

Abstract
The enzyme prephenate dehydratase (PDT) converts prephenate to phenylpyruvate in L-phenylalanine biosynthesis. PDT is allosterically regulated by L-Phe and other amino acids. We report the first crystal structures of PDT from Staphylococcus aureus in a relaxed (R) state and PDT from Chlorobium tepidum in a tense (T) state. The two enzymes show low sequence identity (27.3%) but the same prototypic architecture and domain organization. Both enzymes are tetramers (dimer of dimers) in crystal and solution while a PDT dimer can be regarded as a basic catalytic unit. The N-terminal PDT domain consists of two similar subdomains with a cleft in between, which hosts the highly conserved active site. In one PDT dimer two clefts are aligned to form an extended active site across the dimer interface. Similarly at the interface two ACT regulatory domains create two highly conserved pockets. Upon binding of the L-Phe inside the pockets, PDT transits from an open to a closed conformation.
Keywords: prephenate dehydratase structure, PDT domain, ACT domain, allosteric regulation, L-Phe binding
INTRODUCTION
Prephenate dehydratase (PDT, EC:4.2.1.51) converts prephenate to phenylpyruvate through dehydration and decarboxylation reactions (Fig. 1A). The enzyme was discovered in 1965 (Cotton and Gibson, 1965) and was initially characterized in bacteria, including several pathogens, as well as in archaea, yeast, fungi and plants (Fig. 2) (Bode et al., 1984; Fiske and Kane, 1984; Jensen et al., 1988; Porat et al., 2004; Warpeha et al., 2006). The PDT plays a key role in the biosynthesis of L-Phe in organisms that utilize the shikimate pathway (Bentley, 1990; Lingens, 1976). PDT is allosterically regulated by L-Phe and other amino acids (Pohnert et al., 1999; Prakash et al., 2005). Generally, in Gram-positive bacteria and archaea, PDT is a monofunctional enzyme, consisting of two domains - a catalytic domain (PDT domain) and a regulatory domain (ACT) (aspartokinase, chorismate mutase domain). In Gram-negative bacteria, PDT exists as a fusion protein with chorizmate mutase (CM), forming a bifunctional enzyme, P-protein (PheA). The CM in the P-protein catalyzes the pericyclic isomerization of chorismate to prephenate that serves as a substrate for PDT. The CM and PDT are essential enzymes for the biosynthesis of aromatic amino acids in microorganisms but are not found in humans. Therefore, both CM and PDT can potentially serve as drug targets against microbial pathogens (Husain et al., 2001).
Figure 1.

The pathway and the enzymatic activity of Sa-PDT. (A) The conversion of prephenate to phenylpyruvate by prephenate dehydratase (PDT). (B) Activity assay for PDT was performed as described by Dopheide et al. (28) and as described in Materials and Methods. The left panel shows the reaction rate at different prephenate concentrations, the enzyme was 0.04 μM and the prephenate concentrations vary between 0.2 – 16 mM. The right panel shows the time course of the prephenate conversion to phenylpyruvate measured for 0.04 μM enzyme and 0.4 mM prephenate. The data were analyzed using the software PRISM. (Prizm Software, Irvine, CA).
Figure 2.

Multiple Sequence Alignment of PDT domains across different species. The molecules used in the alignment include: Sa-PDT (S. aureus PDT, gi:14247687), Ct-PDT (C. tepidum TLS PDT, gi:21647673), Sh-PDT (S. haemolyticus PDT, gi:70726038), Af-PheA (Archaeoglobus fulgidus Phe, gi:2650414), Hi-PheA (Haemophilus influenzae Phe, gi:1172476), Pa-PheA (Pantoea agglomerans Phe, gi:266771), Ec-PheA (E. coli Phe, gi:16130520), At-PheA (Arabidopsis thaliana Phe, gi:2392772), Cs-PDT (Cyanobacterium synechocystis PDT, gi: 1651896), Bs-PDT (B. subtilis PDT, gi:130048), Mt-PDT (M. tuberculosis PDT, gi:15610974), Cg-PDT (C. glutamicum PDT, gi:144987) and Am-PDT (Amycolatopsis methanolica PDT, gi:2499520). Based on the Sa-PDT and Ct-PDT structures, the β-strands and α-helices are indicated with arrows and coils below the appropriate sequences, respectively. The two linker regions between the two PDT subdomains are underscored with green lines.
CMs have been extensively studied, both functionally and structurally (Helmstaedt et al., 2001; Woycechowsky and Hilvert, 2004). Several high resolution structures of CM have been recently reported, including CM structures from Escherichia coli (Lee and Saier, 1983), Bacillus subtilis (Chook et al., 1994), Saccharomyces cerevisiae (Strater et al., 1996; Strater et al., 1997; Xue et al., 1994), Thermus thermophilus (Helmstaedt et al., 2004) and Mycobacterium tuberculosis (Qamra et al., 2006).
Homologues of ACT domains are found fused to a wide range of metabolic enzymes that are regulated by amino acids. It has been shown that dimers of ACT domains bind a specific amino acid leading to regulation of the linked enzyme unit. The ACT domain containing enzymes include D-3-phosphoglycerate dehydrogenase (inhibited by serine), aspartokinase (inhibited by lysine), acetolactate synthase (inhibited by valine), phenylalanine-4-hydroxylase (inhibited by phenylalanine), formyltetrahydrofolate deformylase (activated by methionine and inhibited by glycine) and PDT (inhibited by L-Phe and L-Met), the subject of this study. Structures of several ACT domains have also been determined and investigated, including their amino acid induced allosteric regulation (Liberles et al., 2005). In contrast to CMs and ACTs, PDT domains, although equally important, remain rather poorly characterized and until recently there was no PDT domain structure reported. However, the crystallization of the prephenate dehydratase from M. tuberculosis has been recently reported (Vivan et al., 2006). This lack of structural information hinders the elucidation of the catalytic and regulatory mechanism for both PDT and P-proteins.
PDT is a potential drug target since it is involved in the synthesis of L-Phe and it was identified in Gram-positive Staphylococcus aureus subsp Aureus Mu50 (Sa-PDT) through sequence analysis (Kuroda et al., 2001). S. aureus is the most common cause of staph infections in communities and hospitals. Sa-PDT is highly conserved (98~100% sequence identities) within different strains of S. aureus. However, as of yet there is no functional study reported for this enzyme. The Sa-PDT shares about 70% sequence identities with PDTs from S. haemolyticus, S. epidemidis and S. saprophyticus. When aligned with PDTs or P-proteins from other microorganisms, the sequence identities are lower (about 30%) including PheA from E. coli (which shows 24% identity and 46% similarity).
The green-sulfur eubacterium Chlorobium tepidum TLS is a photolithotrophic oxidizer of sulfur, growing optimally at 48°C. The PDT of the thermophile (Ct-PDT) was identified from its complete genome sequence (Eisen et al., 2002). Unlike most Gram-negative bacteria, Ct-PDT is monofunctional without a fused CM domain. The enzyme is only about 70% conserved in sequence identities within a few Chlorobia. Limited studies of the bacterium have been reported and its PDT is not functionally characterized. The Ct-PDT shares sequence identity of 27.3% with Sa-PDT. When aligned with P-proteins from strains of E. coli, the sequence identities are slightly higher, about 32% identities and 50% similarities.
Here we report the structural and functional characterization of a native Sa-PDT and a Ct-PDT in complex with L-Phe. The two structures are the first of a large prephenate dehydratase family (Pfam00800) found in all three kingdoms of life (Bateman et al., 2004). Both Sa-PDT and Ct-PDT/L-Phe form tetramers (dimer-of-dimers) in the crystal and in solution. Comparison of the two PDT structures revealed many similarities, especially within the highly conserved regions of PDT molecules, and distinctive differences that we believe correspond to the R-state and T-state of PDT. The R-state Sa-PDT showed an “opened” active conformation. The T-state Ct-PDT revealed a conformational change induced by the L-Phe binding on the ACT dimer interface. Upon binding of the effector, the PDT dimer undergoes a conformational change that results in reduced access to the active site.
RESULTS
Protein Expression and PTD Oligomeric States in Solution
The SeMet-labeled, recombined Sa-PDT and Ct-PDT were expressed in the E. coli BL21 (DE3) and purified to homogeneity using His6-tag immobilized metal affinity chromatography columns. Size-exclusion chromatography was used to determine the diffusion coefficient and establish the oligomeric states of the Sa-PDT and Ct-PDT proteins. The elution profile of the size-exclusion chromatography of each PDT showed a single peak. Based on a calibration curve (data not shown), the apparent molecular weights of the eluted Sa-PDT and Ct-PDT were estimated to be 130 kDa and 142 kDa, respectively. The calculated protein molecular weights from their sequences are 30 kDa (120 kDa for the tetramer) and 31 kDa (124 kDa for the tetramer), respectively, including 3 N-terminal vector derived residues (SNA). This data strongly suggests that both Sa-PDT and Ct-PDT are tetramers in solution.
PDT Enzymatic Activity
PDT activity in the conversion of prephenate to phenylpyruvate was measured using the method described by Dopheide et al. (28). The time course and prephenate concentration dependence of Sa-PDT is shown in Figure 1B. The data appears to follow the quasi steady state approximation with a single binding site. The enzyme is quite active but its affinity for prephenate is rather low. At tested concentrations, no product inhibition was observed. The apparent Michaelis-Menten Km for prephenate is 0.71 ± 0.09 mM and Vmax is 0.13 ± 0.04 mM/min (Fig. 1B). In our assay the Sa-PDT shows activity of 1138 units/mg enzyme. These values are similar to that obtained for other PDTs (Dopheide et al., 1972; Prakash et al., 2005). The activity of Ct-PDT is strongly temperature dependent. The enzyme is inactive at room temperature and shows the highest activity at 50°C, which is consistent with the observation that C. tepidum TLS grows optimally at 48°C. Even at 50°C, a higher concentration of both enzyme and substrate were used to measure time course and kinetics. Our preliminary data suggests that Ct-PDT is approximately 20 times slower than Sa-PDT.
Structure Determination
The crystal structures of both Sa-PDT and Ct-PDT were solved using the multiple-wavelength anomalous diffraction (MAD) technique using X-ray diffraction data collected from a single SeMet-labeled protein crystal (Table 1). After refinement, the final R factors (R/Rfree) were 0.246/0.289 for Sa-PDT and 0.246/0.289 for Ct-PDT, respectively (Table 1). The Ramachandran plot analysis showed that most of the residues (87.9% of Sa-PDT and 85.7% for Ct-PDT) are within the most favored region. Other residues lie within additional (11.5% of Sa-PDT and 13.9% of Ct-PDT) and generously allowed (0.7% of Sa-PDT and 0.4% of Ct-PDT) regions. No residues fell into the disallowed region.
Table 1.
Crystallographic Statistics
| Data collection | Sa-PDT | Ct-PDT |
|---|---|---|
| Space group | P21212 | P21212 |
| Unit Cell (Å, °) | a=60.86, b=87.41, c=107.74 | a=86.87, b=127.89, c=56.60 |
| MW Da (residue) | 29473.411 (264) | 31099.37 1 (280) |
| Mol (AU) | 2 | 2 |
| SeMet (AU) | 10 | 8 |
| Wavelength(Å) | 0.97929(peak), | 0.97918(peak), 0.97932(infl.) |
| Resolution(Å) | 46–2.3 | 41–2.3 |
| Number of unique reflections | 25930 2 | 237982 |
| Redundancy | 11.1 | 6.4 |
| Completeness (%) | 98.4(97.8) 3 | 82.8(52.6) 3,4 |
| Wilson B factor (Å2) | 51.2 | 38.4 |
| Rmerge (%) | 6.8(43.9) 3 | 7.2(25.2) 3 |
| I/σ(I) | 56.5(5.5) 3 | 35.5(5.8) 3 |
| Phasing | ||
| RCullis (anomalous) (%) | 69 (peak), 82 (Inflec.) | 75 (peak), 79 (Inflec.) |
| Figure of merit (%) | 26.2 | 29.0 |
| Refinement | ||
| Resolution | 46–2.3 | 41–2.3 |
| Reflections (work/test) | 24598/1291 | 22409/1347 |
| Rcrystal/Rfree (%) | 24.8/28.9 | 21.7/28.5 |
| Rms deviation from ideal geometry Bond length (Å)/angle (°) | 0.023/2.2 | 0.023/2.3 |
| No.of atoms (Protein/HETATM) | 4042/119 | 4364/114 |
| Mean B-value (Å2) (mainchain/sidechain) | 63.73/64.63 | 51.95/61.32 |
| Ramachandran plot statistic (%) Residues in most favored regions Residues in additional allowed regions Residues in generously allowed regions |
87.4 11.9 0.7 |
89.2 10.2 0.6 |
Not including cloning artifact;
Including Bijvoet pairs;
(Last resolution bin, 2.30–2.35 Å);
Crystal diffracted anisotropically (see text)
The Structure of R-state Sa-PDT
Each Sa-PDT consists of an N-terminal PDT domain (residues 1–176) and a smaller C-terminal ACT domain (residues 185–264) (Fig. 3A, B). There are two monomers related by a pseudo 2-fold symmetry in the asymmetric unit. The C-terminal ACT domains of two monomers contact each other with their helices packed in parallel, forming a small hydrophobic interface (buried surface, ~711 Å2) (Fig. 3A).
Figure 3.

The structure of Sa-PDT. (A) Ribbon drawing of the two PDT monomers in one asymmetric unit. Two monomers are related by a pseudo 2-fold symmetry. The linkers between two PDT subdomains are highlighted in green and the cleft between two PDT subdomains of each monomer is marked. In the monomer B (on left side) some loops are missing due to weak electron densities. (B) The scheme of the PDT structure. (C) Structural details of the PDT active site with some conserved residues drawn in ball-and-stick form. Figure 2A and C and other ribbon diagrams in the paper were prepared using the program MOLSCRIPT (Kraulis, 1991).
The larger N-terminal PDT domain is formed by two structurally similar subdomains, PDTa (residues 1–81 and 168–176 of Sa-PDT) and PDTb (residues 85–161 of Sa-PDT) (Fig. 3A, B). The PDTb is inserted within the PDTa via two linkers (residues 82–84 and 162–167). Each of these two subdomains consists of a central β-sheet with four parallel strands and one anti-parallel strand. The β-sheet is sandwiched by three α-helices, one on one side and two on the other. The primary sequences of the two PDT subdomains show less than 10% sequence identity. However, their structures are very similar. They can be superimposed with an rmsd (root mean square deviation) value of 1.68 Å, in which 62 residues out of each subdomain are aligned. Between the two PDT subdomains there is a cleft, which is defined by the loops from PDTa (β1_α1, β2_α2, and β3_α3) and PDTb (β6_α4a, β7_α5, and β8_α6) as well as two linkers between them (Fig. 3C). The solvent accessible cleft hosts the PDT active site as discussed below.
The C-terminal ACT domain of PDT also has an α/β fold with four anti-parallel β-strands and two α-helices on one side of the β-sheet (Fig. 3A, B). The ACT domain appears to be more conserved than PDT domains across different species. The notable feature of the domain in the PDT structure is that it is closely associated with the PDT domain with its β-sheet forming extensive interactions with the PDTa subdomain. In the Sa-PDT the inter-domain interactions include two salt bridges (D66/R220 and D66/K223), three hydrogen bonds (Q70/R230, E78/Q234 and R169/Q234) and multiple hydrophobic contacts as well as some solvent-mediated hydrogen bonds. The salt bridges and two of the hydrogen bonds are highly conserved (Fig. 2). Additionally, there seems to be a well-conserved bulge (Ile260) on the edge of strand β4 of the ACT domain. The strand after the bulge is almost parallel to the edge of strand β4 of the PDTa domain with two main chain hydrogen bonds. The eight residue linker (Gln176-Ala183) between ACT and PDT is also well structured with interactions to both domains.
Based on molecular packing and interactions across monomer/monomer interfaces, the two monomers related by crystallographic 2-fold form a dimer (Fig. 4A). In the dimer, the total buried surface contributed from two monomers is about 3249 Å2, a value much larger than a "typical" protein interface in which the total area buried by the components in the recognition surface is 1600 (±400) Å2 (Lo Conte et al., 1999). Both PDT and ACT domains contribute to the interactions across the dimerization interface, including 16 hydrogen bonds but no salt bridges. The residues involved in the hydrogen bonds are E36, K39, E54, N55, N62, Y122, Y124, H165, N205, R220, T224 and Y229. Other residues including F35, I63, I108, L200, I217 and K223 contribute to hydrophobic interactions across the dimer interface. A PDT dimer can be treated as a combination of a PDT domain dimer and an ACT domain dimer (Fig. 4A). The buried molecular surface from the PDT dimer is about 1447 Å2 while the value from the ACT dimer is about 637 Å2.
Figure 4.

PDT tetramers, (A) Ribbon drawings of a Sa-PDT tetramer. The right side view is related to the left side view by a 90º rotation around the horizontal axis. The tetramer was generated from the two PDT monomers in an asymmetric unit as shown in Figure 3A and their 2-fold symmetry related molecules. The tetramer is a dimer of dimer formed from two symmetric dimers, the AA dimer on the right side and the BB dimer on the left side. Some loops in the BB dimer are missing due to weak electron densities. The residue T168 from the catalytic triplet (TRF) and the residue F231 critical to L-Phe binding were drawn in ball-and-stick form on the right side, the AA dimer shows the proposed catalytic sites and the potential L-Phe binding sites, respectively. There are four equivalent catalytic sites and four L-Phe binding sites in one tetramer. (B) Ribbon drawings of a Ct-PDT/L-Phe tetramer. The right side view is related to the left side view by a 90º rotation around the horizontal axis. The tetramer was generated from a pseudo 2-fold PDT dimer found in an asymmetric unit (AB dimer, on the right-side) and its symmetric 2-fold dimer (A’B’ dimer, on the left side). The bound L-Phe amino acid, the residue T171 of TRF triplet and the residue F231 within L-Phe binding pocket were drawn in ball-and-stick form to show their positions on the right-side dimer. Both PDT tetermers are expected to be the same in solution, in a symmetry of a 222 point group.
The formation of a PDT dimer aligns two ACT domains, forming an anti-parallel helical array packed onto two parallel β-sheets (Fig. 4). This type of ACT dimer is common in the ACT containing enzyme structures (Kaplun et al., 2006; Kotaka et al., 2006; Schuller et al., 1995). We noticed that the ACT dimers available at the PDB were generally in a closed conformation, in which two ACT domains are arranged to form an anti-parallel 4-helix array packed onto an anti-parallel 8-strand super β-sheet. However, in Sa-PDT, the ACT dimer is an open conformation, in which there is no direct interaction between the two β-sheets of the two ACT domains (Fig. 5A). The open conformation creates solvent accessible cavities at the interface between the two ACT domains (Fig. 5B).
Figure 5.
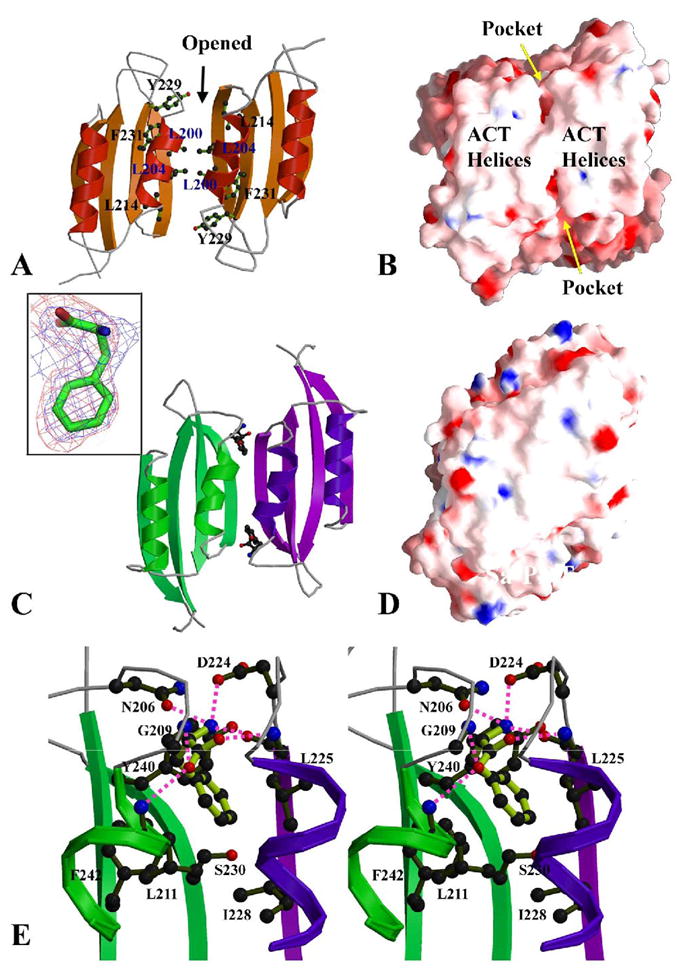
ACT dimer and L-Phe binding sites. (A) The ACT dimer of Sa-PDT in an open conformation. The L-Phe binding pockets are located at the dimer interface. There is no interaction between the two β-sheets of the ACT domains and it creates two L-Phe binding pockets at the interface of the two ACT domains. Some residues contributing these pockets are drawn in ball-and-stick form. (B) The electrostatic potential surface representations of the Sa-PDT ACT dimer, showing the helical array on the ACT domains. The openings of two symmetric pockets for postential L-Phe binding are indicated. (C) The ACT dimer of Ct-PDT, showing a closed conformation with two L-Phe amino acids bound. The two 4-strand β-sheets are closed up to form an 8-strand super β-sheet. The two L-Phe effectors reside in the pockets between the helical array and the 8-strand super β-sheet. The insert shows one L-Phe in a 2Fo-Fc omit map (blue) contoured at a 1.0σ level and a Fo-Fc omit map (red) contoured at a 2.0σ level. (D) The electrostatic potential surface representation of the Ct-PDT ACT dimer, showing no openings behind the helical array on the ACT domains. The residues R221 and E264 that form a total of four salt-bridges between two dimers are labeled. (E) The stereo-view of one L-Phe binding site. The residues directly involved in the interactions to the L-Phe are drawn in ball-and-stick form. The interactions are both from ACT domains of the ACT dimer, colored in green and purple, respectively. Hydrogen bonds to L-Phe are drawn in magenta dash lines. Figures 5B and D and other electrostatic potential surface representation figures in the paper were prepared using the program GRASP (Nicholls et al., 1991). The insert of Figure 5C was prepared with the program PyMOL (http://www.pymoL.org).
The formation of a PDT dimer also seems to be the pre-condition to the formation of the PDT tetramer. The surface of the helical array of the ACT dimer is rather hydrophobic (Fig. 5B), containing residues of L209, F210, K245, I249 and L253 from each monomer. With the helical arrays of their ACT domain dimers packed against each other, two PDT dimers form a tetramer (Fig. 4A), a dimer of dimers. The tetramerization creates a cluster of eight helices between two PDT dimers resulting in a total buried surface of about 1700 Å2. Since most of the interactions across the dimer-dimer interface are hydrophobic, we expect the PDT tetramer with approximately 222 point group symmetry to be a rather stable tetramer in the solution, consistent with size-exclusion chromatography results mentioned earlier.
The Structure of T-state Ct-PDT
Similarly, Ct-PDT consists of a PDT domain (residues 1–178) and an ACT domain (residues 191–280) (Fig. 3A, B). There are also two monomers in the asymmetric unit but related by a different symmetry operator, resulting in a pseudo 2-fold dimer. This dimer is similar to the symmetric dimer found in Sa-PDT with its totally buried surface area of 3649Å2 (Fig. 4B), however Ct-PDT is in closed conformation.
The structure of Ct-PDT contains L-Phe bound to ACT domains (Fig. 5C). The electron density for the ligand is very clear and unambiguous. This suggests that the L-Phe has been trapped by Ct-PDT during protein expression in E. coli and remained associated during purification and crystallization. Although Sa-PDT and Ct-PDT have a low primary sequence identity (27%), their structures and quaternary organization are the same. Their individual PDT and ACT domains can be aligned with rmsd values of 1.75Å (162 aligned out of 176/177 a.a. with sequence identity of 25.9%) and 1.35 Å (78 aligned out of 81/81 a.a. with sequence identity of 24.3%), respectively. Similarly, their PDT subdomains can be aligned with rmsd values of 1.54 Å and 1.64 Å for PDTa (80 aligned out of 88/92 a.a. with sequence identity of 30.0%) and PDTb (75 aligned out of 81/76 a.a. with sequence identity of 24.0%), respectively. However, compared to Sa-PDT the Ct-PDT showed considerable conformational changes across the PDT dimer interface as well as changes resulting from domain-domain movement, especially between PDT and ACT domains as discussed later.
In contrast to the open conformation of the ACT dimer observed in Sa-PDT (Fig. 5A, B), the ACT domain dimer of Ct-PDT is in a closed conformation (Fig. 5C, D), in which the two 4-strand β-sheets from the two ACT domains close up the opening between them and merge into an 8-strand super β-sheet. At the ACT dimer interface, where two bound L-Phe amino acids were located in the two predominantly hydrophobic pockets in a head-in-first mode (Fig. 5C, E). The benzene ring of each L-Phe interacts with L225 and L228 from one ACT domain, and L211, S230, Y240 and F242 from another ACT domain (Fig. 5E). The residues that form a hydrophobic pocket are either identical or highly conserved across different species in the PTD family. The side chain of residue S230 at the bottom of the pocket forms a hydrogen bond to the main chain amino group (I228) of the other monomer, contributing to the formation of the closed conformation of the ACT domain dimer (Fig. 5E).
The α-carboxyl group of the amino acid forms three non-specific hydrogen bonds to main chain amide groups, one (to L225) from one ACT domain and two (to G209 and L211) from the second ACT (Fig. 5E). It also forms a specific hydrogen bond to the residue D224 from the first ACT. The α-amino group of the L-Phe forms three hydrogen bonds to two ACT domains, one non-specific to the main carboxyl group of L225 of the first ACT, two specific to D224 from the first ACT and N206 from the second ACT (Fig. 5E). The residues N206 and N224 are well conserved in different PDTs (Fig. 2).
The pseudo-symmetric Ct-PDT dimer in the asymmetric unit also forms a tetramer that has a smaller surface area of 1513 Å2 (Fig. 4B). The overall organization of the tetramer is just like the Sa-PDT tetramer (Fig. 4A). Across the Ct-PDT tetramer interface, in addition to hydrophobic interactions, there are also four salt bridges between R221 and E264 (Fig. 5D), and a few hydrogen bonds involving R213 and N261. These interactions seem to be unique to Ct-PDT (Fig. 2).
DISCUSSION
The Sa-PDT and Ct-PDT structures described above represent the PDT in its active R-state and L-Phe inhibited T-state. Both enzymes convert prephenate to L-Phe, although they show different temperature optima and activity. The Sa-PDT structure corresponds to an open active site conformation and the Ct-PDT structure with bound allosteric inhibitor corresponds to the closed active site state. Ascertaining the structural relationship between the catalytic and allosteric sites is of vital importance in understanding how chemical information is transmitted from the effector site to the active site.
The PDT active site
Based on the two PDT structures described earlier, we propose that the PDT active site is located in the cleft between two PDT subdomains (Fig. 3), the structural data are supported by sequence analysis and mutagenesis data. A multiple sequence alignment of PDTs map the majority of highly conserved residues to subdomain PDTa, and some to PDTb (Fig. 2). The highly conserved T168, R169, F170 and P52 of Sa-PDT lie at the bottom of the cleft between PDTa and b (Fig. 3C). The side chains of T168 and F170 reside in the cleft and are both solvent accessible. The T168 hydrogen bonds to an ethylene glycol molecule (not shown in the figure), which shows solvent accessibility of the highly conserved residues in the active site. The charged side chain of R169 points away from the cleft bottom, forming multiple salt bridges with two other highly conserved glutamic acids, E54 and E78 on the back of the cleft. The R169 also forms a hydrogen bond with the residue Q234 from the ACT domain (Fig. 3C). In the latter position Q, E or D are found, suggesting that the ability to make this hydrogen bond is conserved in the PDT family (Fig. 2). In addition, three highly conserved residues N55, Q110 and N166, form a hydrogen bond bridge between two PDT subdomains (Fig. 3C). The two highly conserved glycines, G7 and G59 are in the loops (β1-α1 loop and β3-α3 loop, respectively), which form the wall of the cleft on the PDTa subdomain side. These glycines may provide some flexibility allowing adjustment of residue conformation in the cleft during ligand binding. It is remarkable that all N-termini of α-helices (α1 -β6) of the PDT domain point into this cleft filled with several water molecules, providing the equivalent of ~3.0 positive charges (Hol et al., 1978). This may be an important component of the environment for substrate binding since prephenate has two carboxylates and is negatively charged at neutral pH.
The site-directed mutagenesis of E. coli P-protein PDT domain revealed that the residues T278, N160, Q215, and S208 are critical for PDT activity (Zhang et al., 2000). They are equivalent to T168, N55, Q110 and S103 in Sa-PDT (Fig. 2). All of these residues are associated with the cleft between the two PDT subdomains. Similar mutagenesis data for Corynebacterium glutamicum PDT (Cg-PDT) confirmed the role of T168, R169 and F170 equivalent residues in Cg-PDT in substrate binding and/or catalytic activity (Hsu et al., 2004). The residue T168 was also considered to be important in PDT catalytic activity in other early studies (Davidson et al., 1972; Jetten and Sinskey, 1995). All these data suggested that the cleft and associated conserved residues form the active site of prephenate dehydratase with T168 being perhaps the key catalytic residue.
Among highly conserved residues in the PDT domain, G7, D66 and L68 are the only ones that do not directly contribute to the active site. G7 is near the entrance to the cleft. D66 forms a bifurcated salt bridge with a highly conserved R220 and a partially conserved K223 from the C-terminal regulatory ACT domain while L68 is involved in hydrophobic interactions with the ACT domain. Therefore, most of the highly conserved residues in the PDT domain are either related to the catalytic cleft or involved in the interaction with the ACT domain as discussed below.
A structural similarity search of the PDT domain of Sa-PDT using the DALI server (Holm and Sander, 1995) resulted in some structural folds that generally consist of two similar α/β subdomains with one inserted within the other. The top hits include: i) the regulatory domain of LysR-type transcriptional regulator from Ralstonia eutropha (PDB id: 1IZ1, Z score: 7.9, rmsd: 4.1, aligned residues: 145, sequence identity: 8%) (Muraoka et al., 2003), ii) the catalytic domain of ATP phosphoribosyltransferase from M. tuberculosis (1NH7, 7.4, 8.0, 116, 7%) (Cho et al., 2003), iii) the catalytic domain of porphobilinogen deaminase from E. coli (1PDA, 7.3, 4.0, 149, 8%) (Louie et al., 1992), iv) the periplasmic Lysine/Argininine/Ornithine-binding protein from Salmonella typhimurium (1LST, 7.0, 4.3, 122, 10%) (Oh et al., 1993) and v) the ligand-binding core of glutamate receptor ion channel from Synechocystis (1II5, 7.0, 4.4, 135, 10%) (Mayer et al., 2001). Together with the PDT domain of Sa-PDT and Ct-PDT, these proteins show very similar overall fold and subdomain organization with very little primary sequence identity. At the same time they all have the ligand binding site residing in the cleft between two subdomains. The relatively low scores in their overall structural alignments with the Sa-PDT domain usually resulted from the variation of the relative orientation of the two subdomains. However, these proteins show highly diversified biochemical functions involved in very different cellular processes, underscoring the ability of this fold to be tailored to many biochemical or cellular functions.
The location of the L-Phe binding site in PDT is similar to the effectors binding sites observed in several other ACT domain containing enzymes that are involved in binding amino acids or other small ligands. They allosterically regulate the activity of the associated catalytic domain (Aravind and Koonin, 1999; Chipman and Shaanan, 2001; Liberles et al., 2005) through the feedback mechanism. A structural similarity search of the ACT domain of Sa-PDT using the DALI server (Holm and Sander, 1995) found similar ACT domains in several different proteins with higher Z-scores and lower rmsd values as compared to the PDT domain homologue search. The top hits include enzymes involved in amino acid metabolism (Z score: 8.2–10.0, rmsd: 1.9–3.0, aligned residues: 71–80, sequence identity: 5–18%). For example, in the structure of E. coli D-3-phosphoglycerate dehydrogenase (3PDGH), an enzyme that catalyzes the first step of L-Ser biosynthesis, the effector L-Ser binds at the interface of two ACT domains (ACT dimer) of a 3PDGH tetramer (Schuller et al., 1995). In the structure of the E. coli acetohydroxyacid synthase (AHAS) regulatory subunit, a similar effector binding site at the interface of the ACT dimer of AHAS was also proposed for valine, an inhibitor of the enzyme that catalyzes the first common step in the biosynthesis of the three branched-chain amino acids (Kaplun et al., 2006). Moreover, recently reported crystal structures of E. coli aspartokinase III (AKIII) provide more specific information about effector binding sites (Kotaka et al., 2006). AKIII catalyzes an initial commitment step in the aspartate pathway and is allosterically inhibited by lysine. The structure of AKIII has two ACT domains at its C-terminus and the dimerization of AKIII forms two back-to-back ACT dimers. In the structure of lysine-sensitive AKIII two lysine molecules are bound in the pockets formed at the ACT dimeric interface near the N-termini of two central α-helices (Kotaka et al., 2006). These structures may correspond to the closed state of Ct-PDT. However, since the catalytic domains of these enzymes are very different, it is difficult to generalize how the ACT domains regulate enzymatic function.
The analysis of L-Phe interaction with ACT domains in the structure Ct-PDT indicates that most interactions are likely to be non-specific. This explains why other amino acids such as Met can also bind inside the pocket, regulating the catalytic activities of this enzyme. The second feature of the L-Phe binding is that the amino acid simultaneously binds to two ACT domains at the ACT dimer interface. The two bound L-Phe, at two nearly symmetric positions across the ACT dimer interface, seem to pull two 4-strand β-sheets closer to form an 8-strand super β-sheet, resulting in a closed conformation of the ACT dimer (Fig. 5C, E).
As discussed earlier, the residues involved in L-Phe binding are highly conserved across different species. Therefore, L-Phe binding mode unveiled in this study is also likely conserved. Large hydrophobic L-amino acid (like L-Met) should also be able to create very similar interactions. Site-directed mutagenesis and isothermal titration microcalorimetry studies of E. coli P-protein suggested that the two highly conserved regions in the ACT domain, corresponding to sequence motifs GALV and ESRP affected L-Phe binding and feedback inhibition (Pohnert et al., 1999). The GALV and ESRP equivalent sequences in Sa-PDT (G198LLV and E218SRP) or Ct-PDT (G209SLF and E229SRP) are located on the interface between two ACT domains. Some of these residues (L211 and S230 of Ct-PDT) are directly involved in the interaction with L-Phe. The residue S230 also contributes to the formation of closed conformation of the ACT dimer as discussed earlier. In the study of deregulated, feedback-control-resistant Amycolatopsis methanolica PDT (Am-PDT) mutants that completely lost PDT sensitivity to L-Phe inhibition and L-Tyr activation, several residues were identified: A125, A198, N200, L209 and H227 in Am-PDT (corresponding to Y124, I190, D195, L204 and L222 in Sa-PDT, and A123, L204, N206, L215 and S233 in Ct-PDT) (Fig. 2) (Kloosterman et al., 2003). N200 of Am-PDT, like N206 of Ct-PDT, forms a hydrogen bond to the α-amino group of the L-Phe. A198 of Am-PDT is close to the hydrophobic portion of the L-Phe binding pocket. The highly conserved L209 (Am-PDT) contributes to the helix-helix interactions between the two ACT domains in the PDT dimer.
The L-Phe binding site in PDT is at the dimer interface and the PDT dimer seems to be critical for the function of the enzyme. Comparison of Sa-PDT and Ct-PDT structures show how binding of the L-Phe effector changes ATC dimer conformation. The question is how the changes in the ACT dimer are propagated to the active site and block PDT enzymatic activity. In each PDT structure, the PDT dimer remarkably aligns and connects two clefts containing catalytic residues, forming an extended active site (Fig. 6A, B). In the active Sa-PDT, it creates a large cavity with two T168 from opposite PDTa domains at the bottom of the cavity separated by ~21 Å (Fig. 6B). Interestingly, there is a ridge formed by two β3_α3 loops within the cavity (Fig. 6B), suggesting that the two catalytic sites may communicate during the reaction. In the dimer two D126 residues partially block the opening to the central part of the cavity. Right below the D126 residues there are two E58 from the ridge, mentioned earlier, contributing negative electrostatic potential and making the entrance to the active site negatively charged (Fig. 6A). The substrate prephenate has two carboxylates and is also negatively charged and must overcome the repulsing effect of the D126/E58 double pair. Early mutational data has shown that the substitution of the D126 equivalent, the residue E232 to Ala in the PDT domain of E. coli P-protein increased enzyme activity 3.5 times (Zhang et al., 2000). It is therefore suggestive that the decrease in the size of the amino acid side chain and the elimination of the negative charge makes access to the active site cavity easier for the substrate. In the cavity, aside from T168 and F170 discussed earlier, there are also additional residues that are solvent accessible. These residues are mostly hydrophobic and they include P52, I83, F85, I104, P106 and P144 (Fig. 6B). A cluster of such amino acids may help to orient the substrate in the active site.
Figure 6.
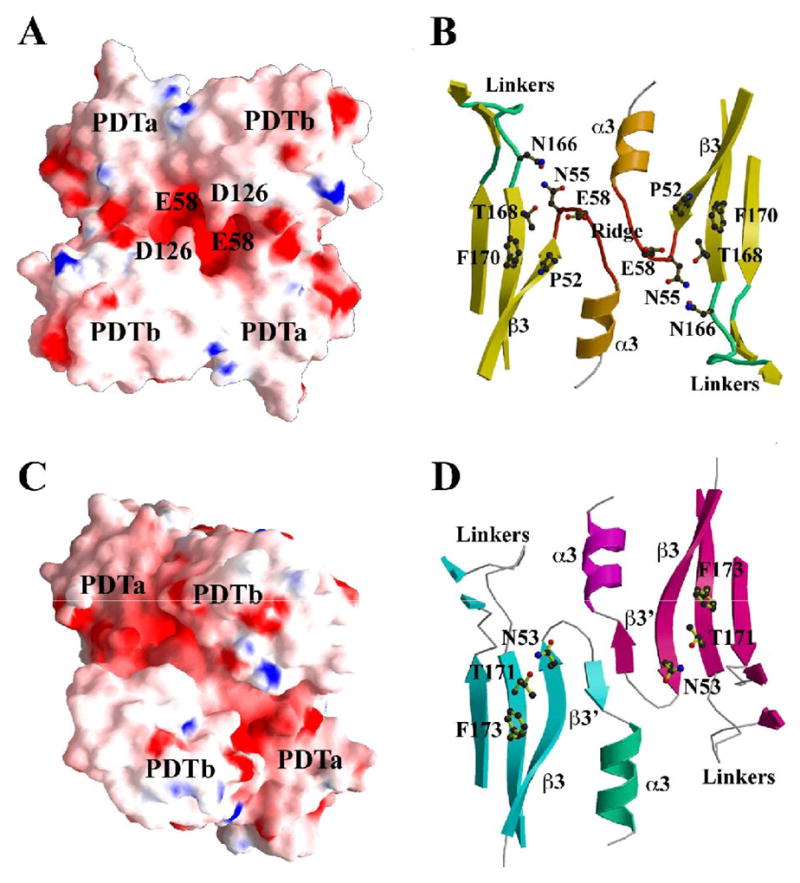
PDT domain dimer and catalytic sites (A) The electrostatic potential surface representations of a PDT domain dimer of Sa-PDT, showing the central opening to the extended catalytic site. The residue E58 under the opening and the residue D126 at the entrance of the opening are labeled. Figure 6A and Figure 5B are related by approximately 180º rotation. (B) The extended catalytic site of the Sa-PDT domain dimer. The dimer integrates two individual catalytic sites represented by the residues T168 and F170 from TRF triplet. Between two active sites, there is a ridge formed by two β3-α3 loops. The residues N55 and N166 form a hydrogen bond. The linkers between the two PDT subdomains were drawn in green. (C) The electrostatic potential surface representations of the PDT domain dimer of Ct-PDT. Figure 6C and Figure 5D are related by approximately 180º rotation. The two PDTb subdomains push toward each other, closing up the central opening (Figure 6A) while the two PDTa subdomains pull away from each other. (D) The extended catalytic site of the Ct-PDT. It shows the flattening of the ridge between two active sites observed in a native Sa-PDT structure (Figure 6B). The β3-α3 loop in Sa-PDT forms a short strand (β3’). The two active sites represented by the residues T171 and F173 that form the catalytic triplet (TRF) are widely open to each other. The highly conserved residue N53 moves its sidechain upward, forming a hydrogen bond with the catalytic residue T171.
Comparison of the PDT active site in open and closed (inhibited) states can reveal how chemical information is transmitted from the effector site to the active site. The bound L-Phe is ~21 Å away from the important catalytic residue T171 and cannot influence the active site directly. However, binding L-Phe introduces a number of major conformational changes in PDT both on a global and local scale that change relative orientation of domains and change the access to the active site. The residues contributing to the extended active site across the PDT dimer interface, including β3_α3 loop, β3 strand and α3 helix from each PDT are the most conserved in the PDTa subdomain (Fig. 2). Remarkably, it is in this very region where one of the most significant structural changes is observed upon L-Phe binding. The ridge formed by two β3_α3 loops found in Sa-PDT is flattened to a strand-like motif and the two catalytic sites are widely open to each other in Sa-PDT (Fig. 6D). Accompanying this change, the highly conserved residue N55 of Sa-PDT (N53 of Ct-PDT), which forms a conserved hydrogen bond to R220 from the ATC domain in the opposite chain in Sa-PDT (Fig. 6B), turns around and forms a hydrogen bond with N169 and T171 in Ct-PDT (Fig. 6D). The T171 is one of the amino acids from the catalytic TRF triplet. This may be the key change that locally affects catalytic properties of PDT and results in the inhibition of PDT.
Based on our data we propose a mechanism for the allosteric regulation of PDT by L-Phe (Fig. 7). The PDT is composed of an ACT regulatory dimer and PDT catalytic domain dimer, with extensive interactions across ACT/PDT domains. Upon two L-Phe residues binding at the ACT dimer interface, the domain dimer rearranges from an open (R) to a closed (T) conformation. These conformational changes are propagated through ACT/PDT interface to the PDT catalytic domain with global and local consequences. The two PDTb subdomains push toward each other while two PDTa subdomains move away from each other. The PDT subdomains movement may be simplified as a relative rotation of the PDT domain dimer onto the ACT dimer (Fig. 7). The movement results in a split of the large opening to the PDT extended catalytic site at the center of the PDT dimer (Fig. 6A) into two smaller openings (Fig. 6C). It is therefore suggestive that the splitting of the cavity results in a reduction of the access of prephenate and the release of phenylpyruvate to and from the catalytic sites. On the local level, side chains in the active site rearrange, potentially impacting the ability to carry out catalytic function. Since both the allosteric site and the catalytic site are associated with the PDT dimer interface, the PDT dimer seems to be a basic biological unit of PDT. However, the significance of the PDT tetramer found in both structures and solution needs further investigation.
Figure 7.
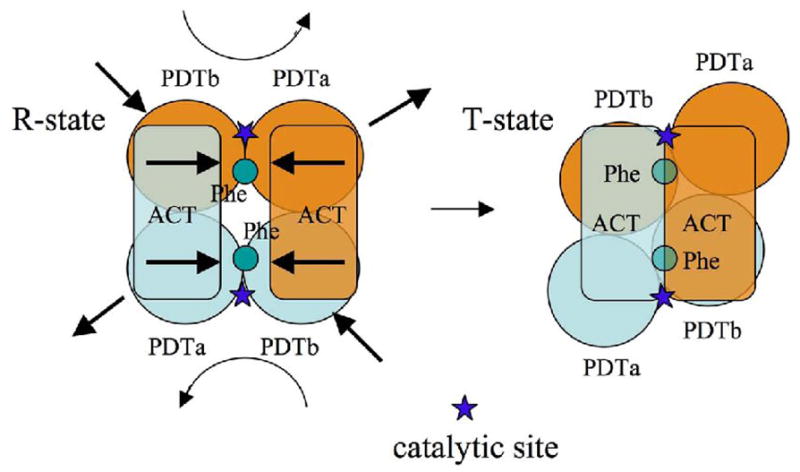
A schematic drawing of the allosteric regulation mechanism of PDT by its effector and the conformational changes to PDT domains.
EXPERIMENTAL PROCEDURES
Preparation of Recombinant PDT proteins
Both Sa-PDT and Ct-PDT genes were cloned in the pMCSG7 vector (Stols et al., 2002) and overexpressed in E. coli BL21 (DE3). The cells were grown at 37°C in seleno-methionine (SeMet) containing enriched M9 medium. The initial L-Phe concentration in the medium was about 1.2 mM. The protein expression was induced with 1 mM IPTG. After induction, the cells were incubated at 20°C overnight. The cells were then harvested, resuspended in 5 volumes of lysis buffer (50 mM HEPES pH 8.0, 500 mM NaCl, 10 mM imidazole, 10 mM β-mercaptoethanol, and 5% v/v glycerol), and frozen at −20°C. The frozen cells were thawed and lysed by sonication after the addition of protease inhibitors (Sigma, P8849) and lysozyme at 1 mg/ml. The lysate was purified by centrifugation for 20 minutes, followed by filtration through 0.45 μm and 0.22 μm in-line filters (Gelman).
The immobilized metal affinity chromatography was performed for purification according to the standard protocol described previously (Kim et al., 2004). His6-tag was cleaved using the recombinant TEV protease expressed from the vector pRK508 (Kapust and Waugh, 2000) (a gift from Dr. D. Waugh, NCI). The protease was added to the target protein in a ratio of 1:30 and the solution was incubated at 4°C for 72 hours. The proteins with His6-tag removed were then purified using a 3 ml Ni-NTA chelating column (Qiagen). Each recombinant protein includes the vector-derived sequence SNA at the N-terminus. The protein buffer was exchanged to crystallization buffer (20 mM HEPES pH 8.0, 250 mM NaCl, and 2 mM dithiothreitol (DTT)) during concentration.
Protein Crystallization
The PDT proteins (1 mM) were screened for crystallization conditions with the help of the Mosquito robot (TTP Labtech) using the sitting drop vapor diffusion technique in a 96-well CrystalQuick plate (Greiner). Initial screening of Sa-PDT only yielded a few small crystals. Therefore, the conditions were modified using the hanging drop vapor diffusion technique to produce crystals suitable for X-ray diffraction data collection. The data were collected from crystals grown at 16°C in the buffer including 3 M sodium chloride, 0.1 M sodium citrate, pH 5.0. Prior to data collection, crystals were treated with a cryoprotectant complemented from the crystallization buffer with the addition of 25% (v/v) glycerol. In the presence of 5 mM L-Phe, Sa-PDT failed to crystallize under the same crystallization conditions. Ct-PDT crystallized in the initial screen under the condition of 30% (v/v) PEG400, 0.1 M acetate, 0.2 M calcium acetate, and pH 4.5. Relatively smaller Ct-PDT crystals also formed with additional 5 mM L-Phe under the same conditions. The cryoprotectant buffer for the Ct-PDT crystals included 10% (v/v) additional glycerol.
Diffraction Data Collection
Diffraction data were collected at 100 K at the 19ID beamline of the Structural Biology Center at the Advanced Photon Source at Argonne National Laboratory using the program SBCcollect. Multiple-wavelength anomalous diffraction (MAD) data sets were collected at the wavelengths near selenium absorption peaks and inflection points from SeMet labeled Sa-PDT and Ct-PDT protein crystals (Table 1). Ct-PDT crystals diffracted anisotropically with resolution limits better than 2.1Å while completeness was better than 95% at 2.9Å. The data set was cut off at 2.3Å with completeness of 53% and I/σ of about 5.8. All diffraction data were processed and scaled with HKL3000 suite (Table 1) (Minor et al., 2006).
Structure Determination and Refinement
Both Sa-PDT and Ct-PDT crystal structures were determined using MAD phasing. Se sites were located using the program SHELXD (Schneider and Sheldrick, 2002) and they were used for phasing with the program MLPHARE (1994). After density modification (DM) (1994), partial models of 206 residues (Sa-PDT) or 338 residues (Ct-PDT) without side chains were obtained from automatic model building trials using the program RESOLVE (Terwilliger, 2003). All of the above programs are integrated within the program suite HKL-3000 (Minor et al., 2006). From the initial partial models, each PDT structure was manually built using the program COOT (Emsley and Cowtan, 2004). After cycles of model building, the structures were refined using the program REFMAC (Brèunger et al., 1998) (Table 1). It is worthwhile to mention that in the case of Sa-PDT, the electron densities for the PDT domain of one monomer were rather weak and had breaks at several loop regions, implying some partial disordering. In the case of Ct-PDT, the densities for two bound L-Phe molecules were well defined even in the maps calculated from initial phases.
Determination of PDT Oligomeric State
Size exclusion chromatography was performed on a Superdex-200 26/60 column (Amersham Pharmacia Biotech). The column was pre-equilibrated with a buffer (20 mM HEPES pH 8.0, 250 mM NaCl, 2 mM DTT) and calibrated with pre-mixed protein standards (chymotrypsinogen A (25 kDa), albumin (67 kDa), aldolase (158 kDa), and Blue Dextran (2,000 kDa)). The chromatography was carried out at 4°C at a flow rate of 2 mL/min. The calibration curve of Kav versus log molecular weight was prepared using the equation , where Ve = elution volume for the protein, Vo = column void volume, and Vt = total bed volume.
PDT Activity Assay
The SA-PDT activity was determined by the conversion of prephenate to phenylpyruvate according to the method described previously (Dopheide et al., 1972). The 1 ml reaction mixture containing 20 mM HEPES (pH 8.0), 250 mM NaCl, 0.2 mM prephenate, 0.04 μM enzyme was incubated at room temperature for 1 to 60 min. The reaction was stopped by removing 50 μl of reaction mix into 200 μl 2N NaOH at each time period. The absorbance was measured at 320 nm. The assay was linear over a 3-min time period. One unit of activity was defined as the amount of substrate (in μM) converted by 1 μM of PDT per min (sodium phenypyruvate was used as the standard) under the assay conditions. The Km and reaction rate were determined using varying amounts of substrate (0.2 mM to 16 mM) under the same assay conditions for 3 min. The apparent Km and Vmax were calculated from the rate of phenylpyruvate production using Lineweaver-Burk analysis (Fersht, 1985). The data were fitted with the software PRISM (Prizm Software, Irvine, CA). The Ct-PDT activity was analyzed using a similar assay. After initial detection of little activity at room temperature, the reaction mixture was incubated at 50ºC and an elevated concentration of both the enzyme and substrate were used.
Acknowledgments
The authors wish to thank members of Structural Biology Center at Argonne National Laboratory for their help with data collection at the 19ID beamline and Andrea Cipriani for help in preparation of this manuscript. This work was supported by National Institutes of Health grant GM074942 and by the U. S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357. The coordinates of Sa-PDT and Ct-PDT/L-Phe have been deposited in Protein Data Bank (PDB) under the accession codes 2QMW and 2QMX, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aravind L, Koonin EV. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. Journal of molecular biology. 1999;287:1023–1040. doi: 10.1006/jmbi.1999.2653. [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR. The Pfam protein families database. Nucleic acids research. 2004;32 :D138–141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley R. The shikimate pathway--a metabolic tree with many branches. Critical reviews in biochemistry and molecular biology. 1990;25:307–384. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- Bode R, Melo C, Birnbaum D. Absolute dependence of phenylalanine and tyrosine biosynthetic enzyme on tryptophan in Candida maltosa. Hoppe-Seyler's Zeitschrift fèur physiologische Chemie. 1984;365:799–803. doi: 10.1515/bchm2.1984.365.2.799. [DOI] [PubMed] [Google Scholar]
- Brèunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta crystallographica. Section D, Biological crystallography. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- CCP4. The CCP4 suite: programs for protein crystallography. Acta crystallographica. Section D, Biological crystallography. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Chipman DM, Shaanan B. The ACT domain family. Current opinion in structural biology. 2001;11:694–700. doi: 10.1016/s0959-440x(01)00272-x. [DOI] [PubMed] [Google Scholar]
- Cho Y, Sharma V, Sacchettini JC. Crystal structure of ATP phosphoribosyltransferase from Mycobacterium tuberculosis. The Journal of biological chemistry. 2003;278:8333–8339. doi: 10.1074/jbc.M212124200. [DOI] [PubMed] [Google Scholar]
- Chook YM, Gray JV, Ke H, Lipscomb WN. The monofunctional chorismate mutase from Bacillus subtilis. Structure determination of chorismate mutase and its complexes with a transition state analog and prephenate, and implications for the mechanism of the enzymatic reaction. Journal of molecular biology. 1994;240:476–500. doi: 10.1006/jmbi.1994.1462. [DOI] [PubMed] [Google Scholar]
- Cotton RG, Gibson F. The biosynthesis of phenylalanine and tyrosine; enzymes converting chorismic acid into prephenic acid and their relationships to prephenate dehydratase and prephenate dehydrogenase. Biochim Biophys Acta. 1965;100:76–88. doi: 10.1016/0304-4165(65)90429-0. [DOI] [PubMed] [Google Scholar]
- Davidson BE, Blackburn EH, Dopheide TA. Chorismate mutase-prephenate dehydratase from Escherichia coli K-12. I. Purification, molecular weight, and amino acid composition. The Journal of biological chemistry. 1972;247:4441–4446. [PubMed] [Google Scholar]
- Dopheide TA, Crewther P, Davidson BE. Chorismate mutase-prephenate dehydratase from Escherichia coli K-12. II. Kinetic properties. The Journal of biological chemistry. 1972;247:4447–4452. [PubMed] [Google Scholar]
- Eisen JA, Nelson KE, Paulsen IT, Heidelberg JF, Wu M, Dodson RJ, Deboy R, Gwinn ML, Nelson WC, Haft DH, Hickey EK, Peterson JD, Durkin AS, Kolonay JL, Yang F, Holt I, Umayam LA, Mason T, Brenner M, Shea TP, Parksey D, Nierman WC, Feldblyum TV, Hansen CL, Craven MB, Radune D, Vamathevan J, Khouri H, White O, Gruber TM, Ketchum KA, Venter JC, Tettelin H, Bryant DA, Fraser CM. The complete genome sequence of Chlorobium tepidum TLS, a photosynthetic, anaerobic, green-sulfur bacterium. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:9509–9514. doi: 10.1073/pnas.132181499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica. Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fersht A. Enzyme Structure and Mechanism. WH Freeman & Co; New York: 1985. [Google Scholar]
- Fiske MJ, Kane JF. Regulation of phenylalanine biosynthesis in Rhodotorula glutinis. Journal of bacteriology. 1984;160:676–681. doi: 10.1128/jb.160.2.676-681.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedt K, Heinrich G, Merkl R, Braus GH. Chorismate mutase of Thermus thermophilus is a monofunctional AroH class enzyme inhibited by tyrosine. Archives of microbiology. 2004;181:195–203. doi: 10.1007/s00203-003-0639-z. [DOI] [PubMed] [Google Scholar]
- Helmstaedt K, Krappmann S, Braus GH. Allosteric regulation of catalytic activity: Escherichia coli aspartate transcarbamoylase versus yeast chorismate mutase. Microbiology and molecular biology reviews : MMBR. 2001;65:404–421. doi: 10.1128/MMBR.65.3.404-421.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hol WG, van Duijnen PT, Berendsen HJ. The alpha-helix dipole and the properties of proteins. Nature. 1978;273:443–446. doi: 10.1038/273443a0. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends in biochemical sciences. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Hsu SK, Lin LL, Lo HH, Hsu WH. Mutational analysis of feedback inhibition and catalytic sites of prephenate dehydratase from Corynebacterium glutamicum. Archives of microbiology. 2004;181:237–244. doi: 10.1007/s00203-004-0649-5. [DOI] [PubMed] [Google Scholar]
- Husain A, Chen S, Wilson DB, Ganem B. A selective inhibitor of Escherichia coli prephenate dehydratase. Bioorganic & medicinal chemistry letters. 2001;11:2485–2488. doi: 10.1016/s0960-894x(01)00485-1. [DOI] [PubMed] [Google Scholar]
- Jensen RA, d’Amato TA, Hochstein LI. An extreme-halophile archaebacterium possesses the interlock type of prephenate dehydratase characteristic of the Gram-positive eubacteria. Archives of microbiology. 1988;148:365–371. doi: 10.1007/BF00411657. [DOI] [PubMed] [Google Scholar]
- Jetten MS, Sinskey AJ. Recent advances in the physiology and genetics of amino acid-producing bacteria. Critical reviews in biotechnology. 1995;15:73–103. doi: 10.3109/07388559509150532. [DOI] [PubMed] [Google Scholar]
- Kaplun A, Vyazmensky M, Zherdev Y, Belenky I, Slutzker A, Mendel S, Barak Z, Chipman DM, Shaanan B. Structure of the regulatory subunit of acetohydroxyacid synthase isozyme III from Escherichia coli. Journal of molecular biology. 2006;357:951–963. doi: 10.1016/j.jmb.2005.12.077. [DOI] [PubMed] [Google Scholar]
- Kapust RB, Waugh DS. Controlled intracellular processing of fusion proteins by TEV protease. Protein Expr Purif. 2000;19:312–318. doi: 10.1006/prep.2000.1251. [DOI] [PubMed] [Google Scholar]
- Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A. Automation of protein purification for structural genomics. J Struct Funct Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman H, Hessels GI, Vrijbloed JW, Euverink GJ, Dijkhuizen L. (De)regulation of key enzyme steps in the shikimate pathway and phenylalanine-specific pathway of the actinomycete Amycolatopsis methanolica. Microbiology (Reading, England) 2003;149:3321–3330. doi: 10.1099/mic.0.26494-0. [DOI] [PubMed] [Google Scholar]
- Kotaka M, Ren J, Lockyer M, Hawkins AR, Stammers DK. Structures of R- and T-state Escherichia coli aspartokinase III. Mechanisms of the allosteric transition and inhibition by lysine. The Journal of biological chemistry. 2006;281:31544–31552. doi: 10.1074/jbc.M605886200. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: A Program to Produce Both Detailed and Schematic Plots of Protein Structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet. 2001;357:1225–1240. doi: 10.1016/s0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- Lee CA, Saier MH., Jr Mannitol-specific enzyme II of the bacterial phosphotransferase system. III. The nucleotide sequence of the permease gene. J Biol Chem. 1983;258:10761–10767. [PubMed] [Google Scholar]
- Liberles JS, Thâorâolfsson M, Martâinez A. Allosteric mechanisms in ACT domain containing enzymes involved in amino acid metabolism. Amino acids. 2005;28:1–12. doi: 10.1007/s00726-004-0152-y. [DOI] [PubMed] [Google Scholar]
- Lingens F. Regulation of aromatic amino acid biosynthesis in microorganisms. Acta Microbiol Acad Sci Hung. 1976;23:161–166. [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. Journal of molecular biology. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- Louie GV, Brownlie PD, Lambert R, Cooper JB, Blundell TL, Wood SP, Warren MJ, Woodcock SC, Jordan PM. Structure of porphobilinogen deaminase reveals a flexible multidomain polymerase with a single catalytic site. Nature. 1992;359:33–39. doi: 10.1038/359033a0. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Olson R, Gouaux E. Mechanisms for ligand binding to GluR0 ion channels: crystal structures of the glutamate and serine complexes and a closed apo state. Journal of molecular biology. 2001;311:815–836. doi: 10.1006/jmbi.2001.4884. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta crystallographica. Section D, Biological crystallography. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Muraoka S, Okumura R, Ogawa N, Nonaka T, Miyashita K, Senda T. Crystal structure of a full-length LysR-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend. Journal of molecular biology. 2003;328:555–566. doi: 10.1016/s0022-2836(03)00312-7. [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- Oh BH, Pandit J, Kang CH, Nikaido K, Gokcen S, Ames GF, Kim SH. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. The Journal of biological chemistry. 1993;268:11348–11355. [PubMed] [Google Scholar]
- Pohnert G, Zhang S, Husain A, Wilson DB, Ganem B. Regulation of phenylalanine biosynthesis. Studies on the mechanism of phenylalanine binding and feedback inhibition in the Escherichia coli P-protein. Biochemistry. 1999;38:12212–12217. doi: 10.1021/bi991134w. [DOI] [PubMed] [Google Scholar]
- Porat I, Waters BW, Teng Q, Whitman WB. Two biosynthetic pathways for aromatic amino acids in the archaeon Methanococcus maripaludis. Journal of bacteriology. 2004;186:4940–4950. doi: 10.1128/JB.186.15.4940-4950.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P, Pathak N, Hasnain SE. pheA Rv 3838c of Mycobacterium tuberculosis encodes an allosterically regulated monofunctional prephenate dehydratase that requires both catalytic and regulatory domains for optimum activity. The Journal of biological chemistry. 2005;280:20666–20671. doi: 10.1074/jbc.M502107200. [DOI] [PubMed] [Google Scholar]
- Qamra R, Prakash P, Aruna B, Hasnain SE, Mande SC. The 2.15 A crystal structure of Mycobacterium tuberculosis chorismate mutase reveals an unexpected gene duplication and suggests a role in host–pathogen interactions. Biochemistry. 2006;45:6997–7005. doi: 10.1021/bi0606445. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta crystallographica. Section D, Biological crystallography. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Schuller DJ, Grant GA, Banaszak LJ. The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Nature structural biology. 1995;2:69–76. doi: 10.1038/nsb0195-69. [DOI] [PubMed] [Google Scholar]
- Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein expression and purification. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- Strater N, Hakansson K, Schnappauf G, Braus G, Lipscomb WN. Crystal structure of the T state of allosteric yeast chorismate mutase and comparison with the R state. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:3330–3334. doi: 10.1073/pnas.93.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strater N, Schnappauf G, Braus G, Lipscomb WN. Mechanisms of catalysis and allosteric regulation of yeast chorismate mutase from crystal structures. Structure (London, England : 1993) 1997;5:1437–1452. doi: 10.1016/s0969-2126(97)00294-3. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods in enzymology. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- Vivan AL, Dias MV, Schneider CZ, de Azevedo WF, Jr, Basso LA, Santos DS. Crystallization and preliminary X-ray diffraction analysis of prephenate dehydratase from Mycobacterium tuberculosis H37Rv. Acta crystallographica. Section F, Structural biology and crystallization communications [electronic resource] 2006;62:357–360. doi: 10.1107/S1744309106006385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warpeha KM, Lateef SS, Lapik Y, Anderson M, Lee BS, Kaufman LS. G-protein-coupled receptor 1, G-protein Galpha-subunit 1, and prephenate dehydratase 1 are required for blue light-induced production of phenylalanine in etiolated Arabidopsis. Plant physiology. 2006;140:844–855. doi: 10.1104/pp.105.071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woycechowsky KJ, Hilvert D. Deciphering enzymes. Genetic selection as a probe of structure and mechanism. European journal of biochemistry / FEBS. 2004;271:1630–1637. doi: 10.1111/j.1432-1033.2004.04073.x. [DOI] [PubMed] [Google Scholar]
- Xue Y, Lipscomb WN, Graf R, Schnappauf G, Braus G. The crystal structure of allosteric chorismate mutase at 2.2-A resolution. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10814–10818. doi: 10.1073/pnas.91.23.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wilson DB, Ganem B. Probing the catalytic mechanism of prephenate dehydratase by site-directed mutagenesis of the Escherichia coli P-protein dehydratase domain. Biochemistry. 2000;39:4722–4728. doi: 10.1021/bi9926680. [DOI] [PubMed] [Google Scholar]