

Abstract
Introduction
Sapacitabine is an orally bioavailable nucleoside analog prodrug that is in clinical trials for hematologic malignancies and solid tumors. The active metabolite of sapacitabine, CNDAC (2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine), exhibits the unique mechanism of action of causing single-strand breaks (SSBs) after incorporation into DNA, which are converted into double-strand breaks (DSBs) when cells enter a second S-phase. CNDAC-induced DSBs are predominantly repaired through homologous recombination (HR). Cells deficient in HR components are greatly sensitized to CNDAC. Therefore, sapacitabine could be specifically effective against tumors that are deficient in this repair pathway.
Areas covered
This review summarizes results from supporting evidence for the mechanisms of action of sapacitabine, its preclinical activities and the current results of clinical trials in a variety of cancers. The novel action mechanism of sapacitabine is discussed, with a view to validate it as a chemotherapeutic drug targeting malignancies with defects in HR.
Expert opinion
Knowledge of CNDAC mechanism identifies tumors that may be sensitized to sapacitabine, thus enabling a personalized treatment strategy. It also creates the opportunity to overcome resistance to current front-line therapies and identify synergistic interactions with known anti-cancer drugs. The results of such investigations may provide rationales for the design of sapacitabine-based clinical trials.
Keywords: ATM, BRCA2, CNDAC, homologous recombination, leukemia and lymphoma, nucleotide excision repair, Rad51, sapacitabine, solid tumor, Xrcc3
1. Overview of sapacitabine and CNDAC
1.1 Nucleoside analogs
As a class of therapeutic agents, nucleoside analogs are more prevalent in the clinical treatment of cancer and viral diseases than other structurally similar groups of drugs. It is remarkable, however, that nucleosides with closely related structures vary so broadly with respect to cellular metabolic pathways and mechanisms of action (Table 1). Presumably because of the structural differences among analogs, however small, enzymes that govern DNA synthesis and metabolism exhibit different and largely unpredictable affinities for these analogs. Variation is also observed for the spectrum of activity in experimental chemotherapy screens of tumor-bearing mice. Most impressively, it is clear that nucleoside analogs with closely related structures, that share metabolic pathways, and inhibit similar target enzymes, still exhibit a diverse spectrum of anticancer activities in human tumor types in the clinic.
Table 1.
Diversity of mechanisms of action and clinical activities of nucleoside and nucleobase analogs.
| Predominant mechanism | Drug | Clinical activity |
|---|---|---|
| Inhibition of DNA replication | Cytarabine | AML |
| Nelarabine | T-ALL | |
| Inhibition of ribonucleotide reductase & DNA replication | Fludarabine | B-cell malignancies, AML |
| Cladribine | Hairy cell leukemia | |
| Gemcitabine | Solid tumor | |
| Clofarabine | AML, ALL | |
| Activation of DNA MMR to futile cycling | 6-Thioguanine | ALL, AML |
| 6-Mercaptopurine | ALL | |
| Block thymidylate synthase | 5-Fluorouracil | Solid tumors |
| Epigenetic modification | Azacitidine | MDS |
| Decitabine | MDS | |
| Mimic genetic syndromes | Pentostatin | Hairy cell leukemia |
| B-cell malignancies | ||
| RNA incorporation may have a role | Fludarabine | B-cell malignancies |
| Azacitidine | MDS | |
| DNA strand breaks | Sapacitabine/CNDAC | AML, MDS, etc. |
AML: Acute myeloid leukemia; CNDAC: 2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine; MDS: Myelodysplastic syndrome; MMR: mismatch repair; T-ALL: T-cell acute lymphocytic leukemia.
Nucleoside analogs differ greatly in the means by which they cause cell death after they are incorporated into DNA. Cytarabine, fludarabine, clofarabine, gemcitabine and nelarabine are relatively poor substrates for DNA strand extension, causing DNA replication forks to stall. Fludarabine, cladribine, clofarabine and gemcitabine also inhibit ribonucleotide reductase, an action that alters the concentration ratio of normal deoxytriphosphates to the analogs, increasing the likelihood for incorporation of the drug into DNA. Inhibition of thymidylate synthase by 5-fluorouracil nucleotide blocks the de novo pathway of dTTP production which inhibits DNA replication and repair. The nucleobases 6-thioguanine and 6-mercaptopurine are converted to deoxy-nucleotides and incorporated into DNA where they are recognized by the mismatch repair (MMR) sensors. This stimulates mismatch DNA repair to conduct futile cycles resulting in toxic levels of damaged DNA. Once they are incorporated into DNA, decitabine and azacitidine act through the epigenetic mechanism of hypomethylation and re-expression of repressed genes. Pentostatin mimics a form of severe combined immunodeficiency by inhibiting adenosine deaminase, which results in dATP accumulation and an imbalance of dNTP pool. Fludarabine, azacitidine and 5-fluorouracil may have RNA-directed mechanisms as well.
1.2 Structures and unique mechanism among nucleoside analogs
Matsuda et al. set out to design a nucleoside analog that would have a novel mechanism of action after incorporation into DNA. CNDAC (2′-C-cyano-2′-deoxy-1-β-D-arabino-pentofuranosylcytosine) was conceptualized as a mechanism-based DNA self-strand breaking nucleoside [1]. This analog is derivatized with a cyano group in the arabino configuration at the 2-carbon of the sugar moiety of the nucleoside. It was hypothesized that introduction of a cyano group at this position would act as a strong electron-withdrawing moiety to increase the acidity of the 2′-α-proton (Figure 2). It was predicted that phosphorylation of the 3′-hydroxyl group would alter the electronic configuration of the nucleoside, and that this structure would be extremely unstable. This is the action that would occur at the replication fork on addition of a deoxynucleotide to a 3′-terminal CNDAC nucleotide in DNA. Thus, it was envisioned that addition of a deoxynucleotide by a DNA polymerase to a CNDAC moiety in DNA would initiate such instability, causing a break in the DNA strand without inhibiting replication fork progression. Specifically, Matsuda et al. [1] postulated that polymerization beyond a terminal CNDAC nucleotide in DNA would lead to the cleavage of the 3′-phosphodiester linkage by a β-elimination process that would result in the rearrangement of the CNDAC molecule to form 2′-C-cyano-2′,3′-didehydro-2′,3′-dideoxycytidine (CNddC). This nucleoside is unique and therefore represents the signature of this DNA self-strand breaking process. In addition, as CNddC lacks a 3′-hydroxyl group, its presence at the 3′-terminus would function as a de facto DNA chain terminator that would enforce the formation of a single-strand DNA break which could not be repaired by ligation.
Figure 2.

Mechanism of DNA self-strand breaking by CNDAC nucleotide after incorporation.
Several lines of experimental evidence support this hypothesis. Chemical ligation of the 3′-hydroxyl group of CNDAC with N′,N′-carbonyldiimidazole resulted in the production of the signal product, CNddC [2]. Further, a chemically synthesized dinucleoside monophosphate of CNDAC-P-thymidine was shown to be very unstable under basic conditions [3]. Extension of the CNDAC nucleotide in DNA with a primer extension assay employing a bacterial DNA polymerase resulted in the formation of CNddC nucleotide at the 3′-terminus of the primer [4]. Subsequently, Hanaoka et al. [5] used high-pressure liquid chromatography and mass spectrometry to demonstrate the presence of CNddC in hydrolysates of DNA isolated from cells after CNDAC treatment, indicating that β-elimination occurs in intact cells. Finally, it was demonstrated that all detectable [3H]CNddC was at the 3′-terminus, providing proof of the self-strand breaking action of CNDAC nucleotide following incorporation into DNA [6]. Thus, the mechanism of action of CNDAC (Figure 2) is distinct from other clinically active nucleosides.
To achieve oral bioavailability, CNDAC was derivatized with a palmitoyl group at the N4 exocyclic amine; this was designated as CS-682 by Sankyo Co., Ltd., Tokyo, Japan, the original pharmaceutical sponsor [5,7]. The fatty acid side chain on the N4 group of the cytosine moiety improves oral bioavailability and reduces inactivation by deamination. Subsequently, after Cyclacel Pharmaceuticals, Berkeley Heights, NJ, USA, assumed clinical development of the compound in 2003, this was re-designated initially as CYC-682, and subsequently as sapacitabine (Box 1; Figure 1). Thus, all the names indicate the same chemical entity, but identify the respective sources of compound.
Box 1. Drug summary.
| Drug name | Sapacitabine |
|---|---|
| Phase | Phase III |
| Indication | cancer |
| Pharmacology description | DNA strand breaks |
| Route of administration | Oral |
| Chemical structure | N-(1-(2-Cyano-)2-deoxy-β-D-arabino-pentofuranosyl)-N4-palmitoylcytosine |
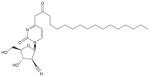 |
|
| Pivotal trial(s) | A Study of Oral Sapacitabine in Elderly Patients With Newly Diagnosed Acute Myeloid Leukemia (SEAMLESS) http://www.clinicaltrials.gov/ct2/show/NCT01303796?term=sapacitabine&rank=2 |
Pharmaprojects – copyright to Citeline Drug Intelligence (an Informa business). Readers are referred to Pipeline (http://informa-pipeline.citeline.com) and Citeline (http://informa.citeline.com).
Figure 1.

Structures of 2′-deoxycytidine, CNDAC and sapacitabine.
1.3 Preclinical studies
As is the case with other deoxycytidine analogs, for example, ara-C, gemcitabine, studies in cell lines demonstrated that CNDAC is phosphorylated to the monophosphate by deoxycytidine kinase [8], albeit with relatively poor efficiency compared with dCyd or the other analogs [9]. Cells lacking this enzyme were greatly resistant to the analog. Also, CNDAC is a substrate for deamination by cytidine deaminase, which generates the inactive uracil derivative CNDAU. The triphosphate accumulates in a concentration-dependent manner [10], and competes with dCTP for incorporation into DNA [6].
CNDAC was demonstrated to have potent antitumor activity in preclinical studies. The antiproliferative effects of CNDAC in terms of IC50 values were more potent than those observed with ara-C [1,11]. The analog showed broad-spectrum activity against tumor cell lines [5,12] and also in the P388 leukemia mouse model [2]. CNDAC was more effective than cytarabine in some human tumor cell lines derived from lung, stomach and osteosarcoma and showed excellent activity against tumor cell lines refractory to cytarabine [13]. However, the orally administered prodrug was more potent against human tumor xenografts than CNDAC or 5-fluorouracil [5]. It was also effective against various human organ tumor xenografts over a wider dose range and with fewer toxicities. CS-682 was also effective against P388 human leukemia cells resistant to a variety of other agents including mitomycin-C, vincristine, 5-fluorouracil and cisplatin in syngeneic mice [5]. Using high-resolution magnetic imaging, Wu et al. demonstrated that CS-682 delayed the growth of orthotopically implanted AX3488 liver tumors, and also delayed their meta-static behavior [14]. The metastatic behavior of an orthotopic model of pancreatic carcinoma was delayed [15], and overall survival of the mice was prolonged by CS-682 [16]. A liposomal formulation of CNDAC showed activity against Meth A sarcoma bearing mice when injected intravenously [17]. The antitumor activity of the liposomally encapsulated formulation was more potent than that of the parent drug CNDAC suggesting that the liposomal preparation enhanced therapeutic efficacy while at the same time reducing toxicity. Sapacitabine in combination with histone deacetylase inhibitors induced an increase in apoptosis and demonstrated significant benefit compared with the single-agent treatments both in vitro and in xenografts of the MV4-11 myeloid leukemia [18].
1.4 Clinical trials
The encouraging activities in preclinical models provided rationale for clinical trials of the bioavailable prodrug formulation. Two multicenter Phase I clinical trials of CS-682 in patients with advanced solid tumors have been reported (Table 2). Two schedules of oral administration were investigated, once daily for 5 days for 4 weeks [19] and once daily on days 1, 3 and 5 for 4 weeks [20]. In the former trial, the drug was investigated in 47 patients with 12 doses that ranged between 1.0 and 67 mg/m2/dose. The dose-limiting toxicity was neutropenia. No objective tumor responses were achieved although 11 patients experienced stable disease. The recommended Phase II dose was 40 mg/m2/dose. In the second trial, CS-682 was given three times per week for 4 consecutive weeks followed by a 2-week rest period [20]. Eleven doses that ranged from 1.5 to 120 mg/m2/day were investigated. Significant hematologic toxicities (mainly neutropenia) occurred at dose levels between 90 and 120 mg/m2/day. Six patients experienced stable disease. The recommended Phase II doses were schedule-dependent; 30 mg/m2/dose (daily times 5 days schedule) and 160 mg/m2/dose (every other day for 3 doses). Non-hematologic toxicities rarely exceeded grade 1 or 2 according to the NCI (National Cancer Institute) common toxicity criteria.
Table 2.
Phase I clinical trials of sapacitabine in solid tumors and hematologic malignancies.
| Disease | Patients | Schedule | Recommended Phase II dose | Dose-limiting toxicity | Ref. |
|---|---|---|---|---|---|
| Advanced solid tumors | 47 | Daily × 5 days for 4 weeks | 40 mg/m2/dose | Neutropenia | [18] |
| Advanced solid tumors | 40 | Days 1, 3 & 5, weekly for 4 weeks | 160 mg/m2/dose | Myelosuppression | [19] |
| Relapsed acute leukemia & MDS | 35 | Twice a day for 7 days Every 3 – 4 weeks | 325 mg/dose | Gastrointestinal | [22] |
| Relapsed acute leukemia & MDS | 12 | Twice daily for 3 days/week × 2 weeks | 425 mg/dose | Gastrointestinal | [22] |
Each of these trials was complemented by extensive pharmacokinetic investigations. These studies demonstrated the bio-availability of CS-682. Administered orally at the maximum tolerated dose of 40 mg/m2 on the daily times 5 days schedule, the peak plasma concentration of 4.1 ± 1.2 ng/ml (approximately 8 nM) was observed at 2.0 h [19]. The Cmax of CNDAC of 27 ± 14 ng/ml (approximately 93 nM) was achieved at 2.6 h. The inactive deamination product CNDAU reach maximum plasma concentrations of 74 ± 33 ng/ml (254 nM) at 2.9 ± 1.1 h and was eliminated with a terminal half-life of 2.1 h. When administered on the three times a week schedule at the maximal tolerated dose (MTD) of 160 mg/m2/dose, the peak CS-682 levels of 8.8 ± 3.5 ng/ml (18 nM) were reached at 2.8 ± 1.4 h, whereas the maximum CNDAC concentration was 62.5 ± 26.6 ng/ml (165 nM) after 2.7 ± 1.4 h [20]. The CNDAU peak was 310 ng/ml (1065 nM) at 3.3 ± 1.1 h. Thus, the metabolism and pharmacokinetic characteristics CNDAC in blood achieved by oral administration of CS-682 (sapacitabine) is similar to those of the clinically active cytosine nucleoside analogs, cytarabine and gemcitabine, although metabolic clearance by deamination occurred at a lesser rate.
A preliminary report of a Phase I study of sapacitabine in 22 patients with refractory solid tumors or lymphoma on a schedule of twice a day administration for 14 days every 21 days indicated a Phase II dose of 30 mg/m2/dose [21]. Using a second formulation and flat dosing, the recommended Phase II doses were 50 mg dose on the twice a day for 14 days schedule (n = 7 patients) and 75 mg/dose on a twice a day for 7 days schedule (n = 9 patients). Again, myelosuppression was dose limiting. Thirteen patients experienced stable disease.
A formulation of the parent nucleoside CNDAC, designated as TAS-109 by Taiho Pharmaceutical Co., Ltd., Tokyo, Japan, was evaluated in patients with refractory solid tumors on continuous infusion schedules of either 14 days followed by 7 days of rest, or over 7 days and then 7 rest days [22]. Fifteen patients were entered on the 14-day schedule at three dose rates (2, 3 and 4 mg/m2/day). Myelosuppression was the dose-limiting toxicity and the maximum tolerated dose was 2 mg/m2/day. Fourteen patients were treated at 3 and 4 mg/m2/day on the 7-day schedule. Again, myelosuppression was dose limiting; the MTD was 3 mg/m2/dose. Stable disease was observed in several patients on each trial. Pharmacokinetic studies demonstrated linear relationships between dose rate and both steady-state CNDAC concentrations and the AUC (area under the curve) of the drug on the 14-day schedule. Steady-state CNDAC plasma concentrations at MTD doses were 2.1 ng/ml (7 nM) and 3.26 ng/ml (11 nM) on the 14- and 7-day schedules, respectively.
Because of its unique mechanism of action, ease of administration, tolerability and its defined dose-limiting toxicity of neutropenia in solid tumors, sapacitabine was an interesting agent to investigate in leukemia. A Phase I trial of sapacitabine in 47 patients with relapsed/refractory acute leukemia and myelodysplastic syndrome (MDS) resistant to cytarabine therapy demonstrated clinical responses in this poor prognosis population [23] (Table 2). Using flat dosing, sapacitabine was escalated in six dose levels from 75 to 375 mg twice daily for 7 days (n = 35 patients) and from 375, 425 and 475 mg twice daily for 3 days on days 1, 2, 3, 8, 9 and 10 (n = 12 patients). The dose-limiting toxicities were gastrointestinal symptoms in both schedules. The MTDs were 375 mg twice daily for 7 days and 425 mg twice daily on the split schedule. The overall response rate and complete remission rate were 28 and 9%, respectively.
The activity of sapacitabine in MDS and acute myeloid leukemia (AML) is being defined further in ongoing Phase II clinical trials in patients over 70 years of age with previously untreated AML or after their first relapse, and in patients with MDS who are refractory to hypomethylating agents. The study design is a three-arm randomized trial of sapacitabine administered orally either (Arm A) at the flat dose of 200 mg twice a day for 7 days every 3 – 4 weeks, Arm B at a higher dose of 300 mg on the same schedule or Arm C at a flat dose of 400 mg administered twice daily for 3 days/week for 2 weeks, every 3 – 4 weeks [24,25]. The most current report on the AML study indicates that 20 patients have been entered on each arm. The overall response rates are 45, 25 and 35% for the respective schedules with complete remission rates of 10, 10 and 25%, respectively. The MDS trial has entered 61 patients with overall response rates of 24, 35 and 10%, for the respective arms. Two complete responses have been observed on Arm A [26]. These trials are continuing to maturity.
Trials of sapacitabine in combinations with established agents have recently been initiated. A schedule alternating decitabine (20 mg/m2) daily for 5 days and sapacitabine (300 mg) administered orally twice a day for 3 days/week for 2 weeks at 4-week intervals has been evaluated in 21 previously untreated AML patients over age 70 years (median 76 years) [27]. Three of the 16 patients with > 60 days of follow-up achieved complete remissions, 2 had partial remissions and 1 had hematological improvement.
These results demonstrate that the metabolic pathways observed in model systems are active in humans, and that several schedules of CS-682/sapacitabine administered orally generate plasma concentrations of the CNDAC that reduce clonogenicity in cell lines and primary AML cells in vitro [28]. Importantly, the initial clinical trials in hematologic malignancies have demonstrated responses in patients who have failed prior treatment with cytarabine or decitabine. Thus, cross-resistance among these drugs does not appear to be prevalent, providing rationale for combination strategies.
2. CNDAC: mechanism of action
After incorporation of CNDAC triphosphate into the DNA, the β-elimination process results in the formation of CNddC, a de facto DNA terminator at the 3′-end of a single-stranded nick. This lesion, which is novel among nucleoside analogs, initiates subsequent responses at both cellular and molecular levels.
2.1 Cell cycle effects
While many nucleoside analogs interfere with DNA replication causing an arrest of cell cycle progression at the S-phase, the unique action of CNDAC is associated with an arrest in the G2-phase in a wide range of cell lines [29]. Central to the DNA damage and repair responses are sensors, in particular, the phosphatidylinositol 3-kinase-related protein kinase family, which includes DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related protein (ATR). Multiple approaches have been used to define the role of DNA damage sensors including genetically paired cell lines, pharmacologic inhibitors and gene knockdown by siRNA. ATR and DNA-PK, but not ATM, have been shown to be responsible for the G2 checkpoint activation by CNDAC [30].
It has been demonstrated that CNDAC activates the G2 checkpoint through the canonical Chk1-Cdc25C-Cdk1/CyclinB1 signaling pathway. This G2 checkpoint can be abrogated by inhibitors of Chk1 kinase, such as UCN-01 [29], CHIR-124 and CHIR-600 [31]. Dysregulation of the G2 checkpoint permits cell cycle progression through mitosis and results in a transient arrest in the G1-phase before cells undergo apoptosis. However, clinically relevant concentrations of CNDAC are less than those needed to induce cell cycle arrest in model systems, although great enough to prevent minimal colony formation in cell lines and primary AML cells [28]. Thus, G2 arrest is a cellular response to CNDAC-induced DNA damage, but it does not necessarily provide survival advantage. These latter findings stimulated a search for alternative mechanisms of CNDAC-induced cytotoxicity.
2.2 DNA damage repair mechanisms
CNddC, the rearranged analog generated in β-elimination process after CNDAC incorporation, lacks a 3′-hydroxyl group. Therefore, it is not a substrate for repair by ligation, nor can it be extended without processing to remove the chain-terminating analog. This functionally poisons the repair process until CNddC is removed. CNDAC-induced single-strand breaks (SSBs)/nicks, generated in DNA replication (first S-phase), can be processed and converted into double-strand breaks (DSBs) when cells go through a second S-phase [28]. Distinguished from the two-ended DSBs caused by other genotoxic agents, such as γ-irradiation and topoisomerase II inhibitors, CNDAC-induced DSBs are by nature one-ended at the collapsed replication fork (Figure 3). It has been revealed that distinct repair mechanisms are involved in cellular responses to CNDAC-induced SSBs and DSBs. They cooperate in maintenance of genetic integrity after CNDAC treatment, and therefore could be potential drug resistance mechanisms.
Figure 3.

Conversion of CNDAC-induced single-strand break into double-strand break after a second DNA replication.
2.2.1 Nucleotide excision repair
Excision of CNddC prior to addition of dCMP would be required for sealing of the nick by ligation. Efforts have been focused on the possible participation of the excision repair mechanisms that are responsible for repair of other forms of DNA damage, namely base excision repair (BER), MMR and nucleotide excision repair (NER). Using paired cell lines proficient or deficient in proteins involved in these repair pathways, as well as pharmacological inhibitors, it was demonstrated that deficiency in either BER or MMR has no effect on clonogenic survival of cells treated with CNDAC [32]. By contrast, defect in the key NER endonuclease XPF, which forms a complex with ERCC1 and acts on the 5′ side of the lesion, renders cells four- to five-fold more sensitive to CNDAC. Furthermore, it has been shown that transcription-coupled, but not global-genome NER, is responsible for removal of CNddC at the 3′-end of the nick [32]. Thus, it is likely that this form of repair is initiated when a transcription complex encounters the CNddC-terminated nick in the DNA sugar-phosphate backbone.
2.2.2 Homologous recombination
If not repaired, CNDAC-induced SSBs will be transformed into more lethal DSBs during a subsequent round of DNA replication [28]. Mammalian cells have developed two major mechanisms for repairing DSBs, that is, non-homologous end-joining (NHEJ) and homologous recombination (HR). The NHEJ pathway is dependent on DNA-PK [33], where as HR is initiated by ATM [34,35]. Ionizing radiation-induced two-ended DSBs are repaired largely by the NHEJ mechanism. By contrast, CNDAC-induced one-ended DSBs are repaired mainly through HR, as demonstrated by clonogenic assays in HR proficient and defective cells, as well as biochemical and cytogenetic evidence [28]. Deficiency in ATM, Rad51D (a paralog of Rad51) or either of the two Rad51-interacting proteins, Xrcc3 and Brca2, sensitizes cells to CNDAC as much as 100-fold. Figure 4 summarizes cellular response to CNDAC actions and the roles of important proteins in the repair pathways discussed above. In contrast to the mechanism of G2 checkpoint activation, neither DNA-PK nor ATR is essential for clonogenic survival after CNDAC. However, ATM and the HR components (including Rad51 and its associated proteins, Xrcc3 and Brca2) are indispensible for survival. While the transcription-coupled nucleotide excision repair (TC-NER) pathway functions in concert with HR, it plays a less significant role, likely because the most challenging damage caused by CNDAC is DSBs. The NHEJ pathway does not make a substantial contribution to repairing CNDAC-induced DNA damage.
Figure 4.

A model for DNA repair proteins in response to sapacitabine/CNDAC-induced DNA damage.
While HR makes a critical contribution to the repair of CNDAC-induced DNA damage, other repair mechanisms may also participate. Recent studies have demonstrated the Fanconi anemia (FA) pathway proteins which have been known for their role in interstrand crosslink repair [36], also make key contributions to DSB repair as well as to aspects of cell cycle regulation and replication fork stability [37,38].
3. Tumor candidates for sapacitabine treatment
Because HR is the major pathway for repair of CNDAC-induced DSBs, defects in this pathway would be expected to result in significant sensitization to CNDAC. The combination of genetic deficiencies with the drug’s unique action mechanism would create synthetic lethal conditions in cancers. Therefore, tumors that are deficient in HR repair function could be good candidates for sapacitabine therapy. Four components in the HR pathway, namely ATM, Rad51, Xrcc3 and Brca2, have been shown to be critical for survival after CNDAC. Loss of or deficiency in any of these repair proteins leads to 20- to 100-fold sensitivity to CNDAC in vitro [28]. We will discuss various malignancies with known defects in HR and how sapacitabine-based chemotherapy may be personalized at the bedside.
3.1 ATM-deficient cancers
ATM kinase, one of the PIKK family members, plays a critical role in DNA damage repair and surveillance of genetic integrity. Loss of ATM function is linked with increased genetic instability and cancer susceptibility. About 10% of ataxia telangiectasia (AT) homozygotes develop cancer, mostly lymphoid malignancies [39]. In AT patients, B-cell non-Hodgkin’s lymphoma (B-NHL) is the most frequent B-cell malignancy, whereas the frequency of T-cell malignancy is estimated to be four- to fivefold greater than B-cell malignancy [40]. The ATM gene is mapped to 11q22.3 [41]. Loss of chromosome material in this region frequently occurs in a range of sporadic malignancies. Deletion of the long arm of chromosome 11 [del(11q22-23)] is a common chromosomal aberration observed in hematologic malignancies. Detection of del(11q22-23) in interphase cells by fluorescence in situ hybridization (FISH) (Figure 5) has become a routine test in hematopathology practice [42]. Tumors with del (11q22-23) can be further characterized either by DNA sequencing or ATM functionality assays in order to check if the second allele of ATM gene remains intact. Frequently, the residual allele is mutated, which results in complete loss of ATM function [43]. ATM non-functional malignancies are defective in HR, thereby becoming extremely sensitive to CNDAC. Thus, this subgroup of cancer patients may be selected for sapacitabine treatment.
Figure 5. Location of DNA repair genes on chromosome 11.

Arrow indicates the FISH probe used for detection of 11q deletion.
3.1.1 Chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL), the most common leukemia in the western hemisphere, is characterized by remarkable clinical heterogeneity. Del(11q22-23) is found in 10 – 20% of CLL patients, and has been identified as a marker for poor prognosis [44]. CLL with 11q deletion can be divided into two subgroups based on the integrity of the residual ATM allele: 64% with one intact ATM allele (mono-allelic ATM defect, ATM functional) and 36% with mutation (biallelic ATM defects, ATM non-functional). The latter CLL patients have defective responses to cytotoxic chemotherapeutics in vitro and a poorer clinical outcome [43]. Although remarkable progress has been made in the treatment of CLL during the last decade [45], relapses remain problematic and development of drug resistance is a major challenge in curing CLL. Emerging information suggests that the prevalence of del(11q-22-23) is increased in patients who fail to maintain their response to chemoimmunotherapy (Keating, personal communication, 2011). The investigations by Austen et al. [43] indicate that the mutation rate in the residual ATM allele may also increase following therapy. Thus, a substantial subgroup of these patients may lack ATM function, predicting that they would selectively benefit from sapacitabine therapy. Clinical investigations have recently been initiated to evaluate these possibilities (Wierda, Clinical Trials.gov identifier: NTC01253460).
3.1.2 Mantle cell lymphoma
Mantle cell lymphoma (MCL) is a rare type of B-cell lymphoma (6%), which is characterized by the chromosomal translocation t(11;14)(q13;q32) and consequent over-expression of cyclin D1 [46,47]. Importantly, del(11q22-23) is of high frequency in MCL, detected in 46% (37 out of 81) of cases [48,49]. More recent investigations [50,51] showed over 50% of MCLs with del(11q22-23) carry mutations in the second ATM allele, which inactivates the PI-3 kinase domain or leads to truncated ATM protein. In addition, biallelic ATM mutations were identified in MCLs without 11q deletions [51]. This subtype of MCLs with somatic biallelic ATM mutations may benefit from sapacitabine therapy.
3.1.3 T-cell prolymphocytic leukemia
T-cell prolymphocytic leukemia (T-PLL) is a rare aggressive mature T-cell leukemia, similar to the mature T-cell leukemia found in AT patients, in which ATM inactivation is primarily by small deletions or insertions. Complete or partial deletion of chromosome 11q is very common in sporadic T-PLL. In fact, both ATM alleles can be disrupted by deletion and/or point mutation in T-PLL cases identified in non-AT individuals [52]. T-PLL was the first sporadic lymphoid tumor in which ATM inactivation was demonstrated [47]. The loss of function mutations in the remaining ATM allele are mainly missense mutations in T-PLL, different from the mutation pattern in AT [39]. Vorechovsky et al. reported about 50% sporadic T-PLLs (17 out of 32 cases) have biallelic ATM mutations [53]. It is not clear whether constitutional AT heterozygosity is associated with predisposition or susceptibility to T-PLL [39]. Similar to ATM-deficient CLL and MCL, the T-PLL cohort with ATM inactivation could respond favorably to sapacitabine.
3.1.4 T-cell acute lymphocytic leukemia
AT patients frequently develop T-cell acute lymphocytic leukemia (T-ALL), but there is no evidence for somatic ATM mutation in sporadic T-ALL [54]. Bradshaw et al. examined 13 T-ALL samples and found one sample harboring a deletion insertion with the RGYW motif at the breakpoint in ATM. This is the first known deleterious mutation detected in ATM in T-ALL [55].
3.1.5 Non-small cell lung cancer
About 85 – 90% of lung cancers are non-small cell lung cancer (NSCLC). Yang et al. [56] provided the first epidemiologic evidence that ATM sequence variants associate with susceptibility to NSCLC. Further studies are warranted to define how the risk-conferring variants might act through downregulating the functions of ATM.
3.1.6 Head and neck cancer
Ai et al. reported that ATM promoter is hypermethylated in 25% (total 100 cases) of squamous cell carcinoma of the head and neck, which accounts for 80 – 90% of head and neck tumors [57]. Hypermethylation of the ATM promoter is significantly correlated with poor prognosis [57]. Lee et al. showed that lower ATM mRNA expression is correlated with poor outcome of laryngeal and pharyngeal cancer patients [58]. Additional investigations are needed to determine the functionality of ATM in these tumors.
3.2 BRCA-deficient cancers
Two breast cancer susceptibility genes have been identified: the BRCA1 gene is located on chromosome 17p12-21 and BRCA2 on 13q12.3. Brca1 and Brca2 proteins have multiple biological functions, especially participation in a pathway (so-called ‘BRCA pathway’) [59] mediating repair of DNA DSBs. Deleterious mutations in BRCA1/2 genes have been detected in solid tumors (including breast, ovarian, prostate and pancreatic cancer), as well as hematologic malignancies. Potential therapeutic benefit with sapacitabine is discussed below.
3.2.1 Breast cancer
Breast cancer is a particular threat for women all over the world. The incidence in American women is about 10%, resulting in more than 40,000 deaths every year. About 5–10% breast cancer cases are hereditary, among which 30–50% are caused by mutations in BRCA1 and BRCA2. Familial breast cancer is inherited in a dominant autosomic manner [60]. Breast tumors from BRCA1 mutation carriers are predominantly of basal-like subtype, that is, triple-negative (negative for estrogen receptor, progesterone receptor and HER2) [61]. Triple-negative breast cancer (TNBC) is more prevalent in premenopausal African-American women; occurs at an earlier age than other types of breast cancer. BRCA1 gene could be down-regulated in basal-like breast cancer via epigenetic silencing (promoter methylation) or other mechanisms [62]. By contrast, tumors from BRCA2 mutation carriers are mostly of luminal subtype and have a high histological grade [61]. Expression of Brca2, which is cell cycle dependent, is high in the thymus and testis and relatively high in the mammary gland and ovary [63]. Male BRCA2 mutation carriers have significantly increased risk for breast cancer, while cancer risk in male BRCA1 mutation carrier is not as profound. Brca1 has an integral function in HR [64], although its specific role in repair of CNDAC-induced DNA damage remains to be defined. Thus, it is likely that sapacitabine will benefit familial breast cancer patients, female or male, with BRCA1 or BRCA2 mutations.
3.2.2 Ovarian cancer
Ovarian cancer is the sixth most common cancer in women and the second most common gynecologic malignancy across the world, with a death toll of 14,500 each year. Similar to breast cancer, about 7% of ovarian cancer cases are hereditary due to mutations in BRCA1 and BRCA2 genes. Women with BRCA1 mutations have a higher risk of ovarian cancer (60% by age 70) than those with BRCA2 mutations [63]. Recent genomic analyses of 489 cases of advanced-stage, high-grade serous ovarian carcinoma identified that 20% samples had either germline or somatic mutations in BRCA1/2, and that additional 11% lost BRCA1 expression through DNA hypermethylation [65]. While the implication for sapacitabine in the treatment of BRCA1-mutated ovarian cancer needs to be determined, it is reasonable to predict the advantage of sapacitabine therapy in ovarian cancers with BRCA2 mutations.
3.2.3 Prostate and pancreatic cancer
In addition to breast cancer, male BRCA1/2 mutation carriers have an increased risk for prostate and pancreatic cancer [66]. Prostate cancer in male BRCA mutation carriers presents a more aggressive phenotype than the matched control [67]. It is possible to expand the anticancer spectrum of sapacitabine to male prostate and pancreatic cancer harboring BRCA mutations.
3.2.4 Hematologic malignancies
In addition to solid tumors, deficiencies in Brca1 and Brca2 are also indicated in hematologic cancers [59]. Reduced expression of Brca1 due to promoter hypermethylation was reported to be frequent in AML with cytogenetic abnormalities and in therapy-related AML [68]. Therefore, sapacitabine might be useful for leukemia and lymphoma subtypes with deleterious BRCA mutations, in addition to those with ATM inactivation.
3.2.5 Non-small cell lung cancer
NSCLC is the most prevalent malignancy worldwide. It has been reported that expression level of BRCA1 mRNA is reduced in a subgroup of NSCLC patients [69]. Recent study using immunohistochemistry-based approach has verified that 11 – 19% of primary NSCLC specimens from two independent cohorts (total 302 patients) are deficient in Brca1 [70] although the genetic/epigenetic cause has not been determined. The distribution of Brca1 deficiency was more prevalent in non-squamous NSCLC (34/130; 26.2%) than in the squamous subtype (15/172; 8.7%). These Brca1-immunodeficient NSCLC patients could be considered for sapacitabine therapy.
3.3 Rad51-downregulated cancers
Rad51 is a key recombinase in HR repair of DSBs that interacts with both BRCA1 and BRCA2. A single-nucleotide polymorphism (SNP) in the 5′ untranslated region (UTR) of RAD51 gene, 135G→C, affects RAD51 splicing within the 5′ UTR, thereby altering RAD51 expression [71]. This SNP has been identified as a modifier of breast cancer risk in BRCA2 mutation carriers. Two independent studies suggest that 135G→C variant is clinically correlated with increased risk of breast cancer in BRCA2 mutation carriers, specifically in raising breast cancer risk at younger ages [71,72]. In hypoxic cancer cells, Rad51 level is downregulated via transcriptional repression, as determined by analyses of RAD51 gene promoter activity and stabilities of mRNA and protein. This is not associated with the cell cycle profile or with expression of hypoxia-inducible factor. HR capacity is suppressed as a consequence of the hypoxia-mediated decrease in Rad51 expression [73]. More recently, increased expression of EZH2, a polycomb protein, has been linked to Rad51 down-regulation in hypoxic breast tumor initiating cells [74]. Rad51 mutated mouse is embryonic lethal [75]. So far, there are no human cancers identified with Rad51 mutations. Recent integrated genomic analyses revealed hypermethylation of RAD51C gene in 3% of high-grade ovarian carcinoma samples [65]. Since cells lacking Rad51D are 50-fold more sensitive to CNDAC in clonogenic survival assays [28], we speculate that CNDAC/sapacitabine will have therapeutic benefits preferentially in tumors with altered Rad51 levels under hypoxic conditions or due to polymorphism.
3.4 Xrcc3 and cancer
Xrcc3 is recruited to DNA DSBs early and independent of Rad51 [76]. It interacts with Rad51C, another Rad51 paralog [77], in addition to Rad51 and is required for assembly of Rad51 complexes in vivo [78]. XRCC3 and Rad51 cooperatively modulate the progression of replication forks on damaged vertebrate chromosomes [79]. To date, no human disease has been linked to inactivation of XRCC3 yet. A naturally occurring mutation (D213N) in an ATP-binding domain of Xrcc3 ablates its function but does not cause cancer susceptibility [80]. Polymorphisms in XRCC3 gene may be associated with increased cancer risk. The correlation between Xrcc3 variant and cancer risk has been actively studied in epidemiology. However, it remains controversial collectively based on statistical results from various types of cancers [81–90].
3.5 Cancer with loss of PTEN
Phosphatase and tensin homolog (PTEN) has a nuclear function of transcriptionally regulating RAD51 gene in addition to its well-known function of inhibiting the PI3K-Akt pathway. PTEN null cells exhibit spontaneous DNA DSBs [91,92]. HR function could be compromised due to loss of PTEN. For example, 36% of glioblastomas present homozygous deletion in PTEN [93–95], which sensitizes them to agents that impact the BER pathway via a conditional lethal mechanism [94]. Glioblastoma, which is often refractory to treatment and has very poor survival rate, is one of the most common high-grade (aggressive) astrocytomas. Recent genomic analyses of high-grade ovarian cancer reported 7% cases with focal deletion or mutation in PTEN gene [65]. These subtypes of glioblastoma and ovarian carcinoma with defective HR capacity due to PTEN loss might be responsive to sapacitabine.
4. Conclusion
Sapacitabine has presented encouraging anticancer activity in both preclinical and clinical investigations. In particular, recent clinical trials demonstrated its efficacy against hematologic malignancies. Sapacitabine and its active metabolite, CNDAC, are distinguished from other nucleoside analogs by the unique action mechanism of inducing DNA strand breaks after incorporation into DNA. CNDAC-caused SSBs are transformed into DSBs during a second cycle of DNA replication. In addition to TC-NER, this appears to participate in repair of SSBs generated in the first replication, HR functions as the major mechanism of repairing DSBs, the lethal form of DNA damage induced by CNDAC. Dependence of cancer cells on the HR pathway to repair CNDAC-induced damage generates the opportunity to preferentially kill tumors with deficiencies in HR function. We hypothesize that a wide range of cancers that have defects in HR capability due to various genetic traits, both hematologic malignancies and solid tumors, may be selectively sensitized to sapacitabine therapy. We have suggested potential candidates for sapacitabine treatment, based on HR deficiency in these tumors. Future trials of sapacitabine-based individualized chemotherapies could test this postulate.
5. Expert opinion
CNDAC and its prodrug, sapacitabine, are unique among nucleoside analogs due to the DNA-strand-breaking mechanism of action. The past or ongoing preclinical and clinical trials indicate that sapacitabine is a safe and promising chemotherapeutic drug for a range of malignancies. The fact that repair of CNDAC-induced damage does not rely on p53 status [28] suggests a broad spectrum of cancer types for sapacitabine therapy. The identification of HR pathway as the major repair mechanism for CNDAC-induced DSBs has provided rationale for clinical application of sapacitabine in HR defective tumors. Incidence of cancer with gene alterations in HR components could be very significant. For example, approximately 50% of high-grade serous ovarian cancer has been demonstrated to have altered HR genes, including BRCA1/2, PTEN, Rad51C and the FA core complex [65].
We have speculated that cancers with deficiency in ATM and BRCA1/2 or downregulation of Rad51 and its interacting proteins are good candidates for sapacitabine therapy. This hypothesis is being tested in a clinical trial of the combination of sapacitabine–cytoxan–rituximab (SCR) for CLL patients with del(11q22-23), substituting fludarabine with sapacitabine in order to overcome resistance to the front-line fludarabine–cytoxan–rituximab (FCR) regimen (Weirda, ClinicalTrials.gov identifier: NCT01253460). This type of research and trials have the potential to direct use of sapacitabine toward personalized treatment for cancer subtypes bearing defects in HR repair mechanism. This strategy matches a genetic lesion in DNA repair to the drug mechanism to generate a tumor-specific therapeutic action. The sapacitabine-induced lesion is not a substrate for BER, a mechanistic feature that distinguishes the synthetic lethal condition created by sapacitabine in ATM-deficient CLL from that created by PARP (poly ADP ribose polymerase) inhibition and a genetic lesion in a second DNA repair pathway [96].
Knowledge of the unique mechanism of action of CNDAC creates the opportunity to identify synergistic interactions with known anticancer drugs. Clonogenic assays have been used to study combinations of CNDAC with agents targeting the BER (temozolomide and PARP inhibitor), NER (bendamustine, 4-hydroperoxycyclophosphamide and platinum-based drugs) as well as HR pathway (ATM inhibitor and imatinib), and these combinations presented either synergistic or additive effects [97] (Liu et al., unpublished data). The results of such investigations may be used to guide the design of clinical trials, and to indicate appropriate biomarkers as end points to be evaluated for validation of the postulated interactions.
Article highlights.
Sapacitabine and its active metabolite, CNDAC, are distinguished among nucleoside analogs by the unique action mechanism of inducing a DNA single-strand break (SSBs) after incorporation into DNA. Double-strand breaks (DSBs), regarded as the lethal form of DNA damage, are subsequently generated during a second cycle of DNA replication.
Transcription-coupled nucleotide excision repair (TC-NER) participates in repair of CNDAC-induced SSBs, while homologous recombination (HR) plays a major role in repairing DSBs, as evidenced by sensitization of cells bearing defects in components of these pathways. The two DNA repair pathways work in concert in cellular responses to CNDAC, and are potential resistance mechanisms.
The dependency of cancer cells on the HR pathway to repair sapacitabine or CNDAC-induced DNA damage creates the opportunity to preferentially sensitize tumors with deficiency in HR function, providing a basis for targeted therapy in subgroups of cancer patients.
A wide range of hematologic malignancies and solid tumors that have defects in HR capability due to various genetic traits may be selectively targeted by sapacitabine-containing therapy. Potential candidates for sapacitabine treatment are discussed.
The authors hypothesize that cancers with deficiencies in ATM and BRCA1/2 or downregulation of Rad51 and its interacting proteins are attractive candidates for sapacitabine therapy. This postulate could be tested with sapacitabine-containing clinical trials.
Acknowledgments
The authors are grateful to Dr. David Blake, Cyclacel Pharmaceuticals, Ltd., for critical reading of the manuscript.
Footnotes
This box summarizes key points contained in the article.
Declaration of interests
Investigations from the authors’ laboratories were supported by Grants CA28596, CA81534 and CA100532 from the National Cancer Institute, Department of Health and Human Services. The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1••.Matsuda A, Nakajima Y, Azuma A, et al. Nucleosides and nucleotides. 100. 2′-C-cyano-2′-deoxy-1-beta-D-arabinofuranosyl-cytosine (CNDAC): design of a potential mechanism-based DNA-strand-breaking antineoplastic nucleoside. J Med Chem. 1991;34(9):2917–19. doi: 10.1021/jm00113a034. This paper describes the rationale for the mechanism of action of CNDAC, the synthesis of the compound and the first evidence of cytotoxicity and antitumor activity. [DOI] [PubMed] [Google Scholar]
- 2.Azuma A, Hanaoka K, Kurihara A, et al. Nucleosides and nucleotides. 141. Chemical stability of a new antitumor nucleoside, 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine in alkaline medium: formation of 2′-C-cyano-2′-deoxy-1-beta-D-ribo-pentofuranosylcytosine and its antitumor activity. J Med Chem. 1995;38(17):3391–7. doi: 10.1021/jm00017a023. [DOI] [PubMed] [Google Scholar]
- 3.Hayakawa Y, Kawai R, Otsuki K, et al. Evidence supporting the activity of 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentafuranosylcytosine as a terminator in enzymatic DNA-chain elongation. Bioorg Med Chem Lett. 1998;8(18):2559–62. doi: 10.1016/s0960-894x(98)00459-4. [DOI] [PubMed] [Google Scholar]
- 4.Matsuda A. 2′-C-Cyano-2′-deoxy-1-beta-D-arabinofuranosyl-cytosine(CNDAC): a mechanism-based DNA-strandbreaking antitumor nucleoside. Nucleosides Nucleotides. 1995;14:461–71. [Google Scholar]
- 5•.Hanaoka K, Suzuki M, Kobayashi T, et al. Antitumor activity and novel DNA-self-strand-breaking mechanism of CNDAC (1-(2-C-cyano-2-deoxy-beta-D-arabino-pentofuranosyl) cytosine) and its N4-palmitoyl derivative (CS-682) Int J Cancer. 1999;82(2):226–36. doi: 10.1002/(sici)1097-0215(19990719)82:2<226::aid-ijc13>3.0.co;2-x. This paper provides evidence for a β-elimination reaction following incorporation into the DNA of a cell, and introduces the investigation of sapacitabine, the prodrug of CNDAC. [DOI] [PubMed] [Google Scholar]
- 6•.Azuma A, Huang P, Matsuda A, et al. 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine: a novel anticancer nucleoside analog that causes both DNA strand breaks and G(2) arrest. Mol Pharmacol. 2001;59(4):725–31. doi: 10.1124/mol.59.4.725. This paper demonstrates the termination of DNA by the β-elimination product CNddC, and the association of this action with a G2 arrest. [DOI] [PubMed] [Google Scholar]
- 7.Kaneko M, Koga R, Murayama K, et al. Synthesis and antitumor activity of a novel antitumor nucleoside 1-(2-C-cyano-2-deoxy-beta-D-arabino-pentofuranosyl)-N4-palmitoylcytosine (CS-682) Proc Am Assoc Cancer Res. 1997;38:679. [Google Scholar]
- 8.Obata T, Endo Y, Tanaka M, et al. Development and biochemical characterization of a 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine (CNDAC)-resistant variant of the human fibrosarcoma cell line HT-1080. Cancer Lett. 1998;123(1):53–61. doi: 10.1016/s0304-3835(97)00402-3. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida R, Matsuda T, Watanabe T, et al. A case of gallbladder cancer which completely responded to gemcitabine. Gan To Kagaku Ryoho. 2010;37(9):1771–3. [PubMed] [Google Scholar]
- 10•.Azuma A, Huang P, Matsuda A, et al. Cellular pharmacokinetics and pharmacodynamics of the deoxycytidine analog 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine (CNDAC) Biochem Pharmacol. 2001;61(12):1497–507. doi: 10.1016/s0006-2952(01)00617-7. This paper demonstrates the metabolism of the triphosphate of CNDAC in cell lines. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka M. Enhancement of the cytotoxicity of cytosine arabinoside by interleukin-3. Jpn J Cancer Res. 1992;83(2):194–9. doi: 10.1111/j.1349-7006.1992.tb00086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serova M, Galmarini CM, Ghoul A, et al. Antiproliferative effects of sapacitabine (CYC682), a novel 2′-deoxycytidine-derivative, in human cancer cells. Br J Cancer. 2007;97(5):628–36. doi: 10.1038/sj.bjc.6603896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka M, Matsuda A, Terao T, et al. Antitumor activity of a novel nucleoside, 2′-C-cyano-2′-deoxy-1-beta-D-arabinofuranosylcytosine (CNDAC) against murine and human tumors. Cancer Lett. 1992;64(1):67–74. doi: 10.1016/0304-3835(92)90024-p. [DOI] [PubMed] [Google Scholar]
- 14.Wu M, Mazurchuk R, Chaudhary ND, et al. High-resolution magnetic resonance imaging of the efficacy of the cytosine analogue 1-[2-C-cyano-2-deoxy-beta-D-arabino-pentofuranosyl]-N(4)-palmitoyl cytosine (CS-682) in a liver-metastasis athymic nude mouse model. Cancer Res. 2003;63(10):2477–82. [PubMed] [Google Scholar]
- 15.Katz MH, Bouvet M, Takimoto S, et al. Selective antimetastatic activity of cytosine analog CS-682 in a red fluorescent protein orthotopic model of pancreatic cancer. Cancer Res. 2003;63(17):5521–5. [PubMed] [Google Scholar]
- 16.Katz MH, Bouvet M, Takimoto S, et al. Survival efficacy of adjuvant cytosine-analogue CS-682 in a fluorescent orthotopic model of human pancreatic cancer. Cancer Res. 2004;64(5):1828–33. doi: 10.1158/0008-5472.can-03-3350. [DOI] [PubMed] [Google Scholar]
- 17.Asai T, Kurohane K, Shuto S, et al. Antitumor activity of 5′-O-dipalmitoylphosphatidyl 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine is enhanced by long-circulating liposomalization. Biol Pharm Bull. 1998;21(7):766–71. doi: 10.1248/bpb.21.766. [DOI] [PubMed] [Google Scholar]
- 18.Green SR, Choudhary AK, Fleming IN. Combination of sapacitabine and HDAC inhibitors stimulates cell death in AML and other tumour types. Br J Cancer. 2010;103(9):1391–9. doi: 10.1038/sj.bjc.6605922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Gilbert J, Carducci MA, Baker SD, et al. A Phase I study of the oral antimetabolite, CS-682, administered once daily 5 days per week in patients with refractory solid tumor malignancies. Invest New Drugs. 2006;24(6):499–508. doi: 10.1007/s10637-006-8219-0. These two papers present the initial Phase I clinical trials of sapacitabine in solid tumors, and provide detailed pharmacokinetic evaluations including estimates of its bioavailability. [DOI] [PubMed] [Google Scholar]
- 20•.Delaunoit T, Burch PA, Reid JM, et al. A phase I clinical and pharmacokinetic study of CS-682 administered orally in advanced malignant solid tumors. Invest New Drugs. 2006;24(4):327–33. doi: 10.1007/s10637-006-5392-0. These two papers present the initial Phase I clinical trials of sapacitabine in solid tumors, and provide detailed pharmacokinetic evaluations including estimates of its bioavailability. [DOI] [PubMed] [Google Scholar]
- 21.Tolcher A, Calvo E, Carmona T, et al. Phase I study of sapacitabine, an oral nucleoside analogue, in patients with refractory solid tumors or lymphomas [abstract #463]. The 18th EORTC-NCI-AACR Symposium on “Molecular Targets and Cancer Therapeutics”; 2006. [Google Scholar]
- 22.Sankhala KK, Takimoto CH, Mita AC, et al. Two phase I, pharmacokinetic (PK) and pharmacodynamic (PD) studies of TAS-109, a novel nucleoside analogue with 14 days and 7 days continuous infusion (CI) schedules. ASCO Meeting Abstracts. 2008;26(15 Suppl):2577. [Google Scholar]
- 23•.Kantarjian H, Garcia-Manero G, O’Brien S, et al. Phase I clinical and pharmacokinetic study of oral sapacitabine in patients with acute leukemia and myelodysplastic syndrome. J Clin Oncol. 2010;28(2):285–91. doi: 10.1200/JCO.2009.25.0209. This is the first report of a Phase I clinical trial of sapacitabine in hematologic malignancies, which indicated encouraging outcomes in AML and MDS in elderly patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kantarjian HM, Garcia-Manero G, Luger S, et al. A Randomized Phase II Study of Sapacitabine. An oral nucleoside analogue, in elderly patients with AML previously untreated or in first relapse. ASH Annual Meeting Abstracts. 2009;114(22):1061. [Google Scholar]
- 25.Garcia-Manero G, Luger S, Venugopal P, et al. A Randomized Phase II Study of Sapacitabine. An oral nucleoside analogue, in older patients with myelodysplastic syndrome (MDS) refractory to hypomethylating agents. ASH Annual Meeting Abstracts. 2009;114(22):1758. [Google Scholar]
- 26.Garcia-Manero G, Luger S, Venugopal P, et al. A Randomized Phase II Study of Sapacitabine. An oral nucleoside analogue, in older patients with MDS refractory to hypomethylating agents. ASH Annual Meeting Abstracts. 2010;116(21):1857. [Google Scholar]
- 27.Ravandi F, Faderl S, Cortes JE, et al. Phase I/II study of sapacitabine and decitabine administered sequentially in elderly patients with newly diagnosed acute myeloid leukemia. ASCO Meeting Abstracts. 2011;29(15 Suppl):6587. [Google Scholar]
- 28••.Liu X, Wang Y, Benaissa S, et al. Homologous recombination as a resistance mechanism to replication-induced double-strand breaks caused by the antileukemia agent CNDAC. Blood. 2010;116(10):1737–46. doi: 10.1182/blood-2009-05-220376. This article illustrates that CNDAC-induced DNA strand breaks are converted to double strand breaks during a subsequent replication cycle. Importantly, it demonstrates that HR is the major pathway responsible for repair of such double strand breaks, thereby providing the basis of sapacitabine selectivity for tumors lack the function of this DNA repair pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Guo Y, Li Y, et al. Molecular basis for G2 arrest induced by 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine and consequences of checkpoint abrogation. Cancer Res. 2005;65(15):6874–81. doi: 10.1158/0008-5472.CAN-05-0288. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Matsuda A, Plunkett W. Ataxia-telangiectasia and Rad3-related and DNA-dependent protein kinase cooperate in G2 checkpoint activation by the DNA strand-breaking nucleoside analogue 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine. Mol Cancer Ther. 2008;7(1):133–42. doi: 10.1158/1535-7163.MCT-07-0416. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Sampath D, Tseng J, et al. Abrogation of S-phase and G2 cell cycle checkpoints by small molecule inhibitors of the DNA damage kinase, Chk1 [abstract #1682] Proc Am Assoc Cancer Res. 2005;46:396–7. [Google Scholar]
- 32•.Wang Y, Liu X, Matsuda A, et al. Repair of 2′-C-cyano-2′-deoxy-1-beta-D-arabino-pentofuranosylcytosine-induced DNA single-strand breaks by transcription-coupled nucleotide excision repair. Cancer Res. 2008;68(10):3881–9. doi: 10.1158/0008-5472.CAN-07-6885. This article reveals transcription-coupled NER as a mechanism for repairing CNDAC-induced nicks in DNA. This pathway functions in concert with the HR pathway in cellular responses to DNA damage caused by CNDAC or sapacitabine. [DOI] [PubMed] [Google Scholar]
- 33.Meek K, Gupta S, Ramsden DA, et al. The DNA-dependent protein kinase: the director at the end. Immunol Rev. 2004;200:132–41. doi: 10.1111/j.0105-2896.2004.00162.x. [DOI] [PubMed] [Google Scholar]
- 34.Beucher A, Birraux J, Tchouandong L, et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. Embo J. 2009;28(21):3413–27. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrison C, Sonoda E, Takao N, et al. The controlling role of ATM in homologous recombinational repair of DNA damage. Embo J. 2000;19(3):463–71. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19(24):2925–40. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 37.D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362(20):1909–19. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24(16):1680–94. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boultwood J. Ataxia telangiectasia gene mutations in leukaemia and lymphoma. J Clin Pathol. 2001;54(7):512–16. doi: 10.1136/jcp.54.7.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taylor AM, Metcalfe JA, Thick J, et al. Leukemia and lymphoma in ataxia telangiectasia. Blood. 1996;87(2):423–38. [PubMed] [Google Scholar]
- 41.Gatti RA, Berkel I, Boder E, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. 1988;336(6199):577–80. doi: 10.1038/336577a0. [DOI] [PubMed] [Google Scholar]
- 42.Ma SK, Wan TS, Au WY, et al. Chromosome 11q deletion in myeloid malignancies. Leukemia. 2002;16(5):953–5. doi: 10.1038/sj.leu.2402442. [DOI] [PubMed] [Google Scholar]
- 43.Austen B, Skowronska A, Baker C, et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol. 2007;25(34):5448–57. doi: 10.1200/JCO.2007.11.2649. [DOI] [PubMed] [Google Scholar]
- 44.Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–16. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 45.Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–74. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- 46.Monni O, Knuutila S. 11q deletions in hematological malignancies. Leuk Lymphoma. 2001;40(3–4):259–66. doi: 10.3109/10428190109057924. [DOI] [PubMed] [Google Scholar]
- 47.Stankovic T, Stewart GS, Byrd P, et al. ATM mutations in sporadic lymphoid tumours. Leuk Lymphoma. 2002;43(8):1563–71. doi: 10.1080/1042819021000002884. [DOI] [PubMed] [Google Scholar]
- 48.Stilgenbauer S, Schaffner C, Winkler D, et al. The ATM gene in the pathogenesis of mantle-cell lymphoma. Ann Oncol. 2000;11(Suppl 1):127–30. [PubMed] [Google Scholar]
- 49.Stilgenbauer S, Winkler D, Ott G, et al. Molecular characterization of 11q deletions points to a pathogenic role of the ATM gene in mantle cell lymphoma. Blood. 1999;94(9):3262–4. [PubMed] [Google Scholar]
- 50.Camacho E, Hernandez L, Hernandez S, et al. ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood. 2002;99(1):238–44. doi: 10.1182/blood.v99.1.238. [DOI] [PubMed] [Google Scholar]
- 51.Schaffner C, Idler I, Stilgenbauer S, et al. Mantle cell lymphoma is characterized by inactivation of the ATM gene. Proc Natl Acad Sci USA. 2000;97(6):2773–8. doi: 10.1073/pnas.050400997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stilgenbauer S, Schaffner C, Litterst A, et al. Biallelic mutations in the ATM gene in T-prolymphocytic leukemia. Nat Med. 1997;3(10):1155–9. doi: 10.1038/nm1097-1155. [DOI] [PubMed] [Google Scholar]
- 53.Vorechovsky I, Luo L, Dyer MJ, et al. Clustering of missense mutations in the ataxia-telangiectasia gene in a sporadic T-cell leukaemia. Nat Genet. 1997;17(1):96–9. doi: 10.1038/ng0997-96. [DOI] [PubMed] [Google Scholar]
- 54.Luo L, Lu FM, Hart S, et al. Ataxia-telangiectasia and T-cell leukemias: no evidence for somatic ATM mutation in sporadic T-ALL or for hypermethylation of the ATM-NPAT/E14 bidirectional promoter in T-PLL. Cancer Res. 1998;58(11):2293–7. [PubMed] [Google Scholar]
- 55.Bradshaw PS, Condie A, Matutes E, et al. Breakpoints in the ataxia telangiectasia gene arise at the RGYW somatic hypermutation motif. Oncogene. 2002;21(3):483–7. doi: 10.1038/sj.onc.1205105. [DOI] [PubMed] [Google Scholar]
- 56.Yang H, Spitz MR, Stewart DJ, et al. ATM sequence variants associate with susceptibility to non-small cell lung cancer. Int J Cancer. 2007;121(10):2254–9. doi: 10.1002/ijc.22918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ai L, Vo QN, Zuo C, et al. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol Biomarkers Prev. 2004;13(1):150–6. doi: 10.1158/1055-9965.epi-082-3. [DOI] [PubMed] [Google Scholar]
- 58.Lee KW, Tsai YS, Chiang FY, et al. Lower ataxia telangiectasia mutated (ATM) mRNA expression is correlated with poor outcome of laryngeal and pharyngeal cancer patients. Ann Oncol. 2011;22(5):1088–93. doi: 10.1093/annonc/mdq569. [DOI] [PubMed] [Google Scholar]
- 59.Friedenson B. The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer. 2007;7:152. doi: 10.1186/1471-2407-7-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferla R, Calo V, Cascio S, et al. Founder mutations in BRCA1 and BRCA2 genes. Ann Oncol. 2007;18(Suppl 6):vi93–8. doi: 10.1093/annonc/mdm234. [DOI] [PubMed] [Google Scholar]
- 61.Fatouros M, Baltoyiannis G, Roukos DH. The predominant role of surgery in the prevention and new trends in the surgical treatment of women with BRCA1/2 mutations. Ann Surg Oncol. 2008;15(1):21–33. doi: 10.1245/s10434-007-9612-4. [DOI] [PubMed] [Google Scholar]
- 62.Fasano J, Muggia F. Breast cancer arising in a BRCA-mutated background: therapeutic implications from an animal model and drug development. Ann Oncol. 2009;20(4):609–14. doi: 10.1093/annonc/mdn669. [DOI] [PubMed] [Google Scholar]
- 63.Tinelli A, Malvasi A, Leo G, et al. Hereditary ovarian cancers: from BRCA mutations to clinical management. A modern appraisal. Cancer Metastasis Rev. 2010;29(2):339–50. doi: 10.1007/s10555-010-9218-3. [DOI] [PubMed] [Google Scholar]
- 64.Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 65.Network TCGAR. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohamad HB, Apffelstaedt JP. Counseling for male BRCA mutation carriers: a review. Breast. 2008;17(5):441–50. doi: 10.1016/j.breast.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 67.Mitra A, Fisher C, Foster CS, et al. Prostate cancer in male BRCA1 and BRCA2 mutation carriers has a more aggressive phenotype. Br J Cancer. 2008;98(2):502–7. doi: 10.1038/sj.bjc.6604132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scardocci A, Guidi F, D’Alo F, et al. Reduced BRCA1 expression due to promoter hypermethylation in therapy-related acute myeloid leukaemia. Br J Cancer. 2006;95(8):1108–13. doi: 10.1038/sj.bjc.6603392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taron M, Rosell R, Felip E, et al. BRCA1 mRNA expression levels as an indicator of chemoresistance in lung cancer. Hum Mol Genet. 2004;13(20):2443–9. doi: 10.1093/hmg/ddh260. [DOI] [PubMed] [Google Scholar]
- 70.Paul I, Savage KI, Blayney JK, et al. PARP inhibition induces BAX/BAK-independent synthetic lethality of BRCA1-deficient non-small cell lung cancer. J Pathol. 2011;224(4):564–74. doi: 10.1002/path.2925. [DOI] [PubMed] [Google Scholar]
- 71.Antoniou AC, Sinilnikova OM, Simard J, et al. RAD51 135G–> C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am J Hum Genet. 2007;81(6):1186–200. doi: 10.1086/522611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levy-Lahad E, Lahad A, Eisenberg S, et al. A single nucleotide polymorphism in the RAD51 gene modifies cancer risk in BRCA2 but not BRCA1 carriers. Proc Natl Acad Sci USA. 2001;98(6):3232–6. doi: 10.1073/pnas.051624098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bindra RS, Schaffer PJ, Meng A, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004;24(19):8504–18. doi: 10.1128/MCB.24.19.8504-8518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang CJ, Yang JY, Xia W, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19(1):86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16(12):7133–43. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Forget AL, Bennett BT, Knight KL. Xrcc3 is recruited to DNA double strand breaks early and independent of Rad51. J Cell Biochem. 2004;93(3):429–36. doi: 10.1002/jcb.20232. [DOI] [PubMed] [Google Scholar]
- 77.Wiese C, Collins DW, Albala JS, et al. Interactions involving the Rad51 paralogs Rad51C and XRCC3 in human cells. Nucleic Acids Res. 2002;30(4):1001–8. doi: 10.1093/nar/30.4.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bishop DK, Ear U, Bhattacharyya A, et al. Xrcc3 is required for assembly of Rad51 complexes in vivo. J Biol Chem. 1998;273(34):21482–8. doi: 10.1074/jbc.273.34.21482. [DOI] [PubMed] [Google Scholar]
- 79.Henry-Mowatt J, Jackson D, Masson JY, et al. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell. 2003;11(4):1109–17. doi: 10.1016/s1097-2765(03)00132-1. [DOI] [PubMed] [Google Scholar]
- 80.Rafii S, Lindblom A, Reed M, et al. A naturally occurring mutation in an ATP-binding domain of the recombination repair gene XRCC3 ablates its function without causing cancer susceptibility. Hum Mol Genet. 2003;12(8):915–23. doi: 10.1093/hmg/ddg102. [DOI] [PubMed] [Google Scholar]
- 81.Shen H, Sturgis EM, Dahlstrom KR, et al. A variant of the DNA repair gene XRCC3 and risk of squamous cell carcinoma of the head and neck: a case-control analysis. Int J Cancer. 2002;99(6):869–72. doi: 10.1002/ijc.10413. [DOI] [PubMed] [Google Scholar]
- 82.Yeh CC, Sung FC, Tang R, et al. Polymorphisms of the XRCC1, XRCC3, & XPD genes, and colorectal cancer risk: a case-control study in Taiwan. BMC Cancer. 2005;5:12. doi: 10.1186/1471-2407-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Winsey SL, Haldar NA, Marsh HP, et al. A variant within the DNA repair gene XRCC3 is associated with the development of melanoma skin cancer. Cancer Res. 2000;60(20):5612–16. [PubMed] [Google Scholar]
- 84.Kuschel B, Auranen A, McBride S, et al. Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet. 2002;11(12):1399–407. doi: 10.1093/hmg/11.12.1399. [DOI] [PubMed] [Google Scholar]
- 85.Auranen A, Song H, Waterfall C, et al. Polymorphisms in DNA repair genes and epithelial ovarian cancer risk. Int J Cancer. 2005;117(4):611–18. doi: 10.1002/ijc.21047. [DOI] [PubMed] [Google Scholar]
- 86.David-Beabes GL, Lunn RM, London SJ. No association between the XPD (Lys751G1n) polymorphism or the XRCC3 (Thr241Met) polymorphism and lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2001;10(8):911–12. [PubMed] [Google Scholar]
- 87.Duan Z, Shen H, Lee JE, et al. DNA repair gene XRCC3 241Met variant is not associated with risk of cutaneous malignant melanoma. Cancer Epidemiol Biomarkers Prev. 2002;11(10 Pt 1):1142–3. [PubMed] [Google Scholar]
- 88.Sanyal S, Festa F, Sakano S, et al. Polymorphisms in DNA repair and metabolic genes in bladder cancer. Carcinogenesis. 2004;25(5):729–34. doi: 10.1093/carcin/bgh058. [DOI] [PubMed] [Google Scholar]
- 89.Webb PM, Hopper JL, Newman B, et al. Double-strand break repair gene polymorphisms and risk of breast or ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2005;14(2):319–23. doi: 10.1158/1055-9965.EPI-04-0335. [DOI] [PubMed] [Google Scholar]
- 90.Jiao L, Hassan MM, Bondy ML, et al. XRCC2 and XRCC3 gene polymorphism and risk of pancreatic cancer. Am J Gastroenterol. 2008;103(2):360–7. doi: 10.1111/j.1572-0241.2007.01615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 92.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27(41):5443–53. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 93.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 94.McEllin B, Camacho CV, Mukherjee B, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly (ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70(13):5457–64. doi: 10.1158/0008-5472.CAN-09-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Network TCGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weston VJ, Oldreive CE, Skowronska A, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116(22):4578–87. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- 97.Liu X, Nowak B, Matsuda A, et al. Mechanism-based drug combinations with the DNA-strand-breaking nucleoside analog CNDAC. Proc Am Assoc Cancer Res. 2011;52:abstract #962. doi: 10.1158/1535-7163.MCT-15-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]