

Abstract
An advanced intermediate in a projected synthesis of pactamycin has been prepared. Early installation of the C1-dimethylurea functionality allows for its participation in a diastereoselective, chelation-controlled addition of organometal nucleophiles to the C5 prochiral ketone. Four of the molecule’s six stereocenters are set with a ketone functional handle provided for subsequent manipulation.
First isolated in 1961 from a fermentation broth of Streptomyces pactum var pactum, pactamycin (1) is a potent, universal inhibitor of translocation.1,2 Through use of its aniline and salicylate moieties, pactamycin mimics a dinucleotide, interacting with stem loops in the 16S RNA, garnering its antitumor, antimicrobial, antiviral, and antiprotozoal activity.3 The unique stereochemically and functionally dense structure was proposed in 19704 and revised after X-ray analysis in 1972.5
Pactamycin’s cyclopentane core features stereogenic carbons at each position with an additional sidechain stereocenter at C7. The molecule is heteroatom-rich and contains urea, salicylate, and aniline functionality about the densely-substituted core. The synthetic challenge posed by this complex target had only received limited attention in the literature with two synthetic studies,6 until the first total synthesis was realized in 2011 by Hanessian and coworkers.7 This Letter details progress on a pactamycin core synthesis, specifically highlighting issues associated with the diastereoselective construction of the C5 tertiary carbinol.
Our retrosynthesis is outlined in Scheme 1. We envisaged access to 1 through selective manipulation of alcohol and amine functinonality at C4 and C3, respectively, from stereochemically-complete core 2.
Scheme 1.

Retrosynthetic Analysis of Pactamycin
The C4 tertiary alcohol and C2/C3 diamine moiety could be installed through allylic and alkene functionalization of cyclopentene 3, which would be constructed employing ring-closing metathesis of diene 4. Prior to cyclopentene formation, we believed the C1, C5, and C7 stereocenters could be completed with high diastereoselectivity in the acyclic form. We proposed formation of the C5 stereocenter via functionalized vinyl addition to methyl ketone 5. The first stereocenter, C1, would be completed employing an enantioselective allylation followed by diastereoselective ketone reduction at C7 of a substrate derived from methyl acetoacetate.
Rather than advancing the unusual C1 dimethyl urea in a protected form, we were intrigued by possible directing or chelating effects that this Lewis base might exert during key bond constructions. To this end, we elected to install this C1 functionality in its final desired form early in the synthesis. Diazo transfer onto methyl acetoacetate with p-acetamidobenzenesulfonyl azide (p-ABSA)8 proceeded in excellent yield to give α-diazo methyl acetoacetate (7, Scheme 2). A Rh-catalyzed N–H insertion reaction adapted from the method reported by Janda9 was then used for installation of the dimethylurea (8). This pronucleophile was subjected to the allylation conditions developed by Ito10 to provide the C1 stereocenter ((±)-9). An unoptimized asymmetric variant of this reaction using the Ito conditions provided enantioenriched (R)-9 (er 92:8, Scheme 3). The allylation was followed by a diastereoselective L-Selectride® reduction which provided the desired diastereomer in 72% yield.10 Silyl protection of the C7 alcohol with TBSOTf proceeded in excellent yield to give methyl ester 10.
Scheme 2.

Diastereoselective Assembly of the C1 and C7 Stereocenters
Scheme 3.

Enantioselective Tsuji-Trost Allylation
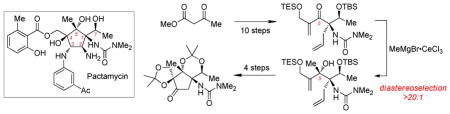
Conversion of the methyl ester 10 to a methyl ketone was unsuccessful under standard conditions (MeMgBr, MeLi, Cp2TiMe2) due to a lack of reactivity attributed to congestion at the adjacent C1 center; however, TMSCH2Li was shown to possess unique nucleophilicity, smoothly providing ketone 11 after a methanol quench (Scheme 4).11 With the desired C5 ketone in hand we tested a variety of nucleophilic additions to evaluate reaction efficiency, diastereoselectively, and influence of the C1 urea during formation of the C4–C5 bond.
Scheme 4.

2-Propenide Addition to a C5 Methyl Ketone
Standard nucleophilic additions to 11 were unsuccessful. Recovered starting material suggested an extreme degree of steric hindrance and/or competitive enolization of the methyl ketone. To overcome this impasse, cerium trichloride was deployed to both activate the ketone and render the organometal nucleophiles less basic.12,13 Isopropenyl Grignard was used as a model nucleophile and, when used with added CeCl3, provided tertiary carbinol 12 in 59% and 2.7:1 diastereoselectivity. Ring-closing metathesis with Grubbs’s second generation catalyst provided cyclopentene 13; nOesy analysis on this rigidified structure revealed the undesired syn relationship between the C5 hydroxyl and C1 urea.14
The conversion of 11 to 12 demonstrated that organometal additions to a hindered α-ureido ketone were feasible and potentially diastereoselective, dictated by the C1/C7 stereodiad. Since the C5 alcohol arises from two consecutive organometal additions to a carbonyl electrophile, it occurred to us that reversing the order of nucleophile additions (functionalized vinyl, then methyl) might deliver the desired stereochemical outcome. To this end, we set out to synthesize a functionalized C5 enone electrophile.
A reduction/oxidation sequence of methyl ester 10 with DIBAL-H and Dess-Martin periodinane proved most reliable in providing aldehyde 14 (Scheme 5). The aldehyde functionality at C5 permitted addition of lithiated 2-bromopropenol15 (15) in good yield to give diol 16.16 A selective triethylsilylation of the primary alcohol followed by Dess-Martin oxidation yielded α,β-enone 17.
Scheme 5.

Methide Addition to a C5 Functionalized Enone
Methyl Grignard addition in the presence of CeCl3 proceeded in excellent yield providing the 1,2-addition product as a single diastereomer (18). Ring-closing metathesis gave cyclopentene 19; nOesy analysis showed the desired stereochemistry at C5, confirming our hypothesis. Notably, this second-generation route allowed for the facile introduction of critical hydroxymethylene functionality at C4.
The stereochemical outcome of C5 ketone nucleophilic additions may be analyzed in the context of the following transition state proposal (20, Scheme 6). A five-membered chelate involving the urea, ketone, and metal (M = CeX2, MgX) could lock the electrophile in a conformation wherein the bulky, branched tert-butyldimethylsilyloxyethyl group shields addition from the Si face providing a modest preference in the methyl ketone case, and exclusive Re facial addition in the functionalized ketone system. This chelation effect has been invoked sporadically in additions to fully substituted cyclic α-amino ketones (to an α-oxo-azaspirocyle,17 and an α-amino cyclobutanone18). Two outcomes flow from this analysis: (1) the identity of nucleophile has little effect on the stereochemical outcome, and (2) the decision to incorporate the dimethylurea early is crucial in achieving the proper C5 stereochemistry via this synthetic route. The enhanced diastereocontrol with the more sterically demanding enone 17 might be consistent with “nonvertical” approach trajectories that accentuate the influence of chiral information in the electrophile.19
Scheme 6.

Proposed Transition State for C5 Nucleophilic
The C3–C4 alkene was expected to provide a useful handle for the elaboration of the remaining functionality in the pactamycin core. Efforts to install an allylic amine or otherwise oxidize the C2 methylene were unsuccessful, so our efforts turned to alkene elaboration. OsO4-catalyzed dihydroxylation of cyclopentene 19 provided syn-diol 21 in good yield as a single diastereomer (Scheme 7).20 Swern oxidation gave the α-hydroxy ketone 22 in 88% yield and avoided competitive oxidative cleavage that was observed with other oxidants (Dess–Martin periodinane, PCC). Diol 21, ketone 22, and their derivatives are expected to be useful intermediates for the introduction of the C2 amine and the C3 aniline.
Scheme 7.

Elaboration of the C3–C4 Alkene
An alternate protecting group scheme was also realized upon treating ketone 22 with 2,2-dimethoxypropane (2,2-DMP) and catalytic acid. Under these conditions, both silyl ethers were cleaved, and the C4 1,3-dioxolane and C1/C7 1,3-dioxane were selectively constructed, conveniently protecting the tetraol (23). Diacetonide 23 was highly crystalline and used for X-ray analysis (Figure 1). The crystal structure confirmed our nOesy-based stereochemical assignments and provided insight into the preferred conformation of functional groups, aiding in the planning of further manipulations.21
Figure 1.

X-ray structure of diacetonide 23.
In conclusion, we have in fifteen steps converted methyl acetoacetate to diacetonide 23, an advanced intermediate in a projected synthesis of pactamycin. Four of the six stereocenters, including the contiguous fully substituted C1/C5/C4 stereotriad, have been established with high levels of stereocontrol. The stereochemical issues associated with the creation of the C5 tertiary alcohol were studied and a stereochemical model for organometal additions to highly substituted α-NH-ketones was advanced. A C3 ketone arose from functionalization of the cyclopentene core and provides a functional handle for subsequent manipulation. Ongoing efforts are focused on α-amination of the C3 ketone and elaboration of the ketone to the C3 aniline functionality.
Supplementary Material
Acknowledgments
The project described was supported by Award R01 GM084927 from the National Institute of General Medical Sciences. J.T.M. acknowledges an ACS Division of Organic Chemistry graduate fellowship. X-ray crystallography was performed by Dr. Peter White (UNC). We thank Andrew Satterfield (UNC) for early exploratory studies.
Footnotes
Supporting Information Available: Experimental details and spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Argoudelis AD, Jahnke HK, Fox JA. Antimicrob Agents Chemother. 1962:191. [PubMed] [Google Scholar]
- 2.Dinos G, Wilson DN, Teraoka Y, Szaflarski W, Fucini P, Kalpaxis D, Nierhaus KH. Mol Cell. 2004;13:113. doi: 10.1016/s1097-2765(04)00002-4. [DOI] [PubMed] [Google Scholar]
- 3.Brodersen DE, Clemons WM, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Cell. 2000;103:1143. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wiley PF, Jahnke HK, MacKellar F, Kelly RB, Argoudelis AD. J Org Chem. 1970;35:1420. doi: 10.1021/jo00830a035. [DOI] [PubMed] [Google Scholar]; (b) Weller DD, Haber A, Rinehart KL, Jr, Wiley PF. J Antibiot. 1978;31:997. doi: 10.7164/antibiotics.31.997. [DOI] [PubMed] [Google Scholar]
- 5.Duchamp DJ. Abstracts, American Crystallographic Association Winter Meeting; Albuquerque, NM. 1972. p. 23. [Google Scholar]
- 6.(a) Knapp S, Yu Y. Org Lett. 2007;9:1359. doi: 10.1021/ol0702472. [DOI] [PubMed] [Google Scholar]; (b) Tsujimoto T, Nishikawa T, Urabe D, Isobe M. Synlett. 2005;433 [Google Scholar]
- 7.Hanessian S, Vakiti RR, Dorich S, Banerjee S, Lecomte F, Del Valle JR, Zhang J, Deschenes-Simard B. Angew Chem, Int Ed. 2011;50:3497. doi: 10.1002/anie.201008079. [DOI] [PubMed] [Google Scholar]
- 8.Davies HML, Cantrell WR, Romines KR, Baum JS. Org Synth. 1992;70:93. [Google Scholar]
- 9.Lee SH, Yoshida K, Matsushita H, Clapham B, Koch G, Zimmermann J, Janda KD. J Org Chem. 2004;69:8829. doi: 10.1021/jo048353u. [DOI] [PubMed] [Google Scholar]
- 10.Kuwano R, Ito Y. J Am Chem Soc. 1999;121:3236. [Google Scholar]
- 11.Mulzer J, Mantoulidis A, Ohler E. J Org Chem. 2000;65:7456. doi: 10.1021/jo0007480. [DOI] [PubMed] [Google Scholar]
- 12.For reviews on organocerium compounds see: Liu HJ, Shia KS, Shang X, Zhu BY. Tetrahedron. 1999;55:3803.Bartoli G, Marcantoni E, Marcolini M, Sambri L. Chem Rev. 2010;110:6104. doi: 10.1021/cr100084g.
- 13.For CeCl3 purification methods see: Dimitrov V, Kostova K, Genov M. Tetrahedron Lett. 1996;37:6787.Imamoto T, Kusumoto T, Tawarayama Y, Sugiura Y, Mita T, Hatanaka Y, Yokoyama M. J Org Chem. 1984;49:3904.
- 14.See Supporting Information for nOesy analysis.
- 15.Snyder SA, Corey EJ. J Am Chem Soc. 2006;128:740. doi: 10.1021/ja0576379. [DOI] [PubMed] [Google Scholar]
- 16.Hegde SG, Myles DC. Synth Commun. 1997;27:2111. [Google Scholar]
- 17.Hilmey DG, Gallucci JC, Paquette LA. Tetrahedron. 2005;61:11000. [Google Scholar]
- 18.Perez-Fernadez M, Avenoza A, Busto JH, Peregrina JM, Rodriguez F. Tetrahedron. 2008;64:9088. [Google Scholar]
- 19.(a) Heathcock CH, Flippin LA. J Am Chem Soc. 1983;105:1667. [Google Scholar]; (b) Lodge EP, Heathcock CH. J Am Chem Soc. 1987;109:2819. [Google Scholar]; (c) Heathcock CH. Aldrichim Acta. 1990;23:99. [Google Scholar]
- 20.Coldham I, Price KN, Rathmell RE. Org Biomol Chem. 2003;1:2111. doi: 10.1039/b303670g. [DOI] [PubMed] [Google Scholar]
- 21.CCDC 878935 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Centre via www.ccdc.cam.ac.uk/data_request/cif
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.