

Abstract
In structural biology, the most critical issue is the availability of high-quality samples. “Structural-biology-grade” proteins must be generated in a quantity and quality suitable for structure determination using X-ray crystallography or nuclear magnetic resonance. The additional challenge for structural genomics is the need for high numbers of proteins at low cost where protein targets quite often have low sequence similarities, unknown properties and are poorly characterized. The purification procedures must reproducibly yield homogeneous proteins or their derivatives containing marker atom(s) in milligram quantities. The choice of protein purification and handling procedures plays a critical role in obtaining high-quality protein samples. Where the ultimate goal of structural biology is the same—to understand the structural basis of proteins in cellular processes, the structural genomics approach is different in that the functional aspects of individual protein or family are not ignored, however, emphasis here is on the number of unique structures, covering most of the protein folding space and developing new technologies with high efficiency. At the Mid-west Center Structural Genomics (MCSG), we have developed semiautomated protocols for high-throughput parallel protein purification. In brief, a protein, expressed as a fusion with a cleavable affinity tag, is purified in two immobilized metal affinity chromatography (IMAC) steps: (i) first IMAC coupled with buffer-exchange step, and after tag cleavage using TEV protease, (ii) second IMAC and buffer exchange to clean up cleaved tags and tagged TEV protease. Size exclusion chromatography is also applied as needed. These protocols have been implemented on multidimensional chromatography workstations AKTAexplorer and AKTAxpress (GE Healthcare). All methods and protocols used for purification, some developed in MCSG, others adopted and integrated into the MCSG purification pipeline and more recently the Center for Structural Genomics of Infectious Disease (CSGID) purification pipeline, are discussed in this chapter.
I. Introduction
During the past decade or so, more than a dozen structural genomics projects in the world have been organized to develop technologies for high-throughput protein production and structure determination by X-ray crystallography and nuclear magnetic resonance (NMR). In USA, the National Institute of Health (NIH) launched the structural genomics program, Protein Structure Initiative (PSI), to develop and optimize new, rapid, integrated methods for cost-effective determination of protein structures involving federal, university, and industry efforts. The long-range goal of PSI is to make the three-dimensional atomic-level structures of most proteins easily obtainable from the knowledge of their corresponding DNA sequences. Since the year 2000, PSI projects produced more than 3000 structures. In the first five years, a pilot project was initiated to develop protein structure determination pipelines(BSGC, CESG, JCSG, MCSG, NESG, NYSGRC, SECSG, SGPP, TBSGC) capable of producing large numbers of protein samples for structural biology applications and the following five years as a production project involving four big centers (JCSG, MCSG, NESG, NYSGRC) with the goal of producing as many unique three-dimensional structures as possible and six small technology developing centers (ATCG3D, CESG, CHTSB, CSMP, ISFI, NY_CMPS) with a goal to improve throughput.
Since the start of the PSI, more than 100,000 clones were generated, over 20,000 proteins were purified, and about 2100 structures were deposited in the PDB from nearly 8000 crystals at an average cost of $70,000 per structure, which is significantly lower than the typical laboratory cost of $250,000. The MCSG alone has deposited 850 structures from about 2500 crystals obtained from 30,000 clones.
Although the procedures and applications are quite different for X-ray crystallography and NMR, both techniques require similar protein sample quality. For crystallography applications, the protein must be soluble, folded properly, and chemically and functionally homogeneous. The protein sample must be free of critical contaminants that may degrade, denature, destabilize, or modify the protein or interfere with its crystallization or structure determination. Protein purity of >95% is typically required. Protein samples must be stable during crystallization trials, be suitable for incorporation of heavy atoms to aid structure determination, and be functionally relevant. The samples must have protein concentrations in the range of 5–25 mg/ml, for testing 200–500 crystallization conditions, growing diffraction-quality single crystals, establishing cryo-conditions, and producing rational heavy atom derivatives. These criteria put certain restrictions on the methods and procedures of sample preparation. For NMR structure determination, protein samples should be similarly pure with homogeneity of 95% or higher, monodisperse, be as high concentration as for crystallography needing about 500 μl at 2–35 mg/ml (or 0.5–2 mM). Proteins also need to be 13C/15N labeled.
In the past several years, the MCSG developed standard operating procedures for protein purification to make protein samples suitable for structure determination using synchrotron-based X-ray crystallography or NMR. These standard operating procedures are based on the following principles:
Every protein is different in structural genomics, where emphasis is on producing more number of unique structures. Effort is to determine the three-dimensional structures of proteins in the closest natural forms keeping in mind the functional aspects of proteins. All proteins are expressed in fusion with a cleavable affinity tag and purified using affinity chromatography and the affinity tag is cleaved off by a specific tagged protease.
Although new technological advances in both NMR and X-ray crystallography require considerably less protein than before, all proteins are overexpressed to have enough material for structural and functional studies.
All procedures need to be simple and efficient to reduce mistakes, to be more reproducible and to save labor and materials. For this, procedures are automated wherever possible and evaluation screens are included to remove protein targets that have no or low chance to yield crystals at early stages to save resources.
Since different proteins are purified by same SOPs, there will be fallouts in each step and some of these can be recovered later by less costly procedures.
II. Protein Constructs and Expression
A. Protein Constructs to Consider
Among many available microbial expression systems, T7 systems under control of lac repressor are most commonly used in the structural biology community to overexpress recombinant proteins. The most frequently used cloning vector is one with an N-terminal 6-His-tag. The tag is removed by a sequence specific protease such as tobacco etch virus (TEV) protease. Advantages of using multiple-histidine-tag in the N-terminus are:
Such proteins can be purified by relatively simple protocols.
His-tag rarely alters expression level, function, or oligomeric states of proteins compared to other commonly used tags such as glutathione-S-transferase (GST), maltose-binding protein (MBP), and S-tag.
His-tag is small enough that crystallization trials can be attempted with or without removing it.
These tags are inserted at the N-terminus because when the tags are removed by the protease, they leave shorter cloning artifact residues, typically three, rather than one at the C-terminus which leaves 6–7 residues.
In MCSG, most of the proteins were cloned in the pMCSG7 vector (Stols et al., 2002; Zhang et al., 2002) and expressed in Escherichia coli BL21(DE3)-Gold (Stratagene) harboring a plasmid that encodes three rare tRNAs (Christendat et al., 2000; Pan and Malcolm, 2000; Studier, 1991; Yee et al., 2003). The pMCSG7 vector allows for the fusion of cleavable His6-tag with the N-terminus of the target protein which upon removal with TEV protease adds three residues (SerAsnAla) to the N-terminus of the target protein.
To complement the N-terminal His-tag, a number of other affinity tags have been used:
GST and MBP tags have been shown to influence expression and solubility of proteins and such tags have been used with some success. However, there are frequent “false positive” cases, that is, an overexpressed, soluble, relatively small target protein fused to a lager GST or MBP became insoluble as soon as the fused partner was removed by a sequence specific protease. There should be a simple step to screen these cases before attempting a scale-up. In MCSG, an N-terminal MBP fusion vector pMCSG19 was developed to solve this problem (Donnelly et al., 2006). The N-terminal MBP is cleaved off in situ by TVMV protease and only if the target protein still remains soluble, it is purified. Other alternatives to improve solubility of a protein include coexpression with chaperones such as GroEL with or without GroES which were used successfully in limited cases (Ribbe and Burgess, 2001; Wynn et al., 2000).
There are also cases where the N-terminal His-tag protein did not bind to an affinity column or the His-tag could not be cleaved completely even after incubation with sequence specific proteases for 2 days. These suggest that the N-termini may not be exposed enough to bind to the column and/or be cleaved. For such proteins, a C-terminal His-tag could be a good alternative, although it leaves a longer cloning artifact.
Often, proteins naturally interact with others to form assemblies in their cellular roles. Alone, some proteins may not maintain their stable conformation and may misfold or aggregate. For such proteins, MCSG has developed several vectors to coexpress (Stols et al., 2007) up to five different proteins at once, although this is not a high-throughput procedure.
There are other affinity tags that have been tested for their efficiencies and adaptability to the structural genomics pipeline (Table I).
Table I.
LIC Vectors Developed in MCSG
| Vector | Base vector | Encoded leader sequence | Use |
|---|---|---|---|
| pMCSG7 | pET21a | N-His-TEV-LICs | Purification |
| pMCSG8 | pMCSG7 | N-His-Sloop-TEV-LICs | Solubility |
| pMCSG9 | pMCSG7 | N-His-MBP-TEV-LICs | Solubility |
| pMCSG10 | pMCSG7 | N-His-GST-TEV-LICs | Solubility |
| pMCSG11 | pACYCDuet-1 | N-His-TEV-LICs | Coexpression |
| pMCSG12 | pACYCDuet-1 | N-His-Sloop-TEV-LICs | Coexpression |
| pMCSG13 | pACYCDuet-1 | N-His-MBP-TEV-LICs | Coexpression |
| pMCSG14 | pACYCDuet-1 | N-His-GST-TEV-LICs | Coexpression |
| pMCSG17 | pMCSG7 | N-Stag-TEV-LICs | Coexpression |
| pMCSG20 | pMCSG7 | N-Stag-GST-TEV-LICs | Coexpression |
| pMCSG16 | pMCSG7 | N-His-AviTag-TEV-LICs | Phage display |
| pMCSG15 | pMCSG7 | LICs-TEV-AviTag-His-C | Phage display |
| pMCSG18 | pMCSG7 | N-His-TEV-LICs-GFP | Screening |
| pMCSG19ABC | pMCSG7 | N-MBP-TVMV-His-TEV-LICs | Purification |
| pMCSG21 | pDONR/zeo | attL1-TEV-LIC-attL2 | Gateway cloning |
| pMCSG26 | pMCSG7 | LICs-C-His | Purification |
| pMCSG30 | pMCSG7 | N-His-TEV-MBP-Linker-LICs | Purification |
B. Protein Expression
A key screening step of small-scale expression and solubility is applied here to assure that clones are effectively overexpressed to produce soluble proteins suitable for large-scale purifications. A problem is that small-scale expression does not always scale up well with parameters such as temperature, culture conditions, and aeration. After a large number of tests with conditions matching large-scale preparations, only a small number of false positives are expected, predicting an 80% accuracy.
For high-level expression of most proteins, BL21(DE3) and its derivatives such as BL21-Codon Plus, Rosetta2, or similar strains carrying additional tRNAs to overcome the codon bias have been successfully used. Protein expression using T7 systems can be induced either with isopropyl-β-d-thiogalactoside (IPTG) or with modified growth medium that allows auto-induction (Studier, 2005). In either case, induction temperature is the most critical growing condition to produce soluble protein.
In MCSG, Pink medium (Stols et al., 2004) has been developed from traditional M9 media for particular applications in structure determination, that is, labeling with 13C and 14N for NMR (Zhao et al., 2004), and Se-Met for X-ray crystallography, combined with high protein expression. Pink media are composed of packets of salts, metal supplements, antibiotics, etc., based on the application and are available commercially (Medicilon).
Typically, bacteria are grown at 37 °C for 3–4 h in 1-l culture in the presence of two antibiotics, ampicillin (100 mg/l) and kanamycin (50 mg/l) agitating at 180 rpm in a 2-l pop-bottle. All the medium is prepared from these packets and the pop-bottles and solutions are not required to be autoclaved (Millard et al., 2003). Protein is induced at 18 °C after cells are grown to OD600 = 1.2 which is significantly higher than 0.4–0.6 used for typical M9 media. After addition of Se-Met and IPTG (0.5–1.0 mM), protein is induced slowly overnight (~16 h) to improve the solubility of the protein with high expression. With this MCSG standard growth procedure, 1 l of culture produces about 20–200 mg of purified protein.
III. Purification
Purification starts from preparation of crude extract (or lysate) followed by two affinity chromatography steps:
IMAC-I (immobilized metal affinity chromatography) using a Ni2+-column
Buffer exchange on a desalting column
His6-tag cleavage using the recombinant TEV protease
IMAC-II using a Ni2+-column
Buffer exchange and protein concentration
Following are detailed procedures used in MCSG.
A. Preparation of Crude
Lysate Extract is prepared by resuspending isolated cell pellets in generally five volumes of a lysis buffer with protease inhibitors (Complete; Roche) and sonication on ice for 3 min in the presence of lysozyme. Lysis buffer (buffer A) includes a small amount of imidazole (10–20 mM) to reduce nonspecific binding of bacterial proteins to Ni-affinity column, 5% glycerol, and 10 mM β-mercaptoethanol (β-ME) to preserve target proteins in stable and reduced forms. The buffer is maintained in pH 8 for optimal binding to Ni-affinity column. A high concentration of salt (500 mM NaCl) also reduces nonspecific binding by impurities and most proteins tend be more soluble at higher concentration of salt. Sonicated samples are clarified by centrifugation at 30,000g for 1 h followed by filtration through 0.45 and 0.22 μm in-line filters.
B. IMAC-I and Buffer-Exchange Steps
All chromatography experiments are performed at 4 °C. The crude extract, typically 15–50 ml, of a target protein is applied slowly with a flow rate of 1 ml/min or lower onto a pre-equilibrated (with buffer A) 5-ml HisTrap chelating HP column (GE Healthcare) charged with Ni2+. The column is then washed with 10 column volumes (CV) of buffer A followed by an additional wash with a 15 CV of buffer A containing 20 mM imidazole. The protein is first eluted in to a 10-ml loop with buffer B containing 250 mM imidazole, then applied on a HiPrep 26/10 desalting column (GE Healthcare) pre-equilibrated with buffer A. Just prior to injecting protein onto the desalting column, 2 ml of 5 mM EDTA in buffer A is injected onto the desalting column to create a slow-moving EDTA zone on the desalting column to sequester any Ni2+ ions released from the chelating column. The buffer-exchange step is run at flow rate 8 ml/min.
The desalting column is washed and re-equilibrated prior to the next purification cycle. The tubing and loop are washed between chromatography steps to avoid crosscontamination. The final peak fractions and all solutions that could contain target protein are collected and analyzed by SDS gel electrophoresis. Throughout the purification process, several parameters including UV absorbance, pressure, flow rate, pH, and ionic strength are monitored. All fractions are analyzed and documented. The entire purification process takes about 8 h for four proteins, depending on the initial sample volumes. The chelating columns are recycled six to seven times using an automated procedure by metal stripping with 50 mM EDTA and charging with 100 mM NiSO4.
C. Affinity Tag Removal by TEV Protease
Three proteases (human thrombin, factor Xa from bovine plasma, and recombinant TEV protease) were tested for efficiency of tag removal using standard protocols suggested by the manufacturers. After evaluating for the efficiency of tag cleavage, level of nonspecific cleavage, optimum temperature, and fraction of successfully processed proteins, the TEV protease was chosen as the most suited for MCSG targets (Table II). TEV protease offers several advantages:
It is highly specific, recognizing a seven-amino-acid sequence.
It shows virtually no nonspecific proteolysis of target proteins.
It is active under a wide range of conditions, including low temperature (4 °C), broad range of pH, and high ionic strength (Dougherty and Parks, 1989).
Table II.
Efficiency of His-Tag Cleavage by TEV Protease
| Percentage of cleavage (%) | 99–80 | 70–50 | 0 |
| Number of proteinsa | 200 | 31 | 8 |
Proteins (total 239) were incubated with 1:50 ratio of protease to target protein at 4 °C for 16–24 h.
The TEV protease, expressed from the vector pRK508 carries noncleavable His6-tag (a gift from Dr. D. Waugh, NCI), purified using a procedure described previously (Kapust and Waugh, 2000) and can be removed from protein samples by IMAC. Moreover, TEV protease was highly effective at removing His6-tags from more than 96% of tested MCSG target proteins (Table II).
In cases where the tag was not cleaved completely, the target protein was recloned to move the His-tag to C-terminal and repurified. AsbF, 3-dehydroshikimate (3-DHS) dehydratase from Bacillus anthracis was repurified in a pMCSG26 C-term His-tag construct. The tag from this protein could be cleaved readily and was crystallized and its structure was determined.
D. IMAC-II and Buffer-Exchange Steps
Proteins purified with IMAC-I and buffer exchange are treated with the His7-tagged TEV protease to remove the His6-tag up to 72 h at 4 °C following the basic protocol (see above). Cleavage is monitored by SDS-PAGE and Coomassie Brilliant Blue R (Amersham Biosciences) staining. After cleavage, the reaction mixture containing target protein (cleaved and some uncleaved), His7-tagged TEV protease, and His6-tag, is applied to a 5-ml Ni-superflow affinity column (GE Healthcare) and is washed with three CV of buffer A. All chromatographic steps are performed at 22 °C. The protein elutes as a flow-through and a part of the wash from the column in lysis buffer with 20 mM imidazole. It is dialyzed in crystallization buffer containing 20 mM HEPES, pH 8.0, 250 mM NaCl, 2 mM DTT prior to setting up trials. The full purification steps including IMAC-I, TEV protease cleavage, and IMAC-II resulted in high purity protein samples, typically 95–98% (Fig. 1).
Fig. 1.
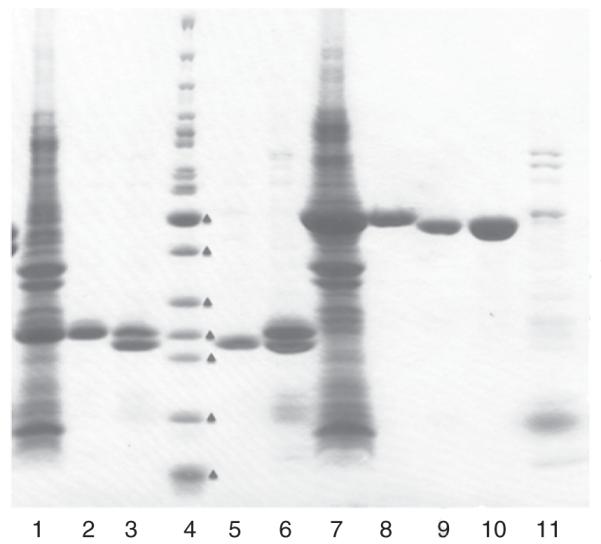
SDS-PAGE of IDP01350 (MW: 23.6 kDa, lanes 1–3, 5, and 6) and IDP01460 (44.6 kDa, lanes 7–11) purified by the process described: lanes 1 and 7—crude extract, lanes 2 and 8—after IMAC-I, lanes 3 and 9—after TEV cleavage, lanes 5 and 10—after IMAC-II, lanes 6 and 11—elution with 250 mM imidazole after IMAC-II, and lane 4—molecular weight markers (EZ-Run Fisher), from the lower to higher one (marked red triangle) 10, 15, 20, 25, 30, 40, 50, 60, 70 kDa. For IDP1350, TEV cleavage was not complete, but after IMAC-II, the cleaved IDP1350 was cleaned well.
E. Platform for Automated Multidimensional Chromatography
MCSG collaborated with Amersham Biosciences (now GE healthcare) to developed AKTA multidimensional automated chromatography (AKTA3D). Initially, it was implemented on purification workstations AKTAexplorer and AKTAfplc to purify six proteins at a time by IMAC-I and buffer-exchange overnight. Now AKTAxpress is well suited for a production line capable of purifying 16 different proteins as it has four systems with each system having five column positions, four for Ni-affinity columns (such as a 5 ml HisTrap) and one buffer-exchange column (HiPrep 26/10 desalting, GE Healthcare). There are five loops available to store protein peak fractions from the Ni-affinity chromatography step before being loaded onto the desalting column. The final purified protein is collected in 1.5–2.0 ml fractions in a 96-well format deep-well block. Both IMAC-I and IMAC-II steps are performed on AKTAxpress workstation units.
F. Size Exclusion Chromatography
In some cases, persistent contaminants must be removed by additional chromatographic step(s) such as size exclusion chromatography or ion exchange chromatography. Size exclusion chromatography as an additional step is more common because it can separate impurities from the target protein as well as large aggregates which elute near the void volume and be applied more broadly. Also the elution profile indicates not only purity but also the primary oligomeric states of the target protein which has biological significance. When size exclusion chromatography is used, it is also essential to examine closely any peak that appears in the later part of the column volume since it might reflect small organic molecules that could be weakly bound ligands (Fig. 2).
Fig. 2.

(A) Size exclusion profile of IDP indicating a small organic molecule eluted near the end of the column volume. (B) Before size exclusion chromatography, the small organic molecule is shown by high absorbance of OD260. (C) After size exclusion chromatography, the species absorbing at 260 nm is removed.
G. On-Column Cleavage
MCSG purification pipeline following two IMAC steps which include IMAC-I, tag cleavage using His-tagged TEV protease, and IMAC-II (Kim et al., 2003) can take 5–6 days to accomplish, including 2–3 days of tag cleavage. To shorten the process, an on-column tag cleavage has been developed to carry out the entire purification procedure on the AKTAx-press chromatography workstation without manual intervention. After a target protein with an affinity tag is applied to an affinity column and washed, a tagged TEV protease is then applied to the same column and incubated for 16–20 h at 30–35 °C. The target protein without the tag is then eluted, while the tag, uncleaved protein, and the protease with the His-tag remain on the column. The temperature and incubation time are controlled by the software and electronics (Fig. 3). This procedure is similar to that described in a previous paper of on-column cleavage using His-tagged TEV protease (Bhikhabhai et al., 2005). However, in this process cleavage is inefficient, and requires a high ratio of protease to protein. The new approach using GST-tagged TEV requires less protease with the same efficiency. The entire process of 2–3 days is automated, and uncleaved protein can be recovered by a simple IMAC-I step, when it cannot be cleaved by the protease. However, in some cases with oligomeric proteins, when the cleavage is not complete due to protein-protein interactions, either some cleaved proteins remain bound in the Ni-column or some uncleaved proteins elute together with cleaved ones causing heterogeneity in the sample.
Fig. 3.

Gadgets used in the on-column cleavage: (A) AKTAxpress with a 26/60 size exclusion column, (B) a 5 ml HisTrap Ni-column wrapped a heat strip, and (C) temperature controller.
IV. Protein Characterization
We have used several methods to characterize protein samples. Table III indicates the method(s) used for the various aspects of protein characterization.
Table III.
Protein Characterization Methods in the MCSG Protocol
| Protein parameter | Method of characterization |
|---|---|
| Purity | SDS-PAGE stained with Coomassie Brilliant Blue |
| Concentration | Coomassie Plus Protein Assay (Pierce, Catalog No. 23236) and UV spectrometry (Nanodrop) |
| Integrity | CD spectrometry (Jasco) |
| Polydispersity | Dynamic light scattering (DynaPro/Wyatt Technology) and Dawn Heleos & QELS (Wyatt Technology) |
| Estimated molecular weight/ size in solution |
Size exclusion chromatography and Dawn Heleos & QELS (Wyatt Technology) |
| Suspected mutations or incorrect proteins |
SDS-PAGE stained with Coomassie Brilliant Blue, Mass spectrometry (ESI-TOF, MALDI-TOF Biflex III, Bruker) |
| Suspected chemical heterogeneity and bound ligands |
Mass spectrometry (ESI-TOF, MALDI-TOF Biflex III, Bruker) |
| Bound ligands | UV/Vis spectrometry |
V. Protein Concentration and Storage
All proteins are concentrated with Centricon Plus Centrifugal Filter Units (Millipore), using molecular weight cutoff as recommended by the manufacturer. Several different membranes are available for low protein binding and the Millipore Ultracel regenerated cellulose low-binding membrane worked well for several hundred proteins in MCSG. Periodically, the protein concentrate needs to be mixed by pipetting as the concentrated proteins are accumulated at the bottom of the tubes and can precipitate. All proteins are flash frozen in ~50 μl aliquots, to avoid the damage from a freeze-thaw cycle, in liquid nitrogen temperature in the storage buffer and stored in an LS6000 liquid nitrogen storage system (Taylor–Wharton) for an extended period of time.
VI. Problems and Recovery/Salvage Procedure to Consider
Since most structural genomics programs use one standard protocol for most of the proteins and supplement it with screens to select fitting samples to their pipelines, there are a number of proteins falling out from various stages. Some of these proteins can be recovered/salvaged to get them back into the pipelines without significant efforts.
A. Refolding
Often recombinant proteins produced in E. coli or other heterologous expression systems accumulate as insoluble inclusion bodies. Generally, more than 50% of clones, a much higher ratio from eukaryotic genes, produce insoluble proteins in E. coli. Some of these can be recovered by recloning into different vectors as described below, and several refolding methods have also been developed (Oganesyan et al., 2005; Vincentelli et al., 2004; Willis et al., 2005). Small proteins with molecular weight less than 25 kDa expressed as inclusion bodies in E. coli, have been refolded by slow dialysis or dilution into a large volume. First, a protein in the inclusion body is dissolved to about 1 mg/ml concentration using a chaotropic solvent such as 6–8 M urea or guanidium HCl in the presence of stabilizing reagents known as osmolites such as trimethylamine oxide, betaine, proline, arginine, glycerol, then slowly these reagents are replaced with the buffer solution of choice by dialysis. More efficiently, the dissolved proteins (His-tagged) in the chaotropic solvent and osmolites are applied to a Ni-affinity column, slowly washed with the buffer without chaotropic solvent and osmolites and eluted with the buffer containing 200–500 mM imidazole. Additional washing steps with mild detergent such as 0.1% TritonX-100 can be included before elution for more efficient folding, although it is difficult to remove detergent due to interactions with protein and its micelle formation. More recently, β-cyclodextrin is used successfully to wash the detergent from an affinity column (Oganesyan et al., 2005; Rozema and Gellman, 1996). After refolding, circular dichroism (CD) or static/dynamic light scattering is necessary to monitor refolding of unfolded analysis proteins. It is also critical to remove protein aggregates by size exclusion chromatography. However, these procedures are hard to automate because most procedures are time consuming, labor intensive, and difficult for multiple samples, that is, applying size exclusion chromatography for each sample, proteins precipitating on columns, etc.
B. Low Solubility
For proteins with yields of less than 5 mg/ml during the purification and concentration, addition of mono- or divalent salts, changing pHs, number of reagents, potential ligands, and various nondetergent sulfobetaines (NDSBs) improve the yield. However, some of additives are cost-prohibitive for large-scale uses.
C. Cloning to Improving Solubility and Expression
To improve solubility and expression level, several fusion tags with/without cleavage sites are used such as GST and MBP mostly at the N-terminus. Problems with such fusion proteins are: for uncleaved proteins the fusion protein, that is, GST influences oligomerization of the target proteins to form dimers or tetramers, and while the tagged protein is in solution, the target protein part is still not folded properly. For those with cleavable fusion tags, the target protein may precipitate as soon as the fusion partner is cleaved off. Still, a number of target proteins are recovered by fused clones to produce eventual three-dimensional structures. MCSG developed MBP fusion vectors (Table I), particularly, pMCSG9 and pMCSG19, without and with tobacco vein mottling virus (TVMV) protease cleavage sites, respectively. With pMCSG9 containing a TEV protease cut site, a target protein, expressed as a MBP fusion, is purified by Ni-affinity chromatography and is free off from the His-tagged MBP fusion by TEV protease. However, pMCSG19 has a MBP fusion with TVMV protease cleavage site, which can be removed in situ by TVMV protease induced together with the target protein. As a result, when bacterial cells are lysed, only the target protein with the His-tag (with TEV protease cut site) are in solution fraction, if it stays in solution after the MBP part is cleaved off.
D. Changing His-Tag Positions
When proteins are incubated with proteases to remove tags after the first affinity chromatography step, some portion of proteins are not cut completely or the proteins may have very low affinity to the column media suggesting that the His-tag and/or the N-terminus are buried in the target protein structure. Quite often, for these cases, the C-terminal His-tag may salvage the situation. One main problem is it leaves a longer (at least 6–7 residues) artifact sequence after the tag is removed. C-terminal His-tag can also help to resolve the cases C-terminal truncations due to incomplete transcriptions or degradation during the purification.
E. Inclusion of ATP in Crude Extract to Remove Copurifying Endogenous GroEL
When proteins are expressed using pMCSG9 and pMCSG19, some endogenous GroEL can copurify with target proteins as evident from SDS gel electrophoresis (Fig. 2). The density of the GroEL (~60 kDa) band is more than 10 times that of the target protein suggesting that the 14-meric GroEL remained complexed with the target protein during the affinity purification step. Perhaps overexpression of a target protein fused with a relatively large protein such as MBP (~50 kDa) might exert on bacteria to induce GroEL to (re)fold the target protein. However, this endogenous GroEL could be readily removed during the IMAC-I by adding about 5 mM of ATP to crude extract or by applying the ATP (in about one column volume) to the affinity column before eluting the target protein with a high concentration (250–500 mM) of imidazole (Fig. 4).
Fig. 4.
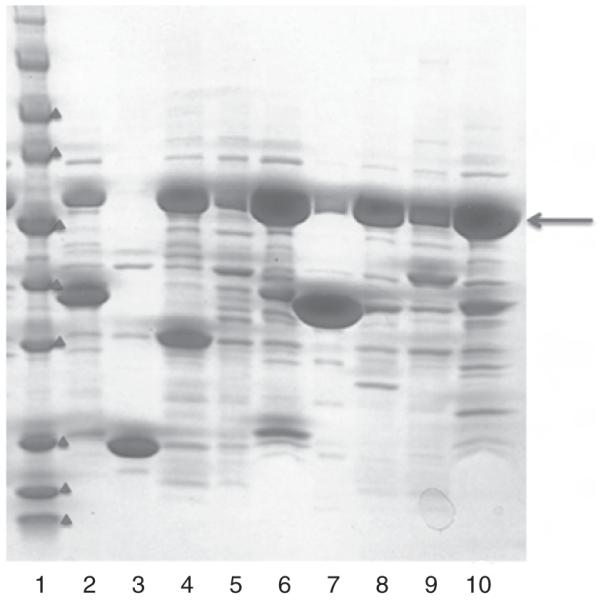
SDS-PAGE showing GroEL, indicated in red arrow near 57 kDa, coeluted with target proteins in the IMAC-I step. Molecular weight marker indicates (in red triangles) 5, 10, 15, 25, 35, 50, 70, 90 kDa.
VII. Conclusion
Recently, SGC et al. (2008) reviewed the protein production and purification methods used by structural genomics centers around the world and presented a consensus purification strategy to serve as a guide to biological and biomedical science community requiring recombinant proteins. In MCSG and CSGID, the purification methods and protocols for structural-biology-grade protein samples using E. coli expression hosts are based on IMAC. All proteins samples are His-tagged either in the N-terminus or C-terminus. After the protein is purified by a Ni-affinity chromatographic step (IMAC-I), the tag is removed by the sequence specific TEV protease, and finally cleaved tags, His-tagged TEV proteases and other residual protein impurities are cleaned by the second IMAC-II. Size exclusion chromatography is also used to further purify and analyze protein samples. Most of these procedures are automated on AKTAfplc, AKTAexplorer, and AKTAxpress purification workstations from GE Healthcare and optimized efficiently for parallel multipurification process. Several additional fusion protein constructs are designed and tested to improve expression and solubility of proteins. Using this high-throughput process, more than 2000 proteins are purified in MCSG and CSGID routinely producing about 200 crystal structures yearly. All methods and protocols developed in MCSG have been made available and applied to many other laboratories successfully including nonstructural genomics.
Acknowledgments
We would like to thank Christine Tesar, Rachele Hendricks, and Grant Shackelford for contributing to the development of protein purification procedures; and Shyamala Rajan for reading and useful comments. This work was supported by National Institutes of Health Grants GM62414, GM074942, and contract No. HHSN272200700058C, and by the U. S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357..
Abbreviations
- PSI
Protein Structure Initiative
- MCSG
Midwest Center for Structural Genomics
- CSGID
Center for Structural Genomics of Infectious Diseases
- IMAC
immobilized metal affinity chromatography
- TEV
tobacco etch virus
- TVMV
tobacco vein mottling virus
- IPTG
isopropyl-β-d-thiogalactoside
- β-ME
β-mercaptoethanol
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetate
- SDS-PAGE
polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate
References
- Accelerated Technologies Center for Gene to 3D Structure (ATCG3D) http://atcg3d.org/default.aspx.
- Berkeley Structural Genomics Center (BSGC) http://www.strgen.org/
- Bhikhabhai R, Sjöberg A, Hedkvist L, Galin M, Liljedahl P, Frigård T, Pettersson N, Nilsson M, Sigrell-Simon JA, Markeland-Johansson C. Production of milligram quantities of affinity tagged-proteins using automated multistep chromatographic purification. J Chromatogr A. 2005;1080:83–92. doi: 10.1016/j.chroma.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Ribbe MW, Burgess BK. The chaperone GroEL is required for the final assembly of the molybdenum-iron protein of nitrogenase. Proc. Natl Acad. Sci. USA. 2001;98:5521–5525. doi: 10.1073/pnas.101119498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Eukaryotic Structural Genomics (CESG) http://www.uwstructuralgenomics.org/
- Center for High-Throughput Structural Biology (CHTSB) http://www.chtsb.org/
- Center for Structures of Membrane Proteins (CSMP) http://csmp.ucsf.edu/index.htm.
- Christendat D, et al. Structural proteomics: Prospects for high throughput sample preparation. Prog. Biophys. Mol. Biol. 2000;73:339–345. doi: 10.1016/s0079-6107(00)00010-9. [DOI] [PubMed] [Google Scholar]
- Donnelly MI, et al. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Express. Purif. 2006;46:446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty WG, Parks TD. Molecular genetic and biochemical evidence for the involvement of the heptapeptide cleavage sequence in determining the reaction profile at two tobacco etch virus cleavage sites in cell-free assays. Virology. 1989;172:145–155. doi: 10.1016/0042-6822(89)90116-5. [DOI] [PubMed] [Google Scholar]
- Integrated Center for Structure and Function Innovation (ISFI) http://techcenter.mbi.ucla.edu/
- Joint Center for Structural Genomics (JCSG) http://www.jcsg.org/
- Kapust RB, Waugh DS. Controlled intracellular processing of fusion proteins by TEV protease. Protein Express. Purif. 2000;19:312–318. doi: 10.1006/prep.2000.1251. [DOI] [PubMed] [Google Scholar]
- Kim Y, et al. Automation of protein purification for structural genomics. J. Struct. Funct. Genomics. 2003;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midwest Center for Structural Genomics (MCSG) http://www.mcsg.anl.gov/
- Millard CS, et al. A less laborious approach to the high-throughput production of recombinant proteins in Escherichia coli using 2-liter plastic bottles. Protein Express. Purif. 2003;29:311–320. doi: 10.1016/s1046-5928(03)00063-9. [DOI] [PubMed] [Google Scholar]
- New York Consortium on Membrane Protein Structure (NY_CMPS) http://www.nycomps.org/
- New York Structural Genomics Research Consortium (NYSGRC) http://www.nysgxrc.org/nysgrc-cgi/index.cgi.
- Northeast Structural Genomics Consortium (NESG) http://www.nesg.org/
- Oganesyan N, et al. On-column protein refolding for crystallization. J. Struct. Funct. Genomics. 2005;6:177–182. doi: 10.1007/s10969-005-2827-3. [DOI] [PubMed] [Google Scholar]
- Pan SH, Malcolm BA. Reduced background expression and improved plasmid stability with pET vectors in BL21 (DE3) BioTechniques. 2000;29:1234–1238. doi: 10.2144/00296st03. [DOI] [PubMed] [Google Scholar]
- Protein Structure Initiative (PSI) http://www.nigms.nih.gov/Initiatives/PSI.
- Rozema D, Gellman SH. Artificial chaperone-assisted refolding of carbonic anhydrase B. J. Biol. Chem. 1996;271:3478–3487. doi: 10.1074/jbc.271.7.3478. [DOI] [PubMed] [Google Scholar]
- Southeast Collaboratory for Structural Genomics (SECSG) http://www.secsg.org/
- Structural Genomics Consortium (SGC) et al. Protein production and purification. Nat. Methods. 2008;5:135–146. doi: 10.1038/nmeth.f.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Structural Genomics of Pathogenic Protozoa (SGPP) http://www.sgpp.org/
- Stols L, et al. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Express. Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- Stols L, et al. Production of selenomethionine-labeled proteins in two-liter plastic bottles for structure determination. J. Struct. Funct. Genomics. 2004;5:95–102. doi: 10.1023/B:JSFG.0000029196.87615.6e. [DOI] [PubMed] [Google Scholar]
- Stols L, et al. New vectors for co-expression of proteins: Structure of Bacillus subtilis ScoAB obtained by high-throughput protocols. Protein Express. Purif. 2007;53:396–403. doi: 10.1016/j.pep.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Studier FW. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking culture. Protein Express. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- TB Structural Genomics Consortium (TBSGC) http://www.doe-mbi.ucla.edu/TB/
- Vincentelli R, et al. High-throughput automated refolding screening of inclusion bodies. Protein Sci. 2004;13:2782–2792. doi: 10.1110/ps.04806004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MS, et al. Investigation of protein refolding using a fractional factorial screen: A study of reagent effects and interactions. Protein Sci. 2005;14:1818–1826. doi: 10.1110/ps.051433205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn RM, et al. GroEL/GroES promote dissociation/reassociation cycles of a heterodimeric intermediate during alpha(2)beta(2) protein assembly. Iterative annealing at the quaternary structure level. J. Biol. Chem. 2000;275:2786–2794. doi: 10.1074/jbc.275.4.2786. [DOI] [PubMed] [Google Scholar]
- Yee A, et al. Structural proteomics: Toward high-throughput structural biology as a tool in functional genomics. Acc. Chem. Res. 2003;36:183–189. doi: 10.1021/ar010126g. [DOI] [PubMed] [Google Scholar]
- Zhang R, et al. Structure of Bacillus subtilis YXKO—A member of the UPF0031 family and a putative kinase. J. Struct. Biol. 2002;139:161–170. doi: 10.1016/s1047-8477(02)00532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, et al. Production in two-liter beverage bottles of proteins for NMR structure determination labeled with either 15N– or 13C–15N. J. Struct. Funct. Genomics. 2004;5:87–93. doi: 10.1023/B:JSFG.0000029205.65813.42. [DOI] [PubMed] [Google Scholar]