effects of OPs
The acute toxicity of OPs in humans has been associated with a host of central nervous system, cardiovascular, respiratory, gastrointestinal, sensory, and motor manifestations which are frequently life threatening (see reviews, Bardin, 1994; Collombet, 2011). Several studies also document long-term neuropsychiatric sequelae in subjects who have experienced acute OP toxicity. These include deficits in signal detection and information processing, sustained attention, memory, sequencing and problem solving, abstraction, flexibility of thinking, and depressed mood (Savage et al., 1988; Rosenstock et al., 1991; Steenland et al., 1994; Dassanayake et al., 2007). Likewise, there is significant evidence to suggest that chronic exposure to OPs can be associated with neurobehavioral abnormalities including anxiety, depression, psychotic symptoms, deficits in short-term memory, learning, attention, information processing, eye-hand coordination and reaction time, and extrapyramidal symptoms (Amr et al., 1997, Salvi et al., 2003, Singh and Sharma, 2000 and Stephens et al., 1995). These symptoms, collectively known as chronic OP-induced neuropsychiatric disorders (COPIND), can occur without antecedent cholinergic symptoms and they do not appear to be dependent on acetylcholinesterase inhibition (Brown and Brix, 1998; Ray and Richards, 2001 and Singh and Sharma, 2000). Prospective animal studies support these clinical and epidemiological observations and indicate that chronic (or repeated) exposures to OPs at levels that are not associated with acute toxicity can result in a variety of neurobehavioral symptoms, particularly cognitive deficits. For example, sustained deficits in delayed matching performance, sensorimotor gating, spatial learning, recognition memory, cognitive flexibility and sustained attention have been reported in association with both pesticide OPs as well as alkylphosphate (nerve agent) OPs (Bushnell et al.., 1991; Terry et al., 2003; Terry et al., 2007, Middlemore-Risher et al., 2010; Terry et al. 2011, Terry et al., 2012). There is also considerable evidence from animal studies to suggest that both prenatal and early postnatal exposure to OPs (at doses not associated with acute toxicity) can result in a variety of protracted cognitive impairments (Levin et al., 2001; Slotkin 2004; Slotkin 2005; Timofeeva et al., 2008) and, further, that these impairments can become more pronounced as the exposed subject ages (Levin et al., 2010).
Interestingly, using electrophysiological and histological methods, Speed et al., 2012 recently reported that in adult mice repeatedly exposed to subclinical doses of chlorpyrifos (3 months earlier), a 50% reduction in synaptic transmission and a corresponding 50% reduction synaptic spine density in pyramidal neurons in the CA1 region of the hippocampus. These data provide an anatomical and neurophysiological basis for the cognitive deficits described above and further support the argument that repeated exposure to relatively low doses of OPs results in persistent damage to the adult mammalian brain.
2.2 OPs and Gulf War Illness
It is now relatively well established that at least one fourth of the United States veterans who served in the 1990–1991 Persian Gulf War are affected by a complex of multiple symptoms known as Gulf War Illness (GWI, Research Advisory Committee on Gulf War Veterans’ Illnesses, 2008). These symptoms, which include unexplained fatigue, respiratory problems, musculoskeletal pain, gastrointestinal distress, skin rashes, and a variety of neurological and neuropsychiatric problems, have also been reported in veterans from other countries who participated in the first Gulf War (United Kingdom, Canada, Australia, and Denmark, reviewed, Iverson et al., 2007). One particularly plausible explanation for several of the neurological-based symptoms (see Golomb, 2008) is exposure to one or more acetylcholinesterase inhibitors (AChEIs). It has been estimated that at least 41,000 military personnel were exposed to insecticides that contained either carbamate or OP-based AChEIs (Fricker et al., 2000; US Department of Defense, 2003) and as many as 100,000 may have been exposed to low (i.e., non-acutely toxic) levels of sarin/cyclosarin following the destruction of an Iraqi munitions storage complex at Khamisiyah, Iraq, in March 1991 (Berardocco, 1997). Among the constellation of chronic symptoms associated with GWI, the deficits in attention, concentration, information processing, etc. (Lange et al., 2001) have much in common with the symptoms that have been attributed to environmental OP exposures in civilians (farmers, sheep dippers, pest control specialists, etc., Amr et al., 1997; Dassanayake et al., 2007; De Silva et al., 2006; Salvi et al., 2003, Singh and Sharma, 2000; Steenland et al., 1994 and Stephens et al., 1995).
It should be noted, however, that despite more than twenty years of research, the etiology of GWI remains unclear. One of the greatest challenges to elucidating the underlying pathobiology of GWI is the confounding nature of the wide variety of factors that either alone or in combination may have contributed to the symptoms. These factors include prophylactic therapeutic measures (multiple vaccinations, pyridostigmine bromide to prevent nerve agent-related toxicity, etc.) and environmental exposures (smoke from oil well fires, infectious organisms, pesticides, nerve agents, etc).
3. OP Targets other than Cholinesterase
OPs are believed to manifest their acute biological actions primarily through inhibiting the various forms of cholinesterase, the degradative enzyme for the neurotransmitter acetylcholine. Toxicity to the target organism is then mediated through elevation of synaptic levels of acetylcholine in tissues innervated by cholinergic neurons, and subsequent overstimulation of postsynaptic cells (reviewed, Ecobichon, 1991). While the inhibition of cholinesterase enzymes undoubtedly plays a key role in the toxicology of OPs, there is now substantial evidence that this mechanism cannot alone account for the wide range of symptoms and disorders that have been reported (Pope, 1999; Duysen, et al., 2001). Due to their highly reactive nature, it is not surprising that OPs might alter the function of a number of other important enzymes and proteins (see reviews, Casida and Quistad, 2005 and Lopachin and Decaprio, 2005). Moreover, it has been suggested that interactions of OPs with non-cholinesterase targets may contribute to the more delayed and persistent effects observed following chronic exposure to OPs (see reviews, Lotti and Moretto, 2005; Costa, 2006). The list of non-cholinesterase targets for OPs (in environmentally relevant doses) is growing and now includes a variety of proteins, receptors, and enzymes (see Table 1 for a referenced list of potential targets and their known or theoretical functions). Prior to the last several years, it was generally believed that OP adducts formed only with the active site of enzymes in the serine hydrolase superfamily. However, analysis of a variety of proteins treated with OPs in vitro by mass spectrometry has also lead to the identification of covalent binding of OPs to tyrosine and lysine residues (Grigoryan et al., 2008,Grigoryan et al., 2009a,b,c). These results suggest that numerous proteins can be modified by OPs.
Table 1.
Non-Cholinesterase Targets of OPs (at physiologically relevant concentrations)
| Target | Description/Functions | Reference (s) |
|---|---|---|
| papain | cysteine protease found in lysozomes | Chaiken and Smith, 1969 |
| carboxylesterase | Serine hydrolase/enzyme that hydrolyzes carboxyl esters | Su et al., 1971; Chanda et al., 1997 |
| adenylyl cyclase | Enzyme that catalyzes the conversion of ATP to cyclic AMP/Important in the G protein signaling cascade | Huff et al., 1994; Song et al., 1997; Auman et al., 2000 |
| Neuropathy target esterase | Phospholipase enzyme important in phospholipid metabolism, neurite outgrowth and process elongation during neuronal differentiation | Lush et al., 1998 |
| acylpeptide hydrolase | Serine protease enzyme that catalyzes the removal of N- acylated amino acids from acetylated peptides role in the coordinated protein-degradation | Richards et al., 2000 |
| M2 muscarinic receptors | Second messenger coupled acetylcholine autoreceptor | Bomser and Casida 2001* |
| fatty acid amide hydrolase | Serine hydrolase enzyme that catabolizes a class of bioactive lipids called the fatty acid amides including endocannabinoids (e.g., anandamide) | Quistad et al., 2001 |
| cannabinoid CB1 receptors | G protein-coupled receptors for endocannabinoids/functions not fully understood, but may play roles in neurotransmitter release, synaptic plasticity, pleasure, appetite, memory/concentration, perception of time, pain tolerance, etc. | Quistad et al., 2002 |
| albumin | Most abundant transport protein in plasma, also regulates the colloidal osmotic pressure of blood | Peeples et al., 2005 |
| transferrin | Glycoprotein that binds and transports iron in the plasma | Grigoryan et al., 2009 |
| kinesin | Motor protein involved in anterograde axonal transport in neurons as well as other cellular functions such as mitosis, meiosis, etc. | Grigoryan et al., 2009 |
| ATP Synthase | Mitochondrial enzyme responsible for synthesizing ATP from ADP and inorganic phosphate | Grigoryan et al., 2009 |
| tubulin | Globular proteins that form microtubules (i.e., polymers of dimerized tubulin which serve structural roles in the cytoskeleton, support intracellular transport, mitosis, etc.) | Jiang et al., 2010 |
OP-phosphorylation of M2 receptors reported by Bomser and Casida 2001 (using chlorpyrifos oxon) was not observed in experiments where paraoxon or a biotinlabeled fluorophosphonate were evaluated (Proskocil et al., 2010).
3.1 Oxidative Stress and Neuroinflammation
It is relatively well accepted that OPs can elicit oxidative stress and DNA damage to cells, particularly at high concentrations (see Soltaninejad and Abdollahi, 2009). In addition, it has been demonstrated that the OP, chlorpyrifos, can evoke lipid peroxidation in the developing rat brain at concentrations that only cause mild signs of systemic toxicity (Slotkin, 2005). Chronic low-level exposure to the OP, dichlorvos in adult rats, has been shown to induce apoptotic neurodegeneration by raising mitochondrial Ca++ levels, impairing mitochondrial complexes I, III and IV activities, and increasing oxidative stress (Kaur et al., 2007). The OP-exposure regimen in this study was also associated with an increase in lipid peroxidation and decreases in the mitochondrial antioxidants glutathione and superoxide dismutase. Low-level repeated exposure to OPs (e.g., chlorpyrifos, acephate, respectively) has also been shown to induce inflammatory responses in cultured astrocytes (i.e., increases in IL-6, GFAP, and p-ERK1/2) (Mense et al., 2006) and to upregulate inflammatory cytokines in vivo (Singh and Jiang, 2003). Notably, inflammatory cytokines have been demonstrated to cause significant impairment in spatial memory (Wenk et al., 2003), although it is important to note that no clear functional link between inflammatory responses and OP-related neurobehavioral deficits has been established to date (reviewed, Rohlman et al., 2011).
3.2 Motor Proteins, the Neuronal Cytoskeleton, and Axonal Transport
The wide variety of long-term neurological and psychiatric symptoms that have been associated with OPs suggest that some basic or fundamental neuronal process was adversely affected during the exposure period. Several observations suggest that OPs might interfere with one such fundamental neuronal process, axonal transport (i.e., another potential source of OP toxicity that might be unrelated to direct effects on cholinesterase enzymes). Axonal transport is responsible for the movement of lipids, mitochondria, synaptic vesicles, mRNAs, enzymes, receptor proteins, growth factors, and other macromolecules to and from a neuron’s cell body through the cytoplasm of its axon (reviewed, Duncan and Goldstein, 2006). Moreover, impairments in axonal transport have been suggested to contribute to the pathology of a wide variety of neurological illnesses (e.g., amyotrophic lateral sclerosis, Alzheimer’s disease, Huntington’s disease, Pick’s disease, progressive supranuclear palsy, see Stokin and Goldstein, 2006 for review). It is noteworthy that many of these illnesses are characterized by deficits in attention, concentration; information processing and memory function (i.e., symptoms that have been identified as OP-related sequelae as well as Gulf War Illness).
One older published study (using a rat optic nerve preparation) indicated that OPs that produce delayed neurotoxicity at relatively high doses (e.g., phenylphosphonothioate esters and tri-o-cresyl phosphate) inhibit fast anterograde axonal transport (Reichart and Abou-Donia, 1980). Additional studies documented accumulations of tubulovesicular profiles within axons prior to degeneration (Abou-Donia and Lapadula, 1990), a pathology that is consistent with stagnation of membrane traffic (Chretien et al., 1981; Souyri et al., 1981). More recent work in our laboratories indicated that repeated exposures to OPs at doses that were not associated with acute signs of toxicity can lead to deficits in axonal transport. Specifically, both anterograde and retrograde transport of vesicles in the sciatic nerves (ex vivo) of rats was significantly reduced after a 14 day exposure period to the commonly used OP, chlorpyrifos (O,O-diethyl O-[3,5,6,-trichloro-2-pyridyl] phosphorothionate) (CPF), and these deficits persisted throughout a 14 day washout period (Terry et al., 2003). Later, time course studies indicated that a significant reduction in axonal transport occurred within 10 hours of a single 18mg/kg s.c. CPF exposure (Terry et al., 2007).
The effects of OPs on axonal transport noted above lead to the next logical objective of indentifying the neurobiological substrate of these effects. Possible candidates include motor proteins such as kinesin and dynein and/or components of the neuronal cytoskeleton (e.g., microtubules). Kinesin superfamily proteins (KIFs) and cytoplasmic dynein serve as motors that move along microtubules carrying the various cargoes described above (e.g., synaptic vesicles, receptor proteins, growth factors). Anterograde axonal transport (i.e., transport in the direction from the cell body to the synaptic terminal) is carried out by KIFs while retrograde transport (from synaptic terminals to the cell body) is primarily carried out by cytoplasmic dynein (reviewed, Hirokawa and Takemura, 2004). Microtubules serve a fundamental role as structural (cytoskeletal) components within neurons and they also function as substrates (or rails) along which motor proteins move. Microtubules are polymers of α- and β-tubulin dimers which have polarity and are organized in special (dense parallel) array within the cell with kinesins (in axons) generally moving towards the (+) end (i.e., toward the synapse), and dynein, generally moving towards the (−) end of the microtubule (toward the cell body). Accordingly, there are several sites where modifications by OPs could disrupt microtubule function such as the motor domains of kinesin and dynein, the polymerization of tubulin, etc., leading to deficits in axonal transport (see Fig 2).
Fig. 2.
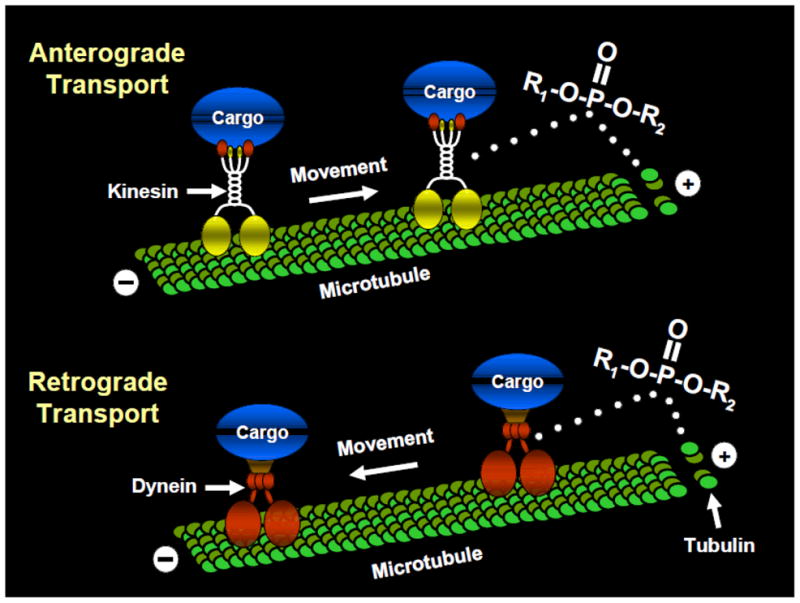
Theoretical targets of organophosphates on proteins involved in axonal transport. Anterograde axonal transport (i.e., transport in the direction from the cell body to the synaptic terminal) is carried out by the motor protein kinesin while retrograde transport (from synaptic terminals to the cell body) is primarily carried out by cytoplasmic dynein. Tubulin polymerizes to form microtubules which serve a fundamental role as structural (cytoskeletal) components within neurons and they also function as substrates (or rails) along which motor proteins move.
The hypothesis that OPs negatively affect kinesin-driven movement of important macromolecules is supported by recent studies in our laboratory. Specifically, using in vitro microtubule motility assays, we observed an increase in the number of locomoting microtubules that detached from kinesin-coated glass when kinesin was preincubated with the OPs chlorpyrifos, chlorpyrifos-oxon, or diisopropylfluorophosphate (Gearhart et al., 2007). These data suggested that OPs might covalently modify kinesin, thereby weakening kinesin-microtubule interactions that are necessary for anterograde axonal transport. This hypothesis has been strengthened by more recent studies where (using the biotin-tagged OP agent, FP-biotin) OP binding to tyrosine in the human kinesin 3C motor domain was demonstrated (Grigoryan et al., 2009).
OP-related effects on tubulin have also been documented. For example, Prendergast and colleagues demonstrated (utilizing a spectrophotomometric method) that chlorpyrifos-oxon inhibits the polymerization of tubulin, and (utilizing organotypic slice cultures of rodent brain and histological methods) caused a marked decrease in the concentration of microtubule associated protein-2. This latter effect was associated with a progressive decrease in neuronal viability in the hippocampus (Prendergast et al., 2007). Utilizing atomic force microscopy, Lockridge and colleagues confirmed that chlorpyrifos oxon disrupts tubulin polymerization and further (utilizing mass spectrometry), that chlorpyrifos oxon covalently binds to tubulin, an effect that is likely responsible for the disruptions in tubulin polymerization (Grigoryan and Lockridge, 2009; Jiang et al., 2010). At present we are unaware of any published studies which demonstrate OP-related alterations in dynein, but this would likely be another important area of investigation.
3.3. Neurotrophins
Evidence that OPs might affect the activity of growth factor-like (neurotrophic) molecules was initially presented over sixteen years ago. Specifically, in adult chickens, Pope et al., 1995 demonstrated that acute poisoning with DFP produced neuropathies and (using soluble extracts of cervical spinal cords from these animals on human neuroblastoma SY5Y cells in culture) a compensatory increase in trophic factors that mediated neuronal repair and neuritic outgrowth (Pope et al. 1995). Over the last several years, the effects of OPs on neurotrophins and their receptors have been investigated from several different perspectives. For example, exposure of neonatal rats to doses of chlorpyrifos and diazinon that were subthreshold for signs of acute toxicity (and including doses that did not inhibit cholinesterase) were associated with marked and regionally selective effects on the expression of specific members of the fibroblast growth factor (FGF) superfamily of neurotrophic factors (Slotkin et al., 2007). Specifically, both OPs markedly suppressed fgf20 expression in the forebrain and fgf2 in the brain stem, while elevating brain stem fgf4 and evoking a small deficit in brain stem fgf22. Early postnatal exposure to chlorpyrifos (at doses not associated with acute toxicity) has also been associated with decreases in nerve growth factor in the rat forebrain (Betancourt and Carr, 2004). Studies in our laboratories indicated that repeated exposures to chlorpyrifos or DFP (again at doses not associated with acute toxicity) can result in protracted alterations in nerve growth factor related signaling proteins. For example, in rats exposed to chlorpyriifos every other day for 30 days and a subsequent 14 day OP-free washout period, significant deficits in the high affinity nerve growth factor receptor TrkA and its activated form (phospho-TrkA) were detected in the prefrontal cortex (Terry et al., 2007). In a similar OP-exposure regimen and washout period, we detected DFP-related elevations in neurotrophins and neurotrophin receptors that have been associated with apoptosis and neuronal death (i.e., proNGF and p75NTR) in the hippocampus as well as deficits in TrkA protein in the basal forebrain and hippocampus (Terry et al., 2011a). Elevations in proNGF might indicate that enzymatic processes involved in the conversion of the proneurotrophin to its mature form might be inhibited by OPs. Likewise, decreases in the phospho-TrkA might indicate that kinases involved in the Trk autophosphorylation process might be adversely affected by OPs (see Fig 3).
Fig. 3.

Theoretical targets of organophosphates on proteins involved in the neurotrophin response. OP-related elevations in proNGF may indicate that enzymatic processes involved in the conversion of the proneurotrophin to its mature form might be inhibited by OPs (top center of figure). Likewise, decreases in the phospho-TrkA might indicate that kinases involved in the TrK autophosphorylation process might be adversity affected by OPs (bottom right). OPs have also been observed to elevate levels of the low affinity p75 neurotrophin receptor (top left) and to decrease levels of the high affinity TrkA receptor (top right).
3.4 Mitochondria
OP effects on mitochondria may be particularly important for studies of low level OP exposure given their fundamental importance to normal neuronal functions including aerobic metabolism, calcium homeostasis, and apoptotic processes (see Hollenbeck and Saxton, 2005 for review). Recent studies in our laboratories suggest that OPs may exert deleterious effects on mitochondria that are not necessarily related to oxidative stress (Middlemore-Risher et al., 2011). In cultured cortical neurons, we observed that exposure to chlorpyrifos and its oxidative metabolite chlorpyrifos oxon resulted in a concentration-dependent decrease in the transport of mitochondria in axons, an increase in mitochondrial length, and a decrease in mitochondrial number (indicative of increased fusion versus fission events). Moreover, these neuronal changes occurred at concentrations of the OPs that did not inhibit cholinesterase activity, they were not blocked by cholinergic receptor antagonists, and they did not appear to be associated with directly toxic effects on mitochondria (as would be suggested by diminished ATP production, alterations in mitochondrial membrane potential, or elevations in superoxide production). From these studies we hypothesized that an underlying mechanism of OP-based deficits in cognition and other neurological functions might involve alterations in mitochondrial dynamics (e.g., placement, morphology, function), and/or their transport in axons (see Fig 4).
Fig. 4.
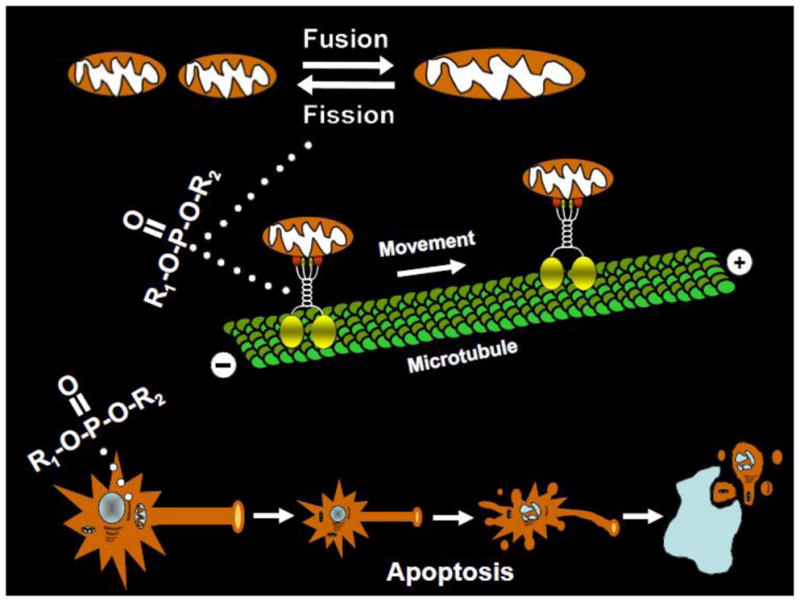
Theoretical targets of organophosphates on mitochondrial function. OPs have been observed in neuronal culture to increase mitochondrial length and to decrease mitochondrial number (indicative of increased fusion versus fission events, see top of figure). OPs have also been observed to decrease the transport of mitochondria in axons (middle of figure) as well as to increase oxidative stress and promote mitochondrial-dependent apoptotic processes (bottom of figure).
3.5 Non-cholinesterase Target Interactions
The OP-related effects on axonal transport, neurotrophin-related signaling proteins, and mitochondria described above leads to logical questions as to how these non-cholinesterase OP targets might interact to result in long term (deleterious) functional changes. Taken separately, OP interactions at any one of these sites could have profound effects on the long-term health of neurons. For example, alterations in axonal transport could lead to impaired trafficking of key macromolecules including receptor proteins, enzymes, growth factors, etc.; OP effects on mitochondria could lead to altered aerobic metabolism and calcium homeostasis; and OP effects on neurotrophin-related signaling proteins could lead to alterations in neuronal plasticity and survival. Interactions among two or more of these targets could (at least theoretically) have even more profound (deleterious) effects.
In the context of OP-related effects on axonal transport and mitochondrial function, a chicken-egg dichotomy appears to arise since each neuronal process requires the other (i.e., their functions are mutually dependent). Specifically, axonal transport is an ATP dependent process that requires intact motor proteins moving along cytoskeletal networks (e.g., polymerized tubulin). Without the appropriate placement of mitochondria, ATP availability and buffering of intracellular Ca++ is compromised resulting in the impairment of axonal transport as well as a variety of other neuronal processes (Chang and Reynolds, 2006). For proper placement, mitochondria, like other organelles and vesicles, require axonal transport (i.e., movement along microtubules via motors of the kinesin family and cytoplasmic dynein, see Hollenbeck and Saxton, 2005 for review).
The neurotrophin response also requires intact axonal transport. Following their release from target tissues neurotrophic factors (e.g., NGF, BDNF) bind to specific receptors (TrKA and TrkB, respectively) on the nerve terminal membrane and are internalized. Vesicles containing neurotrophin-receptor complexes are then transported along microtubules using the dynein motor protein towards the cell body (see reviews, Reynolds et al., 2000, Ginty and Segal, 2002) where their nuclear effects (e.g. CREB phosphorylation) are exerted to promote neuronal plasticity and cell survival. OP-related effects on motor proteins such as dynein would, therefore, theoretically impair this process.
Finally, there are also key interactions between neurotrophins and mitochondria in axons that may be relevant to OP-related effects. Nerve growth factor (NGF) has been observed to regulate the trafficking, localization, and metabolic activity of mitochondria in axons (Chada and Hollenbeck 2003; Verburg and Hollenbeck 2008). BDNF has been shown to increase mitochondrial respiratory coupling in neurons, an effect mediated by the MAP kinase pathway and enhancement of complex I activity (Markham et al., 2004). These effects may be particularly important to synaptic neurotransmission since synaptic mitochondria are much more sensitive to changes in complex 1 activity than non-synaptic or astrocytic mitochondria.
4. Study Limitations and Experimental Strategies
The various studies discussed above provide insight as to how OPs (via non-cholinesterase targets) might affect a number of neuronal processes that could result in long term functional deficits including cognitive impairment. However, there are a number of questions that remain to be answered. For example, a considerable amount of the work related to OP effects on non-cholinesterase targets has been conducted in vitro and as a result it is currently unclear if the reported effects would occur in a similar fashion in vivo. If they do occur in vivo, it would be important to determine if the deleterious effects occur at similar dose levels (i.e., if concentrations of OPs detected in the plasma and brain of treated animals mimic those associated with deleterious effects in vitro). To establish potential causal relationships, it would also be important to determine if cognitive impairments (or some other functional deficits in neuronal function) would be detected in the same animals where OP-related effects on specific non-cholinesterase targets were identified.
4.1 OPs-Effects on Axonal Transport
OP-related alterations in axonal transport have to date been demonstrated in vitro (in cultured neuronal cells) and ex vivo (in sciatic nerves obtained from rats previously exposed to OP). Thus, (as indicated in the paragraph above) it has not been fully established whether this effect occurs in vivo in the living mammalian brain. In preliminary studies conducted in our laboratories (published in abstract form, see Adam et al., 2011) we have demonstrated axonal transport deficits in the brains of living rats previously exposed to chlorpyrifos via manganese-enhanced magnetic resonance imaging (MEMRI) of the projections of the optic nerve to the superior colliculus. Future studies will be designed to establish time course and dose-effect relationships as well as whether these effects may be associated with multiple OPs.
4.2 OPs-Effects on Neurotrophins
OP effects on neurotrophin-related signaling proteins have been demonstrated both in neonatal rats (Slotkin et al., 2007) and adult rats, including those that showed deficits in memory-related task performance (Terry et al., 2007, Terry et al., 2011). However, there are a number of questions left to be answered in this context as well. For example, it is presently unclear if OP-related alterations in neurotrophin signaling proteins can be linked to neuropathological changes (e.g., neurite retraction, apoptotic changes) in regions of the brain known to support information processing (e.g., hippocampus, cortex). As in the case of the axonal transport effects described above additional studies are needed to establish OP time course and dose-effect relationships on neurotrophin function as well as whether these effects might be associated with multiple OPs. It is also currently unclear if the aforementioned OP-related effects on neurotrophins and their receptors are specific for these molecules versus more generalized effects on DNA/RNA synthesis, mRNA stability, cell viability, etc.
4.3 OPs-Effects on Mitochondria
The OP-related morphological changes in mitochondria described above are interesting; however, to date the mechanism of this effect has not been elucidated. OP-related effects on key fusion and/or fission proteins such as mfn2/opa1 or drp1 may be important and should be investigated. Interestingly, the fusion protein mfn2 has been shown to interact with Miro (an essential member of the complex that links mitochondria to kinesin motor proteins) to assist with bidirectional axonal transport of mitochondria (Russo et al., 2009). Importantly, Misko et al. (2010) determined that disruption of mfn2 can selectively alter mitochondrial transport/distribution (a suggested mechanism of peripheral axon degeneration in Charcot Marie Tooth disease, Cartoni and Martinou, 2009). An additional limitation to the aforementioned studies is the lack of a functional correlate to the morphological changes, (e.g., readout on mitochondrial function/activity). To strengthen the argument that OP effects on mitochondrial transport and/or morphology are indeed significant to mitochondrial function and more generally to neuronal function (at the low OP concentrations described), additional experiments are needed (e.g., studies measuring the impact of OPs on mitochondrial complex activity or rate of oxygen consumption as well as neurite outgrowth, calcium homeostasis, and/or apoptotic markers).
5. Potential Therapeutic Strategies
Given the wide variety of behavioral symptoms and neuropathological abnormalities that have been reported in individuals previously exposed to OPs, as well as the large list of potential OP targets, the design of rational therapeutic strategies is challenging. The text below (while speculative in some cases) is provided to highlight a few potential areas of interest and to stimulate discussion. The strategies discussed could potentially apply to the protracted neurological effects associated with either acute or repeated exposures to OPs.
5.1 Cholinergic Ligands
OPs have now been shown to affect multiple proteins and enzymes in addition to cholinesterase, as well as neurotransmitter receptors and signal transduction systems. Within the cholinergic system itself, OPs have been shown to either directly or indirectly affect both muscarinic and nicotinic receptors, choline acetyltransferase activity, high affinity choline uptake, the vesicular acetylcholine transporter, and different kinases involved in cholinergic signal-transduction (see review, Costa, 2006). These observations lead to the logical question of whether cholinergic ligands (e.g., muscarinic and nicotinic agonists, allosteric agents) and/or reversible cholinesterase inhibitors (e.g., donepezil, galantamine) might have therapeutic effects against OP-related sequelae. Reversible cholinesterase inhibitors are widely prescribed for the cognitive dysfunction function of associated with neurodegenerative illnesses (e.g., Alzheimer’s disease).
5.2 Protecting mitochondria from OP-induced oxidative stress
Protecting mitochondria from OP-induced oxidative stress and the associated apoptotic cell death and/or neurodegeneration has also been suggested as another potential therapeutic strategy against the deleterious effects of OPs. To support this hypothesis, the administration of the mitochondria-targeted antioxidant MitoQ attenuated dichlorvos-induced ROS production, increased MnSOD activity and glutathione levels as well as decreased lipid peroxidation, protein and DNA oxidation in rats. MitoQ also suppressed dichlorvos-related DNA fragmentation, cytochrome c release and caspase-3 activity and it attenuated mitochondrial swelling, loss of cristae and chromatin condensation (Wani et al., 2011).
5.3 Drugs that Increase Axonal Transport
Likewise, given the evidence that OP exposure leads to persistent deficits in axonal transport, approaches designed to increase axonal transport in vivo might be worth pursuing. To our knowledge, this strategy has not been rigorously investigated for any human disorder to date. Interestingly, there are reports in the literature of compounds that can increase the rate of axonal transport (e.g., the memory-enhancing agent, salbeluzole, Geerts et al., 1992). However, if OPs impair axonal transport by disrupting tubulin polymerization and/or affecting the function of kinesins/dyneins (as the studies described in section 3.2 suggest), it is currently unclear if salbeluzole (or similar compound) would be able to overcome the OP effect. An alternative strategy would be to design novel compounds that could stimulate the synthesis of dyneins and/or kinesins and/or increase their activity. While dyneins and kinesins have been discussed as potential targets for improving drug delivery for several years (see review, Hamm-Alvarez, 1996), to date, their role as potential targets for increasing axonal transport has not been explored. Emerging data on the regulation of kinesin motor activity by autoinhibitory mechanisms (e.g., cargo binding and/or phosphorylation mechanisms, see review, Verhey et al., 2009) may help to identify novel targets (e.g., kinases and Rab GTPases) for increasing axonal transport. Again, it is unclear if such strategies would be viable in the setting of OP-disrupted tubulin polymerization and/or covalent (OP) modifications of motor proteins.
5.4 Low Molecular Weight Growth Factor-Like Molecules
The aforementioned study by Speed et al., 2012 (see section 2.1), indicated persistent damage in the adult mammalian brain after subclincal exposures to OPs, however, their findings also suggested that the injury primarily involved synaptic spine loss as opposed to neuronal loss. This important observation would support the argument that the adverse changes are treatable (particularly with approaches designed to improve neural plasticity). Moreover, reports of deleterious effects of OPs on neurotrophins led us to speculate that the development of low molecular weight growth factor-like molecules might represent a valid approach to drug discovery for long-term OP-related sequelae. Methods to improve neuronal sensitivity to endogenous neurotrophins (via increasing the number of receptors or their phosphorylation) might also be viable. Notably, we have shown that certain low molecular weight compounds (e.g., nicotine, cysteamine) can enhance growth factor receptor levels (e.g., NGF and BDNF) in vivo in other contexts (see Hernandez and Terry, 2005; Kutiyanawalla et al., 2011).
5.5 Additional Strategies
Several additional (non-cholinergic) treatment approaches for OP-related neurological damage have also been discussed recently and include stereotaxic injection of neural stem or precursor cells into damaged brain regions, methods to enhance neurogenesis by cytokine treatments (see Collombet for review) as well as treatments with general antioxidants (e.g., vitamin E) and glutamate antagonists (e.g., memantine, see Zaja-Milatovic et al., 2009).
6. Conclusion
Substantial evidence now suggests that the canonical (cholinesterase-based) mechanism of OP toxicity cannot alone account for the wide-variety of adverse consequences of OP exposure that have been described, particularly the long-term neuropsychiatric symptoms. OP interactions with proteins involved in fundamental neuronal processes such as axonal transport, neurotrophin support, and mitochondrial function (both oxidation-related processes as well as those that affect their morphology and movement in axons) may explain some of the more protracted effects of OPs. There are, however, many questions that remain to be answered such as whether the variety of OP-related effects on non-cholinesterase targets described in vitro also occur in vivo and if potential causal relationships between OP-related effects on non-cholinesterase targets and cognitive impairments (or other functional deficits in neuronal function) can be more clearly established. Additional studies are also needed to establish time course and dose-effect relationships for individual OPs as well as to determine whether the deleterious effects described to date may be associated with multiple types of OPs. If further experimentation continues to support the significance of the aforementioned non-cholinesterase targets in the protracted effects of OPs, the design of therapeutic strategies that target these interactions (e.g., with compounds such as mitochondria-targeted antioxidants, low molecular weight growth factor-like molecules, drugs that increase axonal transport) may thus prove to be important for novel drug discovery and development.
Acknowledgments
The authors would like to thank Ms. Ashley Davis for her administrative assistance in preparing this article. This author’s laboratory is currently was supported by the National Institute of Environmental Health Sciences (ES012241), the National Institute on Aging (AG029617), and the National Institute on Drug Abuse (DA029127).
Abbreviations
- CPF
chlorpyrifos
- DFP
diisopropylfluorophosphate
- FGF
fibroblast growth factor
- GWI
Gulf War Illness
- NGF
nerve growth factor
- OP
organophosphate
Footnotes
Conflict of interest statement: “The author declares that there are no conflicts of interest.”
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abou-Donia MB, Lapadula DM. Mechanisms of organophosphorus ester-induced delayed neurotoxicity: Type I and Type II. Annu Rev Pharmacol Toxicol. 1990;30:405–440. doi: 10.1146/annurev.pa.30.040190.002201. [DOI] [PubMed] [Google Scholar]
- Adam BL, Beck WD, Middleton C, Joshi R, Yanasak N, Terry AV. Neuroscience Meeting Planner. Washington, DC: Society for Neuroscience; 2011. Manganese enhanced magnetic resonance imaging (MEMRI) for studying axonal transport deficits induced by organophosphates (OP) Abstract/Poster No. 787.16/DD25. Online. [Google Scholar]
- Amr MM, Halim ZS, Moussa SS. Psychiatric disorders among Egyptian pesticide applicators and formulators. Environ Res. 1997;73(1–2):193–9. doi: 10.1006/enrs.1997.3744. [DOI] [PubMed] [Google Scholar]
- Auman JT, Seidler FJ, Slotkin TA. Neonatal chlorpyrifos exposure targets multiple proteins governing the hepatic adenylyl cyclase signaling cascade: implications for neurotoxicity. Brain Res Dev Brain Res. 2000 May 11;121(1):19–27. doi: 10.1016/s0165-3806(00)00021-3. [DOI] [PubMed] [Google Scholar]
- Bardin PG, van Eeden SF, Moolman JA, Foden AP, Joubert JR. Organophosphate and carbamate poisoning. Arch Intern Med. 1994;154:1433–1441. [PubMed] [Google Scholar]
- Barnaby F. The nuclear arsenals and nuclear disarmament. Med Confl Surviv. 1998;14(4):314–20. doi: 10.1080/13623699808409411. [DOI] [PubMed] [Google Scholar]
- Betancourt AM, Carr RL. The effect of chlorpyrifos and chlorpyrifos-oxon on brain cholinesterase, muscarinic receptor binding, and neurotrophin levels in rats following early postnatal exposure. Toxicol Sci. 2004 Jan;77(1):63–71. doi: 10.1093/toxsci/kfh003. [DOI] [PubMed] [Google Scholar]
- Berardocco D. DoD, CIA release Khamisiyah modeling data. GulfNEWS. 1997;1:3. [Google Scholar]
- Bomser JA, Casida JE. Diethylphosphorylation of rat cardiac M2 muscarinic receptor by chlorpyrifos oxon in vitro. Toxicol Lett. 2001 Feb 3;119(1):21–6. doi: 10.1016/s0378-4274(00)00294-0. [DOI] [PubMed] [Google Scholar]
- Brown MA, Brix KA. Review of health consequences from high-, intermediate- and low-level exposure to organophosphorus nerve agents. J Appl Toxicol. 1998 Nov-Dec;18(6):393–408. doi: 10.1002/(sici)1099-1263(199811/12)18:6<393::aid-jat528>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Buckley NA, Roberts D, Eddleston M. Overcoming apathy in research on organophosphate poisoning. BMJ. 2004;329(7476):1231–1233. doi: 10.1136/bmj.329.7476.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burklow TR, Yu CE, Madsen JM. Industrial chemicals: terrorist weapons of opportunity. Pediatr Ann. 2003;32(4):230–234. doi: 10.3928/0090-4481-20030401-06. [DOI] [PubMed] [Google Scholar]
- Bushnell PJ, Padilla SS, Ward T, Pope CN, Olszyk VB. Behavioral and neurochemical changes in rats dosed repeatedly with diisopropylfluorophosphate. J Pharmacol Exp Ther. 1991;256:741–750. [PubMed] [Google Scholar]
- Calvert GM, Karnik J, Mehler L, Beckman J, Morrissey B, Sievert J, et al. Acute pesticide poisoning among agricultural workers in the United States, 1998–2005. Am J Ind Med. 2008 Dec;51(12):883–98. doi: 10.1002/ajim.20623. [DOI] [PubMed] [Google Scholar]
- Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol. 2009 Aug;218(2):268–73. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Casida JE, Quistad GB. Serine hydrolase targets of organophosphorus toxicants. Chem Biol Interact. 2005;157–158:277–83. doi: 10.1016/j.cbi.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Chaiken IM, Smith EL. Reaction of a specific tyrosine residue of papain with diisopropylfluorophosphate. J Biol Chem. 1969 Aug 10;244(15):4247–50. [PubMed] [Google Scholar]
- Chakraborty S, Mukherjee S, Roychoudhury S, Siddique S, Lahiri T, Ray MR. Chronic exposures to cholinesterase-inhibiting pesticides adversely affect respiratory health of agricultural workers in India. J Occup Health. 2009;51(6):488–97. doi: 10.1539/joh.l9070. [DOI] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J Exp Biol. 2003 Jun;206(Pt 12):1985–92. doi: 10.1242/jeb.00263. [DOI] [PubMed] [Google Scholar]
- Chanda SM, Mortensen SR, Moser VC, Padilla S. Tissue-specific effects of chlorpyrifos on carboxylesterase and cholinesterase activity in adult rats: an in vitro and in vivo comparison. Fundam Appl Toxicol. 1997 Aug;38(2):148–57. [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol. 2006 Dec;80(5):241–68. doi: 10.1016/j.pneurobio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Chretien M, Patey G, Souyri F, Droz B. Acrylamide-induced neuropathy and impairment of axonal transport of proteins. II abnormal accumulations of smooth endoplasmic reticulum as sites of focal retention of fast transported pro. Brain Research. 1981;205:15–28. doi: 10.1016/0006-8993(81)90716-2. [DOI] [PubMed] [Google Scholar]
- Collombet JM. Nerve agent intoxication: recent neuropathophysiological findings and subsequent impact on medical management prospects. Toxicol Appl Pharmacol. 2011 Sep 15;255(3):229–41. doi: 10.1016/j.taap.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Cooper J, Hans Dobson. The benefits of pesticides to mankind and the environment. Crop Protection. 2007;26:1337–1348. [Google Scholar]
- Costa LG. Current issues in organophosphate toxicology. Clin Chim Acta. 2006;366:1–13. doi: 10.1016/j.cca.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Cronin AK. Terrorist motivations for biological and chemical weapons use: placing the threat in context. Congressional Research Service, The Library of Congress; March 28.2003. [Google Scholar]
- Dassanayake T, Weerasinghe V, Dangahadeniya U, Kularatne K, Dawson A, Karalliedde L, et al. Cognitive processing of visual stimuli in patients with organophosphate insecticide poisoning. Neurology. 2007;68:2027–30. doi: 10.1212/01.wnl.0000264423.12123.f0. [DOI] [PubMed] [Google Scholar]
- De Silva HJ, Samarawickrema NA, Wickremasinghe AR. Toxicity due to organophosphorus compounds: what about chronic exposure? Trans R Soc Trop Med Hyg. 2006;100:803–6. doi: 10.1016/j.trstmh.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Dharmani C, Jaga K. Epidemiology of acute organophosphate poisoning in hospital emergency room patients. Rev Environ Health. 2005 Jul-Sep;20(3):215–32. doi: 10.1515/reveh.2005.20.3.215. [DOI] [PubMed] [Google Scholar]
- Duncan JE, Goldstein LS. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006 Sep 29;2(9):e124. doi: 10.1371/journal.pgen.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duysen EG, Li B, Xie W, Schopfer LM, Anderson RS, Broomfield CA, et al. Evidence for nonacetylcholinesterase targets of organophosphorus nerve agent: supersensitivity of acetylcholinesterase knockout mouse to VX lethality. J Pharmacol Exp Ther. 2001;299(2):528–35. [PubMed] [Google Scholar]
- Ecobichon DJ. Toxic effect of pesticides. In: AmdurMODoull J, Klaassen CD, editors. Cassarett and Doull’s Toxicology. 4. Pergamon; New York: 1991. pp. 565–622. [Google Scholar]
- Fricker RD, Reardon E, Spektor DM, et al. Volume 12: Pesticide Use During the Gulf War: A Survey of Gulf War Veterans. Santa Monica, CA: RAND; 2000. A Review of the Scientific Literature as It Pertains to Gulf War Illnesses. MR-1018/12-OSD. [Google Scholar]
- Gallo MA, Lawryk NJ. Organic phosphorus pesticides. In: Hayes ER Jr, Laws ER, editors. Handbook of Pesticide ToxicologyR Classes of Pesticides. Vol. 2. San Diego: Academic Press; 1991. pp. 917–1123. [Google Scholar]
- Gearhart DA, Sickles DW, Buccafusco JJ, Prendergast MA, Terry AV., Jr Chlorpyrifos, chlorpyrifos-oxon, and diisopropylfluorophosphate inhibit kinesin-dependent microtubule motility. Toxicol Appl Pharmacol. 2007;218:20–29. doi: 10.1016/j.taap.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Geerts H, Nuydens R, Nuyens R, Cornelissen F, De Brabander M, Pauwels P, et al. Sabeluzole, a memory-enhancing molecule, increases fast axonal transport in neuronal cell cultures. Exp Neurol. 1992 Jul;117(1):36–43. doi: 10.1016/0014-4886(92)90108-3. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol. 2002 Jun;12(3):268–74. doi: 10.1016/s0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- Golomb BA. Acetylcholinesterase inhibitors and Gulf War illnesses. Proc Natl Acad Sci U S A. 2008 Mar 18;105(11):4295–300. doi: 10.1073/pnas.0711986105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan H, Schopfer LM, Thompson CM, Terry AV, Masson P, Lockridge O. Mass spectrometry identifies covalent binding of soman, sarin, chlorpyrifos oxon, diisopropyl fluorophosphate, and FP-biotin to tyrosines on tubulin: a potential mechanism of long term toxicity by organophosphorus agents. Chem Biol Interact. 2008 Sep 25;175(1–3):180–6. doi: 10.1016/j.cbi.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan H, Schopfer LM, Peeples ES, Duysen EG, Grigoryan M, Thompson CM, et al. Mass spectrometry identifies multiple organophosphorylated sites on tubulin. Toxicol Appl Pharmacol. 2009 Oct 15;240(2):149–58. doi: 10.1016/j.taap.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan H, Li B, Xue W, Grigoryan M, Schopfer LM, Lockridge O. Mass spectral characterization of organophosphate-labeled lysine in peptides. Anal Biochem. 2009 Nov 1;394(1):92–100. doi: 10.1016/j.ab.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan H, Li B, Anderson EK, Xue W, Nachon F, Lockridge O, et al. Covalent binding of the organophosphorus agent FP-biotin to tyrosine in eight proteins that have no active site serine. Chem Biol Interact. 2009 Aug 14;180(3):492–8. doi: 10.1016/j.cbi.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan H, Lockridge O. Nanoimages show disruption of tubulin polymerization by chlorpyrifos oxon: implications for neurotoxicity. Toxicol Appl Pharmacol. 2009 Oct 15;240(2):143–8. doi: 10.1016/j.taap.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm-Alvarez SF. Microtubule-based motor proteins: new targets for enhancing drug delivery? Pharm Res. 1996 Apr;13(4):489–96. doi: 10.1023/a:1016025500402. [DOI] [PubMed] [Google Scholar]
- Hancock DB, Martin ER, Mayhew GM, Stajich JM, Jewett R, Stacy MA, et al. Pesticide exposure and risk of Parkinson’s disease: a family-based case-control study. BMC Neurol. 2008 Mar 28;8:6. doi: 10.1186/1471-2377-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden K, Norton M, Darcey D, Ostbye T, Zandi P, Breitner J, et al. Cache County Study, I. Occupational exposure to pesticides increases the risk of incident AD: the Cache County study. Neurology. 2010;74(19):1524–1530. doi: 10.1212/WNL.0b013e3181dd4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrami SA, Ibrahim N. Experiencing chemical warfare: two physicians tell their story of Halabja in Northern Iraq. Can J Rural Med. 2004 Summer;9(3):178–81. [PubMed] [Google Scholar]
- Hernandez CM, Terry AV., Jr Repeated nicotine exposure in rats: effects on memory function, cholinergic markers and nerve growth factor. Neuroscience. 2005;130(4):997–1012. doi: 10.1016/j.neuroscience.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2004;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005 Dec 1;118(Pt 23):5411–9. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff RA, Corcoran JJ, Anderson JK, Abou-Donia MB. Chlorpyrifos oxon binds directly to muscarinic receptors and inhibits cAMP accumulation in rat striatum. J Pharmacol Exp Ther. 1994;269:329–335. [PubMed] [Google Scholar]
- Hughart J. Industrial chemicals and terrorism: human health threat analysis, mitigation and prevention. Agency for Toxic Substances Disease Registry; 1999. [Google Scholar]
- Iversen A, Chalder T, Wessely S. Gulf War Illness: lessons from medically unexplained symptoms. Clin Psychol Rev. 2007;27(7):842–54. doi: 10.1016/j.cpr.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Jiang W, Duysen EG, Hansen H, Shlyakhtenko L, Schopfer LM, Lockridge O. Mice treated with chlorpyrifos or chlorpyrifos oxon have organophosphorylated tubulin in the brain and disrupted microtubule structures, suggesting a role for tubulin in neurotoxicity associated with exposure to organophosphorus agents. Toxicol Sci. 2010 May;115(1):183–93. doi: 10.1093/toxsci/kfq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz KD, Brooks DE. Emergency Medicine. Medscape from WebMD, Medscape, LLC; New Yorl, New York: 2010. Mar 16, [accessed 2011 Sept 1]. Organophosphate Toxicity. Available: http://emedicine.medscape.com/article/167726. [Google Scholar]
- Kaur P, Radotra B, Minz RW, Gill KD. Impaired mitochondrial energy metabolism and neuronal apoptotic cell death after chronic dichlorvos (OP) exposure in rat brain. Neurotoxicology. 2007 Nov;28(6):1208–19. doi: 10.1016/j.neuro.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Kutiyanawalla A, Promsote W, Terry AV, Jr, Pillai A. Cysteamine treatment ameliorates alterations in GAD67 expression and spatial memory in heterozygous reeler mice. Int J Neuropsychopharmacol. 2011 Oct;6(10):e26153. doi: 10.1017/S1461145711001180. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange G, Tiersky LA, Scharer JB, Policastro T, Fiedler N, Morgan TE, et al. Cognitive functioning in Gulf War Illness. J Clin Exp Neuropsychol. 2001 Apr;23(2):240–9. doi: 10.1076/jcen.23.2.240.1208. [DOI] [PubMed] [Google Scholar]
- Levin ED, Addy N, Nakajima A, Christopher ND, Seidler FJ, Slotkin TA. Persistent behavioral consequences of neonatal chlorpyrifos exposure in rats. Dev Brain Res. 2001;130:83–89. doi: 10.1016/s0165-3806(01)00215-2. [DOI] [PubMed] [Google Scholar]
- Levin ED, Timofeeva OA, Yang L, Petro A, Ryde IT, Wrench N, et al. Early postnatal parathion exposure in rats causes sex-selective cognitive impairment and neurotransmitter defects which emerge in aging. Behav Brain Res 2010. 2010 Apr 2;208(2):319–27. doi: 10.1016/j.bbr.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopachin RM, Decaprio AP. Protein adduct formation as a molecular mechanism in neurotoxicity. Toxicol Sci. 2005;86(2):214–25. doi: 10.1093/toxsci/kfi197. [DOI] [PubMed] [Google Scholar]
- Lotti M, Moretto A. Organophosphate-induced delayed polyneuropathy. Toxicol Rev. 2005;24:37–49. doi: 10.2165/00139709-200524010-00003. [DOI] [PubMed] [Google Scholar]
- Lush MJ, Li Y, Read DJ, Willis AC, Glynn P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem J. 1998 May 15;332(Pt 1):1–4. doi: 10.1042/bj3320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macilwain C. Study proves Iraq used nerve gas. Nature. 1993;363:3. doi: 10.1038/363003b0. [DOI] [PubMed] [Google Scholar]
- Markham A, Cameron I, Franklin P, Spedding M. BDNF increases rat brain mitochondrial respiratory coupling at complex I, but not complex II. Eur J Neurosci. 2004 Sep;20(5):1189–96. doi: 10.1111/j.1460-9568.2004.03578.x. [DOI] [PubMed] [Google Scholar]
- Mense SM, Sengupta A, Lan C, Zhou M, Bentsman G, Volsky DJ, et al. The common insecticides cyfluthrin and chlorpyrifos alter the expression of a subset of genes with diverse functions in primary human astrocytes. Toxicol Sci. 2006 Sep;93(1):125–35. doi: 10.1093/toxsci/kfl046. [DOI] [PubMed] [Google Scholar]
- Middlemore-Risher ML, Buccafusco JJ, Terry AV., Jr Repeated exposures to low-level chlorpyrifos results in impairments in sustained attention and increased impulsivity in rats. Neurotoxicology and Teratology. 2010;32:415–24. doi: 10.1016/j.ntt.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlemore-Risher ML, Adam BL, Lambert NA, Terry AV., Jr Effects of chlorpyrifos and chlorpyrifos-oxon on the dynamics and movement of mitochondria in rat cortical neurons. J Pharmacol Exp Ther. 2011 Nov;339(2):341–9. doi: 10.1124/jpet.111.184762. Epub 2011 Jul 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010 Mar 24;30(12):4232–40. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao M, Takatori T, Matsuda Y, Nakajima M, Iwase H, Iwadate K. Definitive evidence for the acute sarin poisoning diagnosis in the Toyko subway. Toxicol Appl Pharmacol. 1997;144 (1):198–203. doi: 10.1006/taap.1997.8110. [DOI] [PubMed] [Google Scholar]
- O’Leary CA. The Kurds of Iraq: recent history, future prospects. Middle East Rev Int Affairs (MERIA) J. 2002;6(4) [Google Scholar]
- [accessed October 17, 2011.];Organization for the Prohibition of Chemical Weapons (OPCW) website. www.opcw.org.
- Peeples ES, Schopfer LM, Duysen EG, Spaulding R, Voelker T, Thompson CM, et al. Albumin, a new biomarker of organophosphorus toxicant exposure, identified by mass spectrometry. Toxicol Sci. 2005 Feb;83(2):303–12. doi: 10.1093/toxsci/kfi023. [DOI] [PubMed] [Google Scholar]
- Pope C, di Lorenzo K, Ehrich M. Possible involvement of a neurotrophic factor during the early stages of organophosphate-induced delayed neurotoxicity. Toxicol Lett. 1995 Jan;75(1–3):111–7. doi: 10.1016/0378-4274(94)03167-6. [DOI] [PubMed] [Google Scholar]
- Pope CN. Organophosphorus pesticides: do they all have the same mechanism of toxicity? J Toxicol Environ Health B Crit Rev. 1999;2:161–181. doi: 10.1080/109374099281205. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Self RL, Smith KJ, Ghayoumi L, Mullins MM, Butler TR, et al. Microtubule-associated targets in chlorpyrifos oxon hippocampal neurotoxicity. Neuroscience. 2007;146(1):330–9. doi: 10.1016/j.neuroscience.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistad GB, Sparks SE, Casida JE. Fatty acid amide hydrolase inhibition by neurotoxic organophosphorus pesticides. Toxicol Appl Pharmacol. 2001 May 15;173(1):48–55. doi: 10.1006/taap.2001.9175. [DOI] [PubMed] [Google Scholar]
- Quistad GB, Nomura DK, Sparks SE, Segall Y, Casida JE. Cannabinoid CB1 receptor as a target for chlorpyrifos oxon and other organophosphorus pesticides. Toxicol Lett. 2002 Sep 5;135(1–2):89–93. doi: 10.1016/s0378-4274(02)00251-5. [DOI] [PubMed] [Google Scholar]
- Ray DE, Richards PG. The potential for toxic effects of chronic, low-dose exposure to organophosphates. Toxicol Lett. 2001;120(1–3):343–51. doi: 10.1016/s0378-4274(01)00266-1. [DOI] [PubMed] [Google Scholar]
- Reichart BL, Abou-Donia MB. Inhibition of fast axoplasmic transport by delayed neurotoxic organophosphorus esters: a possible mode of action. Mol Pharmacol. 1980;17:56–60. [PubMed] [Google Scholar]
- Research Advisory Committee on Gulf War Veterans’ Illnesses. Gulf War Illness and the Health of Gulf War Veterans: Scientific Findings and Recommendations. U.S. Department of Veterans Affairs; Nov 17, 2008. [Google Scholar]
- Reynolds AJ, Bartlett SE, Hendry IA. Molecular mechanisms regulating the retrograde axonal transport of neurotrophins. Brain Res Brain Res Rev. 2000 Sep;33(2–3):169–78. doi: 10.1016/s0165-0173(00)00028-x. [DOI] [PubMed] [Google Scholar]
- Richards PG, Johnson MK, Ray DE. Identification of acylpeptide hydrolase as a sensitive site for reaction with organophosphorus compounds and a potential target for cognitive enhancing drugs. Mol Pharmacol. 2000 Sep;58(3):577–83. doi: 10.1124/mol.58.3.577. [DOI] [PubMed] [Google Scholar]
- Rohlman DS, Anger WK, Lein PJ. Correlating neurobehavioral performance with biomarkers of organophosphorous pesticide exposure. Neurotoxicology. 2011 Mar;32(2):268–76. doi: 10.1016/j.neuro.2010.12.008. Epub 2010 Dec 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstock L, Keifer M, Daniell WE, McConnell R, Claypoole K. Chronic central nervous system effects of acute organophosphate pesticide intoxication. The Pesticide Health Effects Study Group. Lancet. 1991 Jul 27;338(8761):223–7. doi: 10.1016/0140-6736(91)90356-t. [DOI] [PubMed] [Google Scholar]
- Russo GJ, Louie K, Wellington A, Macleod GT, Hu F, Panchumarthi S, et al. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci. 2009 Apr 29;29(17):5443–55. doi: 10.1523/JNEUROSCI.5417-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi RM, Lara DR, Ghisolfi ES, Portela LV, Dias RD, Souza DO. Neuropsychiatric evaluation in subjects chronically exposed to organophosphate pesticides. Toxicol Sci. 2003;72:267–271. doi: 10.1093/toxsci/kfg034. [DOI] [PubMed] [Google Scholar]
- Savage EP, Keefe TJ, Mounce LM, Heaton RK, Lewis JA, Burcar PJ. Chronic neurological sequelae of acute organophosphate pesticide poisoning. Arch Environ Health. 1988 Jan-Feb;43(1):38–45. doi: 10.1080/00039896.1988.9934372. [DOI] [PubMed] [Google Scholar]
- Singh AK, Jiang Y. Lipopolysaccharide (LPS) induced activation of the immune system in control rats and rats chronically exposed to a low level of the organothiophosphate insecticide, acephate. Toxicol Ind Health. 2003 Jul;19(2–6):93–108. doi: 10.1191/0748233703th181oa. [DOI] [PubMed] [Google Scholar]
- Singh S, Sharma N. Neurological syndromes following organophosphate poisoning. Neurol India. 2000;48:308–313. [PubMed] [Google Scholar]
- Song X, Seidler FJ, Saleh JL, Zhang J, Padilla S, Slotkin TA. Cellular mechanisms for developmental toxicity of chlorpyrifos: targeting the adenylyl cyclase signaling cascade. Toxicol Appl Pharmacol. 1997;145:158–174. doi: 10.1006/taap.1997.8171. [DOI] [PubMed] [Google Scholar]
- Slotkin TA. Cholinergic systems in brain development and disruption by neurotoxicants: nicotine, environmental tobacco smoke, organophosphates. Toxicol Appl Pharmacol. 2004;198:132–151. doi: 10.1016/j.taap.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Slotkin TA. Developmental neurotoxicity of organophosphates: a case study of chlorpyrifos. In: Gupta RC, editor. Toxicity of organophosphate and carbamate pesticides. San Diego: Elsevier Academic Press; 2005. pp. 293–314. [Google Scholar]
- Slotkin TA, Seidler FJ, Fumagalli F. Exposure to organophosphates reduces the expression of neurotrophic factors in neonatal rat brain regions: similarities and differences in the effects of chlorpyrifos and diazinon on the fibroblast growth factor superfamily. Environ Health Perspect. 2007 Jun;115(6):909–16. doi: 10.1289/ehp.9901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA. Does early-life exposure to organophosphate insecticides lead to prediabetes and obesity? Reprod Toxicol. 2011 Apr;31(3):297–301. doi: 10.1016/j.reprotox.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltaninejad K, Abdollahi M. Current opinion on the science of organophosphate pesticides and toxic stress: a systematic review. Med Sci Monit. 2009 Mar;15(3):RA75–90. [PubMed] [Google Scholar]
- Souyri F, Chretien M, Droz B. Acrylamide-induced neuropathy and impairment of axonal transport of proteins. I Multifocal retention of fast transported proteins at the periphery of axons as revealed by light microscope. Brain Research. 1981;205:1–13. doi: 10.1016/0006-8993(81)90715-0. [DOI] [PubMed] [Google Scholar]
- Speed HE, Blaiss CA, Kim A, Haws ME, Melvin NR, Jennings M, et al. Delayed reduction of hippocampal synaptic transmission and spines following exposure to repeated subclinical doses of organophosphorus pesticide in adult mice. Toxicol Sci. 2012 Jan;125(1):196–208. doi: 10.1093/toxsci/kfr253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland K, Jenkins B, Ames RG, O’Malley M, Chrislip D, Russo J. Chronic neurological sequelae to organophosphate pesticide poisoning, Am. J Public Health. 1994;84:731–6. doi: 10.2105/ajph.84.5.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens R, Spurgeon A, Calvert IA, Beach J, Levy LS, Berry H, et al. Neuropsychological effects of long-term exposure to organophosphates in sheep dip. Lancet. 1995;345:1135–1139. doi: 10.1016/s0140-6736(95)90976-1. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Goldstein LS. Axonal transport and Alzheimer’s disease. Annu Rev Biochem. 2006;75:607–27. doi: 10.1146/annurev.biochem.75.103004.142637. [DOI] [PubMed] [Google Scholar]
- Su M, Kinoshita FK, Frawley JP, DuBois KP. Comparative inhibition of aliesterases and cholinesterase in rats fed eighteen organophosphorus insecticides. Toxicol Appl Pharmacol. 1971;20:241–249. doi: 10.1016/0041-008x(71)90050-0. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Morita H, Ono K, Maekawa K, Nagai R, Yazaki Y. Sarin Poisoning in Tokyo subway. Lancet. 1995;345(8955):980. [PubMed] [Google Scholar]
- Terry AV, Jr, Stone JD, Buccafusco JJ, Sickles DW, Prendergast MA. Repeated, subthreshold exposures to chlorpyrifos in rats: hippocampal damage, impaired axonal transport and deficits in spatial learning. J Pharmacol Exper Ther. 2003;305:375–384. doi: 10.1124/jpet.102.041897. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Gearhart DA, Beck WD, Truan JN, Middlemore ML, Williamson LN, et al. Chronic, Intermittent Exposure to Chlorpyrifos in Rats: Protracted Effects on Axonal Transport, Neurotrophin Receptors, Cholinergic Markers, and Information Processing. J Pharmacol Exper Ther. 2007;322:1117–1128. doi: 10.1124/jpet.107.125625. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Buccafusco JJ, Gearhart DA, Beck WD, Middlemore-Risher ML, Truan JN, et al. Repeated, intermittent exposures to diisopropylfluorophosphate in rats: protracted effects on cholinergic markers, nerve growth factor-related proteins, and cognitive function. Neuroscience. 2011;10;176:237–53. doi: 10.1016/j.neuroscience.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry AV, Jr, Beck WD, Warner S, Vandenhuerk L, Callahan PM. Chronic impairments in spatial learning and memory in rats previously exposed to chlorpyrfos or diisopropylfluorophosphate. Neurotoxicol Teratol. Neurotoxicol Teratol. 2012 Jan;34(1):1–8. doi: 10.1016/j.ntt.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeeva OA, Roegge CS, Seidler FJ, Slotkin TA, Levin ED. Persistent cognitive alterations in rats after early postnatal exposure to low doses of the organophosphate pesticide diazinon. Neurotoxicol Teratol. 2008;30:38–45. doi: 10.1016/j.ntt.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker Jonathan B. Pantheon Books. 2006. War of Nerves: Chemical Warfare from World War I to Al-qaeda; p. 479. [Google Scholar]
- US Department of Defense. Environmental Exposure Report. Pesticides Final Report. 2003 Available at http://www.gao.gov/htext/d04159.html.
- U.S. Environmental Protection Agency. Pesticides Industry Sales and Usage: 2006 and 2007 Market Estimates. Feb, 2011. [Google Scholar]
- Verburg J, Hollenbeck PJ. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci. 2008 Aug 13;28(33):8306–15. doi: 10.1523/JNEUROSCI.2614-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Gudup S, Sunkaria A, Bal A, Singh PP, Kandimalla RJ, et al. Protective efficacy of mitochondrial targeted antioxidant MitoQ against dichlorvos induced oxidative stress and cell death in rat brain. Neuropharmacology. 2011 Dec;61(8):1193–201. doi: 10.1016/j.neuropharm.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Wenk GL, McGann K, Hauss-Wegrzyniak B, Rosi S. The toxicity of tumor necrosis factor-alpha upon cholinergic neurons within the nucleus basalis and the role of norepinephrine in the regulation of inflammation: implications for Alzheimer’s disease. Neuroscience. 2003;121(3):719–29. doi: 10.1016/s0306-4522(03)00545-1. [DOI] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Gupta RC, Aschner M, Milatovic D. Protection of DFP-induced oxidative damage and neurodegeneration by antioxidants and NMDA receptor antagonist. Toxicol Appl Pharmacol. 2009 Oct 15;240(2):124–31. doi: 10.1016/j.taap.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]