

Abstract
A series of compounds were designed and synthesized as antagonists of cIAP1/2, ML-IAP, and XIAP based on the N-terminus, AVPI, of mature Smac. Compound 1 (GDC-0152) has the best profile of these compounds; it binds to the XIAP BIR3 domain, the BIR domain of ML-IAP, and the BIR3 domains of cIAP1 and cIAP2 with Ki values of 28, 14, 17 and 43 nM, respectively. These compounds promote degradation of cIAP1, induce activation of caspase-3/7, and lead to decreased viability of breast cancer cells without affecting normal mammary epithelial cells. Compound 1 inhibits tumor growth when dosed orally in the MDA-MB-231 breast cancer xenograft model. Compound 1 was advanced to human clinical trials and it exhibited linear pharmacokinetics over the dose range (0.049 to 1.48 mg/kg) tested. Mean plasma clearance in humans was 9 ± 3 mL/min/kg and volume of distribution was 0.6 ± 0.2 L/kg.
Introduction
Apoptosis is a physiological cell-death program that is critical for the maintenance of tissue homeostasis. This process results in the removal of unwanted cells such as those with potentially harmful genomic mutations or alterations in cell-cycle control. Cancer cells, unlike normal cells, are under stress and highly dependent on aberrations in the apoptosis signaling pathways to remain viable. Therefore, drugs that can restore apoptosis in tumor cells might be effective for the treatment of cancer.
Members of the mammalian inhibitor of apoptosis (IAP) family of proteins, including X chromosome-linked IAP (XIAP), cellular IAP 1 (cIAP1), cellular IAP 2 (cIAP2), and melanoma IAP (ML-IAP), are frequently over-expressed in cancer cells,1–5 where they confer protection against a variety of pro-apoptotic stimuli.6–13 The IAP proteins have also been demonstrated to function in the regulation of signal transduction pathways associated with malignancy.14–25In particular, the cIAP proteins regulate TNFα-mediated NF-κB activation via their C-terminal RING ubiquitin E3-ligase domains, which have been shown to ubiquitinate receptor interacting protein (RIP)-1 and NF-κB inducing kinase, NIK.26
Efforts to target the IAP proteins have focused on the design of small molecules that mimic the binding of the endogenous IAP antagonist second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI (Smac/DIABLO)27, 28 to a shallow groove on the surface of select IAP baculoviral IAP repeat (BIR) domains.29 The IAP BIR domains are approximately 80-amino acid zinc-binding domains that are necessary for the anti-apoptotic function of the IAP proteins. The third BIR domain (BIR3) of XIAP is a specific inhibitor of caspase-9,30–33 while the BIR2 domain is necessary for potent inhibition of caspases-3 and -7.34–38 Antagonism of XIAP-mediated inhibition of these caspases is required for efficient caspase-dependent cell death via both the extrinsic death receptor-mediated and the intrinsic mitochondrial-mediated apoptosis pathways.39
The four amino-acid N-terminus of mature Smac (AVPI) is capable of antagonizing XIAP with a binding affinity of approximately 500 nM against the BIR3 domain.40 AVPI also binds with high affinity to the BIR3 domains of cIAP1/2, the single BIR domain of ML-IAP, and with lower affinity to the BIR2 domain of XIAP. In an effort to uncover lead matter capable of mimicking these interactions, we undertook several large high-throughput-screening campaigns. Screening greater than two million compounds for binding to the ML-IAP BIR domain failed to uncover any viable starting points, thus we relied solely on a peptidomimetic approach. Herein we report the design, synthesis, and evaluation of a series of Smac mimetics that were based on the AVPI tetrapeptide. This peptide sequence has served as the lead structure for several reports detailing the evaluation of monovalent and bivalent small-molecule Smac mimetics capable of antagonizing the IAP proteins.41–46 We sought to evolve the peptide into a compound with improved potency, pharmacokinetic properties, and cell-killing characteristics that would allow us to evaluate the effectiveness of IAP antagonism in human clinical trials. We took a systematic approach to evaluate the contributions of each substituent in the P1 through P4 positions using a combination of structure-based design and targeted compound library generation. The efforts culminated in the discovery of Compound 1 (GDC-0152), a potent antagonist of cIAP1/2, ML-IAP, and XIAP and the first compound targeting this class of proteins to enter clinic trials.
Synthesis
Compounds were prepared using either a solid-phase (Scheme 1) or a solution-phase synthesis (Scheme 2, 3). In the solid-phase method, DFPE polystyrene resin was treated with 2, 2-diphenethylamine and sodium cyanoborohydride to provide amine 2. Substituted proline (R1) and valine (R2) derivatives were prepared using HATU/DIPEA and HBTU/HOBt, DIPEA, respectively, to yield3 and4. N-Capped anilines (6 or7) were made using two procedures from intermediate4. In the first method, Fmoc-alanine was coupled to4 followed by a reductive amination with an aldehyde and sodium cyanoborohydride to give enantiomerically pure compound6. Alternatively, using the peptoid synthesis method developed by Zuckermann,474 was coupled with bromopropionoic acid followed by displacement with a primary amine to yield racemic compound 7. The solution synthesis of P4 derivatives (Scheme 2) all utilized carboxylic acid 8. Primary amines with a saturated or unsaturated two-carbon spacer and an aromatic ring (9) were coupled to the carboxylic acid using EDC/HOBt to yield 10. The synthesis of 1 is outlined in Scheme 3. The thiadiazole 11 was coupled to Boc-proline using EDC/HOBt to yield 12. Boc-cyclohexylglycine and Boc-methylalanine were sequentially coupled to 13 using BOP, which, after deprotection with hydrochloric acid, yielded 1.
Scheme 1.

Solid Phase Synthesis of P1, P2, and P3 Derivatives. Reagents and Conditions; a) 2, 2 – diphenethylamine, NaCNBH3; 1%AcOH/DMF. b) FmocPro, HATU; DIPEA/DMF (X = −CH2-). c) Fmoc-Val(R2 = isopropyl), HBTU/HOBt, DIPEA/DMF. d) FmocAla, HBTU/HOBt, DIPEA/DMF. e) Aldehyde, NaCNBH3; 1%AcOH/DMF. f) TFA/H2O. g) bromopropionoic acid, DIC/DMF. h) Primary amine, DMF. i) TFA/H2O
Scheme 2.
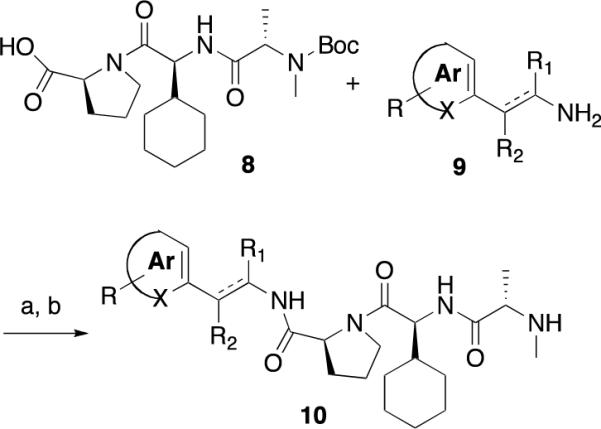
Synthesis of P4 Derivatives. Reagents and Conditions; a) DIC, HOBt, CH2Cl2, 24 hr. b) TFA, CH2Cl2.
Scheme 3.

Reagents and Conditions: a) EDC, HOBt, DIPEA, DMF, 24 hr; b) HCl, dioxane; c) TEA, CH2Cl2; d) Boc-Chg, BOP, DIPEA, DMF 3 hr; e) HCl, dioxane; f) TEA, CH2Cl2; g) Boc-MeAla, BOP, DIPEA, DMF 3hr; h) HCl, dioxane
Results and Discussion
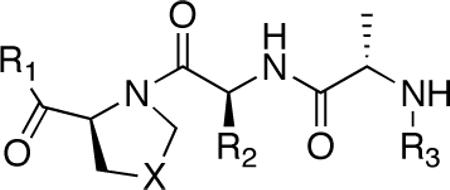
We used an analysis of our previously published 2.3-Å resolution crystal structure of Ala-Val-Pro-2,2-diphenethylamine bound to the BIR domain of ML-IAP48 as our starting point for the design of our compounds. From this structure, we understood that the alanine methyl group in P1 would be difficult to replace as the methyl group was buried within a hydrophobic pocket formed by the side chains of Leu131, Trp134, and Glu143. Additional evidence for the need to maintain this methyl group came from phage-display of naive peptide libraries.49 In that work Ala was the only residue observed at the N-terminus following three sorts of phage libraries of linear peptides against both ML-IAP-BIR and XIAP-BIR3. The other three positions (P2–P4) all tolerated some level of substitution. The N-terminus of the peptide binds in an acidic site on the BIR domains, with the amino group of Ala interacting with the side-chain carboxylates of Asp138 and Glu143 in ML-IAP, Glu314 in XIAP, and Asp320 and Glu325 in cIAP1. Several bond vectors from this nitrogen could be extended to potentially pick up additional binding interactions with the BIR domains. To explore these interactions, libraries of nitrogen-capped derivatives were made, fixing diphenethylamine in P4, proline in P3, and valine in P2 as detailed in Scheme 1. Primary amines and aldehydes were selected for this library by carefully choosing derivatives from randomly generated diversity sets of these compounds that contained functionality that could potentially interact with the BIR domain. Several hundred compounds were made and individually purified by reverse-phase HPLC prior to analysis. The compounds derived from the primary amines (7) were assayed as diastereomeric mixtures and the compounds from the aldehydes (6) were enantiomerically pure. The binding affinities of these compounds for ML-IAP-BIR and XIAP-BIR3 were determined using a fluorescence polarization-based competition assay. No substituent in this position was more effective than a methyl group. For example, as shown in Table 1, replacement of the P1 methyl group with either a cyclopropylmethyl group (6c) or a 2, 3-dihydroxypropyl group (6d) failed to achieve the same level of binding affinity versus either protein.
Table 1.
Structure and Potency of IAP Antagonistsa
 | ||||||
|---|---|---|---|---|---|---|
| Ki(μM)b | ||||||
| Compound | R1 | X | R2 | R3 | XIAP-BIR3 | MLXBIR3SG |
| 6a |  |
CH2 |  |
←CH3 | 0.43c | 0.03c |
| 6b | 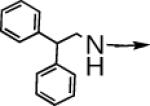 |
S | 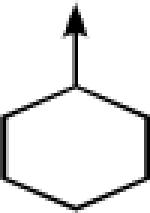 |
←CH3 | 0.19c | 0.03c |
| 6c | 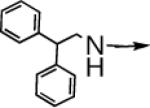 |
CH2 | 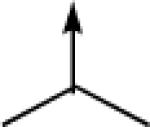 |
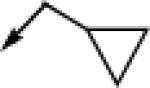 |
7.6 ± 0.5 | 0.46 ± 0.12 |
| 6d | 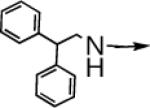 |
CH2 | 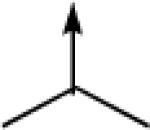 |
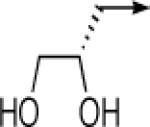 |
0.8 ± 0.2 | 0.08 ± 0.02 |
| 10a | 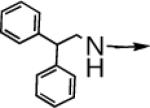 |
CH2 | 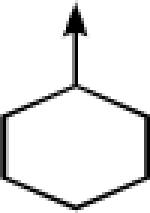 |
←CH3 | 0.41 ± 0.11 | 0.07 ± 0.01 |
| 10b | 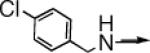 |
CH2 | 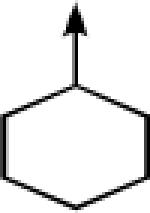 |
←CH3 | 0.7 ± 0.2 | 0.09 ± 0.02 |
| 14 | 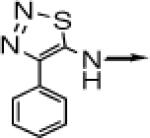 |
CH2 | 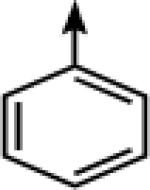 |
←CH3 | 0.035 ± 0.003 | 0.016 ± 0.003 |
| 1 |
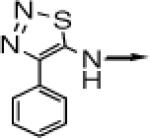 > > |
CH2 | 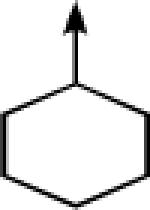 |
←CH3 | 0.028 ± 0.011 | 0.014 ± 0.006 |
Ki determined as described in reference 50.
Average of two measurements.
Single measurement
For the analysis of the P2 position, we fixed methyl-Ala in P1 and proline in P3 utilizing the solid phase synthesis in Scheme 1. The P2 position tolerated the most substitution in our prior peptide-phage selection, thus we anticipated that this area would accommodate a wide range of functionality. The endogenous valine makes van der Waals contact with the β-methylene of Ser133 in ML-IAP, but compared with the other side chains in the sequence this contribution is minimal. Consistent with this observation, very little difference was noted in binding affinity when the valine was replaced with a large hydrophobic substituent such as a cyclohexyl (10a) or t-butyl (data not shown) group. The rotation of bulky P2 side chains is restricted by the presence of a Pro at P3 and these interactions favor a peptide conformation that optimizes the interactions between P2-Pro and the BIR domains. Larger P2 side chains also improve the proteolytic stability of the P2-Pro peptide bond (data not shown).
The proline in P3 productively orients the P1 and P4 substituents. We have published previous efforts to modify this region with a 7–5 bicyclic ring,50 an azabicyclooctane fuzed ring,48 or a thiazoline.51 Substitution directly off the proline ring allows for favorable interactions with Phe148 of ML-IAP or Phe330 of cIAP1-BIR3, but can result in a steric clash with the have selectivity (> 2,000 fold) for cIAP1 over XIAP.52 Given our desire in this work to create a pan-specific compound, we limited our exploration of proline replacements to changes directly in the ring. As illustrated in Table 1, substitution of a methylene with a sulfur is tolerated without any loss of binding affinity (6b).
Having used a solid-phase method to rapidly scan P1–P3, we turned to the solution-phase synthesis outlined in Scheme II to assess the binding contributions of the P4 residue. MeAla, ChGly, and Pro were fixed in the P1, P2 and P3 positions, respectively, and primary amines were added to the terminal carboxylic acid. The peptide-phage data revealed a preference for an aromatic ring in the P4 position over the endogenous isoleucine of Smac. Consistent with this, the crystal structure of Ala-Val-Pro-2,2-diphenethylamine bound to the BIR domain of ML-IAP showed one of the phenyl rings occupying a hydrophobic pocket defined by Thr116, Gly130, Gln132 and the aliphatic portions of Lys121 and Arg123. With this structural information, we limited our initial exploration to primary amines with a one- or two-carbon spacer connected to an aromatic ring. Compounds were selected based on their ability to add additional binding interactions in this well-defined pocket. Of particular interest were the thiadiazole compounds 1 and 14, both of which showed improved binding affinity relative to diphenethylamine. We initiated further studies with compound 1.
To understand the specific interactions resulting in high-affinity interactions, crystal structures were determined of 1 bound to XIAP chimeras of ML-IAP BIR (MLXBIR3SG) and cIAP1 BIR3 (cXBIR3CS) domains (Figure 1). Key interactions are consistent between the two structures including the critical hydrogen-bond interaction with Asp138 in ML-IAP and the corresponding Asp320 in cIAP1. This helps position the α-methyl group of the methyl-Ala in the P1 cavity formed in both complexes. In both examples the thiadiazole heterocycle positions the attached aromatic ring optimally within the hydrophobic P4 pocket.
Figure 1.
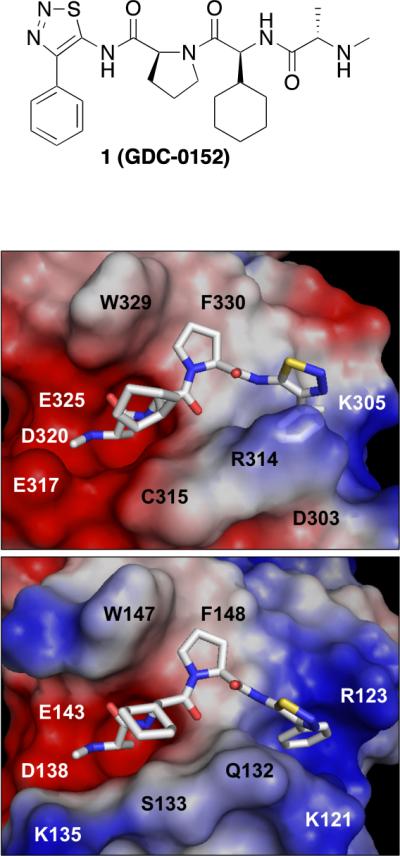
Solvent-accessible surface representation of the peptide-binding site of (A) the 1.79-Å resolution crystal structure of the cIAP1/XIAP chimeric BIR3 domain (cXBIR3CS) (PDB 3UW4), and (B) the 1.71-Å resolution crystal structure of the ML-IAP/XIAP chimeric BIR domain (MLXBIR3SG) (PDB 3UW5), in complex with 1. The protein surfaces are color-coded according to the electrostatic surface potentials: red is negatively charged; blue is positively charged. This figure was produced using the program PyMol (www.pymol.org).
As we desired a pan-selective antagonist we evaluated the thiadiazole (1) for its ability to bind to the BIR domain of ML-IAP and the BIR2 and BIR3 domains of cIAP1, cIAP2 and XIAP using a fluorescence polarization-based competition assay (Figure 2, Table 2). The ML-IAP/XIAP chimeric BIR domain MLXBIR3SG, which retains the native ML-IAP-BIR Smac-peptide-binding site,53 was used as a surrogate for the ML-IAP BIR domain. Compound 1 binds to XIAP-BIR3, cIAP1-BIR3, cIAP2-BIR3, and the single BIR domain of ML-IAP with affinities in the low nanomolar range (Ki < 50 nM). By contrast, the 1 Ki values of 1 for cIAP1-BIR2 and cIAP2-BIR2 are approximately 10 μM.
Figure 2.
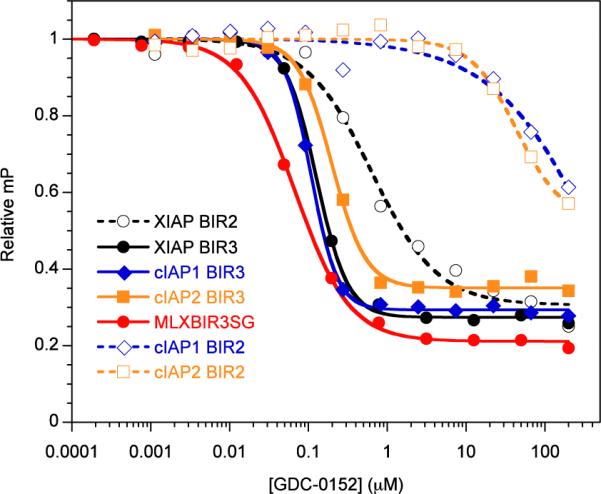
Competitive binding curves for 1 binding to MLXBIR3SG and select BIR domains of XIAP, cIAP1, and cIAP2. The Ki values (Table 1) were calculated from IC50 values as described previously.63
Table 2.
Summary of Ki values (μM) for Compound 1
The ability of 1 to block protein-protein interactions that involve IAP proteins and pro-apoptotic molecules was evaluated by immunoprecipitation and immunoblotting. Using transiently transfected HEK293T cells, 1 was shown to disrupt XIAP binding to partially processed caspase-9 (Figure 3A) and to disrupt the association of ML-IAP, cIAP1, and cIAP2 with Smac (Figure 3B). In melanoma SK-MEL28 cells the endogenous association of ML-IAP and Smac was effectively also abolished by 1 (Supplemental).
Figure 3.

(A) HEK293T cells were transiently transfected with caspase-9 and Flag-tagged XIAP-BIR3 domain or empty vector. Cells were lysed and lysates were incubated with the previously published IAP protein antagonist Compound 14a,48 its non-IAP protein binding stereoisomer, Compound 14a* (a negative control)48, or 1. Immunoprecipitation was performed with anti-Flag antibody and samples were immunoblotted with anti-caspase-9 and anti-Flag antibodies. Whole-cell lysates are shown in the right panels. Astericks denote nonspecific bands from immunoprecipitations.
(B) HEK293T cells were transiently transfected with Smac and Myc-tagged cIAP1, cIAP2, ML-IAP, or empty vector. Cells were lysed and lysates were incubated with the indicated concentrations of 1 and immunoprecipitated with anti-Myc antibody (left panels). Samples were then immunoblotted with anti-Smac and anti-Myc antibodies. Whole-cell lysates are shown in the right panel.
To assess how these interactions with IAP proteins translated into cell killing, compound 1 was tested in a three-day cell viability assay. As shown in Figure 4A, 1 led to a decrease in cell viability in the MDA-MB-231 breast cancer cell line while having no effect on normal human mammary epithelial cells (HMEC). This result was consistent with the moderate cell permeability of 1 in MDCK cells (A − B Papp = 1.8 × 10−6 cm/s).
Figure 4.

(A) MDA-MB-231 breast carcinoma cells (solid symbols) and HMECs (open symbols) were treated with the indicated concentrations of 1. Cell death was assessed using the CellTiter-Glo® luminescent cell viability assay 72 hours following the start of treatment. (B) Effect of 1 (solid symbols) or it's enantiomer (open symbols) on caspase-3 and -7 activities in MDA-MB-231 cells assessed using the Apo-ONE® homogeneous caspase-3/7 assay.
The effect of 1 on the activity of the effector caspases 3 and 7 in the MDA-MB-231 breast cancer cell line was measured to determine the specific mechanism of action. Compound 1 was found to activate caspases 3 and 7 in a dose- and time-dependant manner as shown in Figure 4B. To probe the specificity of the observed effects, the enantiomer of 1 was assessed at the 24-hour time point and found to not activate the caspases at concentrations up to ~10 μM. The potency of the caspase activation correlated well with the cell viability results and provided strong evidence that the cell killing was mediated through the desired apoptosis signaling culminating in caspase-3/7 activation.
Previous studies have demonstrated that Smac mimetics can induce auto-ubiquitination activity and rapid proteasomal degradation of cIAP1.54 Consistent with these studies 1 was shown to induce rapid degradation of cIAP1 in A2058 melanoma cells (Figure 5). Compound 1 effectively induces degradation of cIAP1 at concentrations as low as 10 nM, consistent with its affinity for cIAP1, while it's enantiomer, which does not bind to cIAP1, has no effect following a two-hour exposure at a concentration of 10 μM.
Figure 5.

A2058 cells were treated with increasing amounts of 1 or with 10 mM of its enantiomer for indicated time periods. Cells were lysed and lysates were immunoblotted with anti-c-IAP1 and antibody.
Pharmacokinetic optimization was performed in parallel with the optimization of potency. We focused on the identification of compounds that would enable both preclinical proof of concept in an in vivo efficacy model and testing of IAP antagonism in the clinic. The compounds were not dissolution limited as all had favorable solubility properties. The kinetic solubility of compound 1 was 40 mg/mL at pH = 5.5, 2.0 mg/mL at pH = 6.7, and 0.46 mg/mL at pH = 7.7. Compound 1 had moderate predicted hepatic clearance based on metabolic stability assays conducted using human liver microsomes (data not shown). Additional in vitro ADME evaluation also included an assessment of plasma-protein binding and blood-plasma partitioning. The plasma-protein binding of 1 was evaluated in mouse, rat, rabbit, dog, monkey, and human plasma by equilibrium dialysis using [14C]1. Plasma-protein binding of 1 was moderate and comparable among mice (88–91%), rats (89–91%), dogs (81–90%), monkeys (76–85%), and humans (75–83%) over the range of concentrations investigated (0.1–100 μM); higher plasma-protein binding was observed in rabbits (95–96%). Compound 1 did not preferentially distribute to red blood cells with blood-plasma partition ratios ranging from 0.6 to 1.1 in all species tested.
The in vivo pharmacokinetics for 1 were evaluated initially in mice in order to enable efficacy studies in human-tumor mouse xenograft models. The mono-trifluoroacetic acid salt of 1 was dosed orally in female mice at 100 mg/kg as a solution in phosphate-buffered saline (PBS). Significant exposure was achieved with a Cmax of 53.7 μM, and AUC of 203.5 hr·μM. The in vivo efficacy studies of 1 were performed using human-tumor xenograft mouse models of MDA-MB-231 breast cancer with a variety of dosing schedules and regimens. These in vivo studies demonstrated that 1 has robust anti-tumor activity as a single agent as shown in Figure 6. Dosing once weekly with 10, 50, or 100 mg/kg resulted in significant tumor volume reduction (p-value = < 0.001 for all doses). Similar results were obtained when dosed at 10 mg/kg daily or every third day for three weeks. The initial rate of regression was greatest with doses of 100 mg/kg given once a week, although by Day 21 all groups dosed with 1 had experienced between 85 and 95% inhibition of tumor growth, with four to six complete responses, relative to the vehicle-control group. In mice that experienced less than a partial response (defined as a > 50% but < 100% reduction in tumor volume, compared with the starting tumor volume), tumors resumed growth after the final doses of 1 were administered. Notably, 1 was well tolerated by all groups and by Day 28 all remaining mice had a mean gain in body weight of ≥ 5%. A transient loss in body weight was observed for mice that were given weekly doses of 100 mg/kg, although body weights were similar among all groups on Day 28.
Figure 6.

Efficacy of 1 compared to vehicle control in human-tumor xenograft mouse models of MDA-MB-231 breast cancer. Compounds were dosed as an oral solution at the indicated time intervals and doses. Compound 1 showed significant dose-dependant growth inhibition throughout dosing (p-value = <0.001 for all doses).
The preclinical pharmacokinetics of 1 were investigated in mouse, rat, dog, and monkeys following single-dose intravenous (IV) administration. Following a low IV dose (1 mg/kg), 1 had a high plasma clearance (> 80% of hepatic blood flow) in nude mice, Sprague-Dawley rats, and cynomolgus monkeys and a moderate plasma clearance (52% of hepatic blood flow) in beagle dogs (Table 3). At high doses (100 mg/kg in mice; 20 mg/kg in rats, dogs, and monkeys), mechanisms of clearance from plasma became saturated in mouse and rat, but not in the monkey and dog.
Table 3.
Pharmacokinetics parameters (mean ± SD) of 1 in mouse, rat, dog, and monkey after IV bolus administration.
| Parameter | Nude Mouse | Sprague-Dawley Rat | Beagle Dog | Cynomolgus Monkey |
|---|---|---|---|---|
| Sex | Female | Male | Male | Male |
| N | 27 | 3 | 3 | 3 |
| Low Dose (mg/kg) | 1 | 1 | 1 | 1 |
| CL (mL/min/kg) | 72 | 67 ± 10 | 16 ± 3 | 47 ± 4 |
| Vss (L/kg) | 1.6 | 2.0 ± 0.3 | 1.0 ± 0.2 | 1.60 ± 0.04 |
| t1/2 (hr) | 0.4 | 0.43 ± 0.05 | 0.9 ± 0.1 | 0.44 ± 0.05 |
| Cmax (μM) | 1.4 | 1.1 ± 0.2 | 3.8 ± 0.1 | 1.46 ± 0.04 |
| AUC0–∞ (hr•μM) | 0.5 | 0.51 ± 0.08 | 2.1 ± 0.4 | 0.71 ± 0.06 |
| Sex | Female | Male | Male/Female | Male |
| N | 27 | 3 | 2 | 3 |
| High Dose (mg/kg) | 100 | 20 | 20 | 20 |
| CL (mL/min/kg) | 23 | 27 ± 2 | 19 | 36 ± 6 |
| Vss (L/kg) | 1.7 | 1.7 ± 0.2 | 1.5 | 2.2 ± 0.3 |
| t1/2 (hr) | 2.9 | 0.84 ± 0.06 | 2.0 | 0.77 ± 0.01 |
| Cmax (μM) | 118.6 | 31 ± 4 | 30 | 24 ± 3 |
| AUC0–∞ (hr•μM) | 147 | 25 ± 2 | 36 | 19 ± 3 |
AUC0–∞ = area under the concentration–time curve from time zero to infinity; CL = total plasma clearance; Cmax = highest observed plasma concentration of 1; t1/2 = terminal half-life; Vss = volume of distribution at steady state. Compound 1 hydrochloride salt in 10% hydroxypropyl-β-cyclodextrin was used in the mouse studies; 1-TFA salt in phosphate-buffered saline was used in the rat, monkey, and dog studies.
Human clearance was predicted using in vivo preclinical pharmacokinetic data and allometry adjusted for maximum life potential (Figure 7) or using in vitro-in vivo extrapolation of hepatocyte stability data. Both methods resulted in similar moderate human clearance predictions of 9.6 (allometry) and 10 mL/min/kg (in vitro-in vivo extrapolation). Human volume of distribution was predicted to be moderate at 1.2 L/kg using allometry (Figure 7).
Figure 7.

Prediction of human clearance and volume of distribution of Compound 1 using allometric methods for clearance (CL) and volume of distribution (Vss).
Based on the human PK predictions, the anti-tumor efficacy, and the favorable safety profile observed in preclinical species, compound 1 was advanced for evaluation in humans. In humans, 1 exhibited linear pharmacokinetics over the dose range (0.049 to 1.48 mg/kg) tested (Figure 8). Mean plasma clearance was 9 ± 3 mL/min/kg and volume of distribution was 0.6 ± 0.2 L/kg (Table S2), in good agreement with preclinical predictions.
Figure 8.

Compound 1 pharmacokinetics in humans. (A) Cycle 1 mean (± SD) plasma concentration versus time profiles of Compound 1 by cohort. (B) Plasma exposure (AUCinf) of 1 plotted versus dose.
Conclusion
Using a combination of structure-based design and solid-phase library synthesis we have made a series of compounds that mimic the four-amino acid N-terminus of mature Smac. From these studies, 1 was found to be a potent antagonist of cIAP1, cIAP2, ML-IAP, and XIAP with the ability to disrupt IAP binding to pro-apoptotic proteins such as partially processed caspase-9 and Smac. X-ray crystallographic analysis confirmed that 1 binds to the Smac-binding site of the cIAP1 BIR3 and ML-IAP BIR domains with a binding mode very similar to that observed for Smac-derived peptides. As described previously for Smac mimetics, 1 also induces auto-ubiquitination activity and rapid proteasomal degradation of cIAP1. Cell-based assays confirmed that 1 promoted activation of caspase-3 and -7, resulting in cell killing in a three-day viability assay. Administration of 1 orally led to effective tumor reduction in a human-tumor xenograft mouse model of MDA-MB-231 breast cancer when dosed as a single agent. Predictions of human pharmacokinetics, using in vivo preclinical pharmacokinetic data and allometry or in vitro-in vivo extrapolation of hepatocyte stability data, suggested that 1 would exhibit moderate clearance in humans with a moderate volume of distribution. Based on the described properties as a whole, 1 was selected as the first compound targeting IAP proteins to enter clinic trials. Pharmacokinetics observed in humans were well behaved and consistent with preclinical predictions.
Experimental Section
All chemicals were purchased from commercial suppliers (Aldrich, VWR). Flash chromatography was carried out with RediSep pre-packed SiO2 cartridges on an ISCO Companion chromatography system. NMR spectra were recorded on a Varian Inova 400 system, and referenced to tetramethylsilane. Preparative HPLC was performed on Polaris C18 5 μm column (50 × 21 mm). Low-resolution mass spectra were recorded on a Sciex 15 in ES+ mode. Final compounds was also purified on a Berger Instruments SFC system operating at 100 bar, 35°C; column = Berger Diol 4.6 × 150 mm; with a six-minute gradient of 20–60% MeOH in CO2 flowing 2.35 mL/min (SFCd method) or a Berger Pyridine column 4.6 × 150 mm; with a six-minute gradient of 5–50% MeOH in CO2 flowing 2.35 mL/min (SFCp). Purity analysis of final compounds was performed by a 20 minute HPLC-MS analysis with a Chromasil C18 column (4.6 × 50 mm) with a gradient of 0–90% acetonitrile (containing 0.038% TFA) in 0.1% aqueous TFA (over 17 min, flow = 2 mL/min). All final compounds were purified to > 95% purity.
(S)-tert-butyl 2-(4-phenyl-1,2,3-thiadiazol-5-ylcarbamoyl)pyrrolidine-1-carboxylate (12)
4-phenyl-1,2,3-thiadiazol-5-amine (1.0 g, 5.64 mmol), Boc-L-Proline (1.28 g, 5.94 mmol), EDC (1.14 g, 5.94 mmol), HOBt, (0.8 g, 5.94 mmol) DIPEA (1.04 mL, 5.94 mmol) were dissolved in 5 mL DMF and stirred for 24 hours at room temperature. EtOAc (100 mL) was added to the solution and the organic layer was washed with saturated aqueous NaHCO3 (20 mL) and separated. The aqueous layer was extracted with two 100 mL portions of EtOAc and the organic layers combined. The combined organic layer was washed one time with saturated aqueous NaHCO3 (20 mL), washed twice with saturated brine (20 mL), dried over MgSO4 and concentrated to a solid residue. The crude material was purified by reverse phase HPLC to obtain compound 12 as a 2/1 mixture of rotomers (1.60 g, 76%). 1H NMR (400 MHz, DMSO) δ ppm 11.90 (s, 0.66H), 11.85 (s, 0.33H), 7.68 – 7.75 (m 2H), 7.50 – 7.63 (m, 3H), 4.69 (dd, J = 8.41, 3.42 Hz, 0.33H), 4.63 (dd, J = 8.13, 4.12 Hz, 0.66H), 3.32 – 3.51 (m, 2H), 2.11 – 2.26 (m, 1H), 1.82 – 1.97 (m, 3H), 1.42 (s, 3H), 1.21 (s, 6H). 13C NMR (100 MHz, DMSO) δ ppm 23.30, 23.94, 27.75, 28.01, 29.76, 30.89, 46.52, 46.70, 58.60, 58.73, 78.77, 79.17, 128.68, 128.74, 128.85, 128.94, 130.57, 145.40, 147.68, 152.74, 152.78, 172.37, 173.06. MS (ESI): m/z (M+H)+ 375.1.
(S)-1-((S)-2-cyclohexyl-2-((S)-2-(methylamino)propan-amido)acetyl)-N- (4-phenyl-1,2,3-thiadiazol-5-yl)pyrrolidine-2-carboxamide (1)
Amide 12 (2.32 g, 6.2 mmol) was treated with 4N HCl/dioxane (25 mL) for 30 minutes and the solution was concentrated. TEA/CH2Cl2 (10:90, 20 mL) was added and the solution was stirred for 5 minutes and concentrated to a solid residue. Boc-L-Cyclohexylglycine (1.6 g, 6.2 mmol), BOP (2.7 g, 6.2 mmol) and DIPEA (2.16 mL, 12.4 mmol) in 10 mL DMF were added and stirred for 3 hours at room temperature. EtOAc (100 mL) was added to the solution and the organic layer was washed with saturated aqueous NaHCO3 (20 mL) and separated. The aqueous layer was extracted with two 100 mL portions of EtOAc and the organic layers combined. The combined organic layer was washed one time with saturated aqueous NaHCO3 (20 mL), washed twice with saturated brine (20 mL), dried over MgSO4 and concentrated to a solid residue. The crude reaction mixture was treated with 25 mL 4N HCl/dioxane for 30 minutes. The solution was concentrated, TEA/CH2Cl2 (10:90, 20 mL) was added and stirred for 5 minutes and the solution was concentrated to a solid residue. Boc-N-Methylalanine (1.26, 6.2 mmol), BOP (2.7 g, 6.2 mmol) and DIPEA (2.16 mL, 12.4 mmol) in DMF (10 mL) were added and the solution was stirred for 3 hours at room temperature. EtOAc (100 mL) was added to the solution and the organic layer was washed with saturated aqueous NaHCO3 (20 mL) and separated. The aqueous layer was extracted with two 100 mL portions of EtOAc and the organic layers combined. The combined organic layer was washed one time with saturated aqueous NaHCO3 (20 mL), washed twice with saturated brine (20 mL), dried over MgSO4 and concentrated to a solid residue. The crude reaction mixture was treated with 25 mL 4N HCl/dioxane for 30 minutes. The solution was concentrated, TEA/CH2Cl2 (10:90, 20 mL) was added and stirred for 5 minutes and the solution was concentrated to a solid residue. Boc-N-Methylalanine (1.26, 6.2 mmol), BOP (2.7 g, 6.2 mmol) and DIPEA (2.16 mL, 12.4 mmol) in DMF (10 mL) were added and the solution was stirred for 3 hours at room temperature. EtOAc (100 mL) was added to the solution and the organic layer was washed with saturated aqueous NaHCO3 (20 mL) and separated. The aqueous layer was extracted with two 100 mL portions of EtOAc and the organic layers combined. The combined organic layer was washed one time with saturated aqueous NaHCO3 (20 mL), washed twice with saturated brine (20 mL), dried over MgSO4 and concentrated to a solid residue. The crude material was purified by reverse phase HPLC. The purified material was treated with 25 mL 4N HCl/dioxane for 30 minutes, the solution concentrated to a solid residue and purified by reverse phase HPLC to obtain 1 (2.28 g, 74%) 1H NMR (400 MHz, CDCl3) δ ppm 7.75 (d, J = 7.23 Hz, 2H), 7.62 (d, J = 9.08 Hz, 1H), 7.59 (t, J = 7.69 Hz, 2H), 7.50 (t, J = 7.38 Hz, 1H), 4.91 (dd, J = 8.08, 1.92 Hz, 1H), 4.45 (d, J = 8.23 Hz, 1H), 3.87 (q, J = 8.56 Hz, 1H), 3.58 (q, J = 5.04 Hz, 1H), 3.06 (q, J = 6.92 Hz, 1H), 2.69 (d, J = 12.46 Hz, 1H), 2.45 (s, 3H), 2.03–2.19 (m, 2H), 1.87–2.03 (m, 1H), 1.40–1.69 (m, 1H), 1.35–1.60 (m, 5H), 1.32 (d, J = 10.31 Hz, 3H), 0.79–1.02 (m, 5H). 13C NMR (100 MHz, CDCl3) δ ppm 175.13, 173.84, 168.92, 148.34, 144.87, 130.52, 129.20, 129.23, 128.92, 128.28, 128.27, 60.34, 59.72, 54.58, 47.99, 40.35, 35.21, 29.65, 28.17, 25.87, 25.74, 25.65, 25.63, 25.11, 19.62. MS (ESI): m/z (M+H)+ 499.7
Preparation of Compounds 6a–6d. 2,2-diphenylethylamine (5.3 g, 27 mmol) and NaCNBH3 (1.7 g, 27 mmol) were added to 2-(3,5-dimethoxy-4-formylphenoxy)ethoxymethyl (DFPE) polystyrene resin (5 g, 4.5 mmol) in 1% HOAc/DMF (200 mL) and nitrogen was bubbled through the reaction mixture for 3 days at room temperature. The resin was dried and washed with DMF (2 × 100 mL) and CH2Cl2 (2 × 100 mL) and dried. Fmoc-Pro (4.6 g, 13.5 mmol), HATU (5.1 g, 13.5 mmol) and DIPEA (4.7 mL, 27 mmol) in 1:1 DCM/DMF (100 mL) were added to the resin and nitrogen was bubbled through the reaction mixture for four hours. The resin was dried and washed with DMF (2 × 100 mL) and CH2Cl2 (2 × 100 mL) and dried to yield Resin 3. Resin 3 was treated with 20% piperidine/DMA (100 mL) for 30 minutes and the resin was dried and washed with DMF (2 × 100 mL) and CH2Cl2 (2 × 100 mL) and dried. Fmoc Val (4.6 g, 13.5 mmol), HBTU (5.1 g, 13.5 mmol), and DIPEA (4.7 mL, 27 mmol) were added to 1:1 DCM/DMF (100 mL) and nitrogen was bubbled through the reaction mixture for 3 hours. The resin was dried and washed with DMF (2 × 100 mL) and CH2Cl2 (2 × 100 mL) and treated with 20% piperidine/DMA (100 mL) for 30 minutes. The resin was dried and washed with DMF (2 × 100 mL) and CH2Cl2 (2 × 100 mL) and dried to yield resin 5. The aldehyde (13.5 mmol) and NaCNBH3 (0.85 g, 13.5 mmol) were added to resin 5 in 1% HOAc/DMF (100 mL) and nitrogen was bubbled through the reaction mixture for 3 days at room temperature. TFA and H2O (95:5, 100 mL) were added and nitrogen was bubbled through the solution for 1 hour. The solution was concentrated to a solid residue and purified by reverse phase HPLC to yield the product.
(S)-N-(2,2-diphenylethyl)-1-((S)-3-methyl-2-((S)-2-(methylamino)propanamido)butanoyl)pyrrolidine-2-carboxamide (6a)
1H NMR (400 MHz, CD3OD) δ 7.35 – 7.22 (m, 8H), 7.22 – 7.10 (m, 2H), 4.45 (t, J = 9.5 Hz, 1H), 4.28 – 4.19 (m, 2H), 4.05 (ddd, J = 13.4, 9.0, 7.0 Hz, 1H), 3.92 – 3.83 (m, 1H), 3.84 – 3.73 (m, 1H), 3.67 – 3.53 (m, 2H), 2.64 (d, J = 3.1 Hz, 3H), 2.18 – 2.03 (m, 1H), 2.00 – 1.77 (m, 3H), 1.50 (t, J = 5.6 Hz, 1H), 1.45 (d, J = 7.0 Hz, 3H), 1.05 (d, J = 6.8 Hz, 3H), 0.99 (d, J = 6.7 Hz, 3H), 0.97 – 0.86 (m, 1H). 13C NMR (75 MHz, CD3OD) δ 173.62, 172.64, 172.11, 170.80, 168.78, 168.18, 161.85, 161.40, 158.50, 156.69, 144.64, 142.38, 132.91, 128.70, 128.40, 128.34, 128.29, 128.06, 127.89, 127.47, 126.49, 126.44, 117.95, 83.38, 60.53, 60.43, 57.36, 57.17, 57.07, 56.76, 50.86, 50.58, 44.11, 43.80, 32.03, 31.89, 30.79, 30.43, 29.61, 24.80, 22.13, 18.75, 18.50, 17.54, 17.41, 15.49, 15.29. LC/MS (ESI+): m/z 480 (M+H)
(R)-3-((S)-2-cyclohexyl-2-((S)-2-(methylamino)propanamido)acetyl)-N-(2,2-diphenylethyl)thiazolidine-4-carboxamide (6b)
1H NMR (400 MHz, CDCl3) δ 7.34 – 7.23 (m, 8H), 7.20 (ddd, J = 10.1, 8.6, 6.1 Hz, 2H), 5.03 (d, J = 8.3 Hz, 1H), 4.60 – 4.51 (m, 1H), 4.49 (s, 2H), 4.27 (t, J = 7.9 Hz, 1H), 4.16 (dd, J = 14.0, 7.2 Hz, 1H), 3.90 (s, 1H), 3.73 (ddt, J = 8.8, 6.0, 4.3 Hz, 1H), 3.60 (d, J = 19.8 Hz, 1H), 2.91 (dd, J = 12.0, 7.2 Hz, 1H), 2.63 (d, J = 9.3 Hz, 1H), 2.55 (s, 3H), 1.66 (t, J = 24.0 Hz, 4H), 1.35 (d, J = 5.3 Hz, 3H), 1.28 – 1.07 (m, 4H), 1.08 – 0.85 (m, 3H). 13C NMR (101 MHz, CD3OD) δ 169.00, 168.60, 168.44, 142.63, 142.52, 128.33, 128.23, 127.85, 127.80, 126.29, 126.23, 61.73, 55.62, 55.49, 49.98, 49.42, 42.96, 33.00, 30.59, 28.41, 27.79, 25.66, 25.45, 25.34, 15.70, 15.56. LC/MS (ESI+): m/z 539 (M+H)
(S)-1-((S)-2-((S)-2-(cyclopropylamino)propanamido)-3-methylbutanoyl)-N-(2,2-diphenylethyl)pyrrolidine-2-carboxamide (6c)
1H NMR (400 MHz, CD3OD) δ 7.37 – 7.23 (m, 8H), 7.23 – 7.09 (m, 2H), 4.44 (t, J = 6.7 Hz, 1H), 4.30 – 4.17 (m, 2H), 4.10 – 3.93 (m, 2H), 3.88 – 3.75 (m, 1H), 3.69 – 3.50 (m, 2H), 2.89 (dt, J = 14.4, 7.2 Hz, 1H), 2.76 (dt, J = 12.7, 7.7 Hz, 1H), 2.22 – 2.01 (m, 1H), 2.00 – 1.74 (m, 3H), 1.51 (t, J = 6.3 Hz, 1H), 1.47 (d, J = 7.0 Hz, 3H), 1.13 – 1.01 (m, 3H), 0.98 (d, J = 6.7 Hz, 3H), 0.92 (dd, J = 10.7, 6.8 Hz, 1H), 0.77 – 0.61 (m, 2H), 0.43 – 0.25 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 172.82, 172.74, 172.23, 170.88, 170.70, 168.93, 168.35, 142.56, 142.53, 142.41, 128.41, 128.36, 128.29, 128.06, 127.94, 127.90, 126.52, 126.49, 126.44, 60.34, 60.30, 56.96, 56.64, 55.47, 55.23, 51.14, 51.05, 50.41, 43.98, 43.75, 43.64, 31.86, 31.58, 30.19, 29.46, 24.63, 21.92, 18.56, 18.32, 17.37, 17.19, 15.63, 15.47. LC/MS (ESI+): m/z 538 (M+H)
(S)-1-((S)-2-((S)-2-((S)-2,3-dihydroxypropylamino)propanamido)-3-methylbutanoyl)-N-(2,2-diphenylethyl)pyrrolidine-2-carboxamide (6d)
1H NMR (400 MHz, CD3OD) δ 7.36 – 7.24 (m, 8H), 7.23 – 7.12 (m, 2H), 4.49 – 4.40 (m, 1H), 4.32 – 4.17 (m, 1H), 4.09 – 4.00 (m, 2H), 3.92 – 3.86 (m, 1H), 3.79 (t, J = 14.5 Hz, 1H), 3.67 – 3.55 (m, 3H), 3.55 – 3.45 (m, 1H), 3.21 – 3.11 (m, 1H), 3.02 – 2.89 (m, 1H), 2.10 (dt, J = 14.1, 4.7 Hz, 1H), 2.01 – 1.77 (m, 3H), 1.52 – 1.47 (m, 3H), 1.44 (t, J = 6.1 Hz, 1H), 1.05 (dd, J = 6.8, 2.1 Hz, 3H), 1.00 – 0.96 (m, 3H), 0.93 (dd, J = 12.1, 6.8 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 172.84, 170.86, 169.77, 168.95, 161.54, 142.53, 128.35, 128.30, 128.07, 127.96, 127.90, 127.73, 126.50, 126.44, 67.28, 67.20, 63.94, 60.35, 57.00, 56.70, 55.84, 50.72, 50.44, 48.88, 43.75, 31.61, 30.31, 30.22, 29.46, 24.61, 21.90, 18.56, 18.32, 17.42, 17.39, 17.25, 16.56, 16.41, 15.50, 15.28. LC/MS (ESI+): m/z 540 (M+H)
(S)-1-((S)-2-cyclohexyl-2-((S)-2-(methylamino)propanamido)acetyl)-N-(2,2-diphenylethyl)pyrrolidine-2-carboxamide (10a)
Boc-MeAla-Chg-Pro (8) (50 mg, 0.11 mmol) was diluted with CH2Cl2 (10 mL), and treated with 2, 2-diphenylethylamine (33.67 mg, 0.171 mmol), DIC (0.03 mL, 0.171 mmol) and HOBT (28 mg, 0.171 mmol). The reaction was stirred at room temperature for 1 hour and the reaction mixture was diluted with CH2Cl2 (100 mL), washed with water (2 × 20 mL) and the organic layer was dried, concentrated, and purified by ISCO chromatograpy (50–80% EtOAc/Hexane) to yield the product. The compound was treated with 1:1 TFA and CH2Cl2 at room temperature for 30 minutes. The reaction mixture was diluted with CH2Cl2 (100 mL), washed with water (2 × 20 mL) and the organic layer was dried, concentrated, and purified by HPLC to yield the product. (48 mg, 85%).
1H NMR (500 MHz, DMSO) δ 7.84 (dt, J = 11.0, 5.5 Hz, 1H), 7.81 (d, J = 9.0 Hz, 1H), 7.36 – 7.30 (m, 1H), 7.38 – 7.23 (m, 9H), 7.23 – 7.08 (m, 2H), 4.35 (dd, J = 16.9, 8.4 Hz, 1H), 4.20 – 4.12 (m, 2H), 3.93 (ddd, J = 22.1, 13.6, 7.5 Hz, 1H), 3.69 – 3.58 (m, 1H), 3.54 – 3.48 (m, 1H), 3.48 – 3.41 (m, 1H), 2.91 (q, J = 6.8 Hz, 1H), 2.13 (d, J = 10.5 Hz, 3H), 1.83 – 1.55 (m, 10H), 1.13 (dt, J = 11.3, 8.5 Hz, 2H), 1.06 (t, J = 9.3 Hz, 3H), 1.03 – 0.81 (m, 3H). 13C NMR (500 MHz, DMSO) δ 173.4, 171.1, 169.3, 142.8, 129.1, 128.1, 126.3, 59.2, 54, 50.2, 46.9, 42.8, 34.2, 29.2, 28.9, 27.9, 25.9, 25.7, 25.5, 24.1, 19.1. LC/MS (ESI+): m/z 534 (M+H)
(S)-N-(4-chlorobenzyl)-1-((S)-2-cyclohexyl-2-((S)-2-(methylamino)propanamido)acetyl)pyrrolidine-2-carboxamide (10b)
Boc-MeAla-Chg-Pro (8) (50 mg, 0.11 mmol) was diluted with CH2Cl2 (10 mL), and treated with 4-chlorophenylmethanamine (24 mg, 0.171 mmol), DIC (0.03 mL, 0.171 mmol) and HOBT (28 mg, 0.171 mmol). The reaction was stirred at room temperature for 1 hour and the reaction mixture was diluted with CH2Cl2 (100 mL), washed with water (2 × 20 mL) and the organic layer was dried, concentrated, and purified by ISCO chromatograpy (50–80% EtOAc/Hexane) to yield the product. The compound was treated with 1:1 TFA and CH2Cl2 at room temperature for 30 minutes. The reaction mixture was diluted with CH2Cl2 (100 mL), washed with water (2 × 20 mL) and the organic layer was dried, concentrated, and purified by HPLC to yield the product. (40 mg, 79%). 1H NMR (500 MHz, DMSO) δ 8.46 – 8.32 (m, 1H), 7.86 (d, J = 8.9 Hz, 1H), 7.42 – 7.31 (m, 2H), 7.28 (t, J = 8.4 Hz, 2H), 4.41 (t, J = 8.1 Hz, 1H), 4.31 (dd, J = 8.0, 5.2 Hz, 1H), 4.27 (t, J = 6.8 Hz, 1H), 4.25 – 4.18 (m, 1H), 3.73 (dt, J = 9.5, 6.6 Hz, 1H), 3.59 (dt, J = 9.5, 6.9 Hz, 1H), 3.01 – 2.88 (m, 1H), 2.17 (d, J = 3.3 Hz, 3H), 2.12 – 2.02 (m, 1H), 1.97 (dd, J = 12.5, 6.1 Hz, 1H), 1.89 – 1.75 (m, 2H), 1.66 (ddd, J = 44.7, 26.1, 13.0 Hz, 6H), 1.19 – 1.11 (m, 2H), 1.08 (dd, J = 10.2, 4.9 Hz, 3H), 1.06 – 0.84 (m, 3H). 13C NMR (500 MHz, DMSO) δ 174.1, 171.6, 169.8, 138.6, 131.1, 128.8, 128, 59.5, 59.2, 54.1, 47.1, 41.5, 34.3, 29.3, 28.9, 27.8, 25.9, 25.6, 25.5, 24.6, 19.1. LC/MS (ESI+): m/z 519 (M+H)
(S)-1-((S)-2-((S)-2-(methylamino)propanamido)-2-phenylacetyl)-N-(4-phenyl-1,2,3-thiadiazol-5-yl)pyrrolidine-2-carboxamide (14)
Amide 12 (1.16 g, 3.1 mmol) was treated with 15 mL 4N HCl/dioxane for 30 minutes and the solution was concentrated. TEA/CH2Cl2 (10:90, 15 mL) was added and the solution was stirred for 5 minutes and concentrated to a solid residue. Boc-L-Phenylglycine (0.8 g, 3.1 mmol), 1.35 g (3.1 mmol) BOP and 1.08 mL (6.2 mmol) DIPEA in 10 mL DMF were added and stirred for 3 hours at room temperature. 100 mL EtOAc was added to the solution and the organic layer was washed with saturated aqueous NaHCO3 (20 mL) and separated. The aqueous layer was extracted with two 100 mL portions of EtOAc and the organic layers combined. The combined organic layer was washed one time with saturated aqueous NaHCO3 (20 mL), washed twice with saturated brine (20 mL), dried over MgSO4 and concentrated to a solid residue. The crude reaction mixture was treated with 25 mL 4N HCl/dioxane for 30 minutes. The solution was concentrated, TEA/CH2Cl2 (10:90, 15 mL) was added and stirred for 5 minutes and the solution was concentrated to a solid residue. (0.63 g, 3.1 mmol) Boc-NMethylalanine, BOP (1.35 g, 3.1 mmol) and DIPEA (1.08 mL, 12.4 mmol) in 10 mL DMF were added and the solution was stirred for 3 hours at room temperature. EtOAc (100 mL) was added to the solution and the organic layer was washed with saturated aqueous NaHCO3 (20 mL) and separated. The aqueous layer was extracted with two 100 mL portions of EtOAc and the organic layers combined. The combined organic layer was washed one time with saturated aqueous NaHCO3 (20 mL), washed twice with saturated brine (20 mL), dried over MgSO4 and concentrated to a solid residue. The crude material was purified by reverse phase HPLC. The purified material was treated with 25 mL 4N HCl/dioxane for 30 minutes, the solution concentrated to a solid residue and purified by reverse phase HPLC to obtain (14) (1.19 g, 78%) 1H NMR (400 MHz, CDCl3) δ 7.88 – 7.76 (m, 2H), 7.74 – 7.61 (m, 2H), 7.58 – 7.47 (m, 1H), 7.29 – 7.21 (m, 1H), 7.22 – 7.12 (m, 2H), 7.04 – 6.92 (m, 2H), 5.54 (s, 1H), 5.02 – 4.89 (m, 1H), 3.78 – 3.58 (m, 1H), 3.24 (t, J = 7.6 Hz, 1H), 3.13 (q, J = 6.9 Hz, 1H), 2.60 – 2.51 (m, 1H), 2.47 (s, 3H), 2.12 – 1.80 (m, 3H), 1.25 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 171.92, 168.96, 167.87, 167.62, 167.27, 157.71, 157.48, 138.10, 136.06, 128.62, 128.56, 128.24, 128.04, 127.88, 127.52, 56.23, 55.79, 54.67, 46.76, 46.66, 31.55, 31.15, 30.49, 29.16, 24.61, 21.83, 20.96, 16.09, 15.38. LC/MS (ESI+): m/z 493 (M+H)
X-ray Crystallography
Compound 1 was soaked into crystals of the chimeric BIR domain MLXBIR3SG in complex with the peptide AVPW using protocols described previously.50 Data for the MLXBIR3SG/1 complex were collected at beamline 9–1 of the Stanford Synchrotron Radiation Lightsource. The starting models for refinement of these structures were derived from a 1.3-Å resolution structure of a different peptidomimetic complex (details to be published elsewhere), which was stripped of the peptidomimetic antagonist molecule and all water molecules within 10 Å of it. After one round of refinement of the antagonist-free model, 1 was built into clear difference electron density visible in Fo – Fc maps. New water molecules were picked automatically using the program Arp/wArp,55 and the entire new complex model was subjected to several rounds of positional, anisotropic B-factor, and translation-libration-screw (TLS) refinement using Refmac5.56 The structure has been deposited with the protein data bank and assigned accession code 3UW5.
DNA encoding for a chimeric human cIAP1 composed of cIAP1 residues 266–354 with the mutation C309S and residues 344–354 replaced by residues 336–348 of XIAP (hereafter referred to as cXBIR3CS) was subcloned into the pET15b vector and expressed in E coli strain BL-21(DE3) grown in TB media supplemented with 1% glycerol, and 50 uM Zinc Acetate. Cultures were transformed and grown at 37 °C until an OD600 of 2.0, induced with 0.5 mM IPTG, grown overnight at 16°C, harvested by centrifugation, and lysed in 50 mM Tris-HCl pH 8.3, 300 mM NaCl, 0.5 mM TCEP, 5 mM imidazole with 2 Complete Protease Inhibitor tablets (Roche). Soluble cXBIR3CS was purified using Ni-NTA agarose (Qiagen) affinity chromatography and eluted in 50 mM Tris-HCl pH 8.3, 300 mM NaCl, 0.5 mM TCEP, 300 mM imidazole. Thrombin was added to the cXBIR3CS-containing fractions followed by dialysis for 36 hours at 4 °C against 20 mM Tris-HCl pH 8.5, 200 mM NaC2, 0.2 mM TCEP, 2 mM CaCl2 to remove the his-tag. Uncleaved material was removed by re-passage over the Ni-NTA agarose column. The flow through was concentrated and loaded onto a S-75 sizing column pre-equilibrated in 20 mM Tris-HCl pH 8.3, 200 mM NaCl, 0.2 mM TCEP. Purified cXBIR3CS was concentrated to 4 mg/mL in 50 mM HEPES, pH 7.2, 300 mM NaCl, 0.2 mM TCEP. Compound 1 (50 mM stock solution in DMSO) was added to a final concentration of 1 mM. The cXBIR3CS-Compound 1 complex crystallized by vapor diffusion from hanging drops containing equal volumes of protein and reservoir solution (0.1 M Tris-HCl pH 8.6, 0.5 M Magnesium Formate). Crystals were cryo-protected in 0.1 M Tris-HCl pH 8.5, 0.5 M Magnesium Formate, 3M Sodium Formate. A dataset was measured at the Advanced Light Source (Berkeley) beamline 5.0.2 and processed with the HKL suite of programs. The structure was solved by molecular replacement with the program Phaser using the BIR3 domain of cIAP2 (PDB code 2UVL) as a search model and was refined with Refmac5. The structure has been deposited with the protein data bank and assigned accession code 3UW4.
Refinement statistics for the complex structures are in Table S1.
Binding Assays
Initial Polarization experiments were performed in order to determine dissociation constants (Kd) between IAP protein BIR domains and fluorescent probes. Samples for fluorescence polarization affinity measurements were prepared by addition of serial dilutions of MLXBIR3SG, XIAP-BIR3, XIAP-BIR2, cIAP1-BIR3, cIAP2-BIR3 in polarization buffer (50 mM Tris [pH 7.2], 120 mM NaCl, 1% bovine globulins, 5 mM DTT, and 0.05% octylglucoside) to 5 nM 5-carboxyflourescein (5-FAM) -conjugated AVP-diphenylalanine-AKK (AVP-diPhe-FAM), or serial dilutions of cIAP1-BIR2 and cIAP2-BIR2 in polarization buffer to 5 nM AVPFAK(5-FAM)K (Hid-FAM). The reactions were read after an incubation time of 30 minutes at room temperature with standard cut-off filters for the fluorescein fluorophore (λex = 485 nm; λem = 530 nm) in 384-well black HE96 plates (Molecular Devices Corp.). Fluorescence polarization values were plotted as a function of the protein concentration, and the EC50 values were obtained by fitting the data to a 4-parameter equation using KaleidaGraph software (Synergy software, Reading, PA). The apparent Kd values were determined from the EC50 values.
Inhibition constants (Ki) for the antagonists were determined by addition of the IAP protein constructs to wells containing serial dilutions of the antagonists or the peptide AVPW, and the Hid-FAM probe or AVP-diPhe-FAM probe, as appropriate, in the polarization buffer. Samples were read after a 30-minute incubation. Fluorescence polarization values were plotted as a function of the antagonist concentration, and the IC50 values were obtained by fitting the data to a 4-parameter equation using KaleidaGraph software. Ki values for the antagonists were determined from the IC50 valued.57
Cells, Reagents and Co-immunoprecipatation
HEK293T, melanoma SK-MEL28 and A2058, and breast cancer MDA-MB-231 cells were obtained from ATCC and maintained in the recommended media. Antibodies against Myc (Upstate), Flag (SIGMA), c-IAP1 (R & D Systems), caspase-9 (Cell Signaling), and Smac (ProSci) were purchased, and ML-IAP antibody was described previously.58 zVAD was purchased from Biomol.
HEK293T cells were transiently transfected with the indicated constructs and 40 h after transfection the cells were lysed in NP40 lysis buffer. Lysates were incubated with 0, 1, 10, or 100 mM concentrations of the indicated test materials for 2 h, followed by immunoprecipitation with anti-Flag or anti-Myc antibodies for 3 h, SDS-PAGE and immuno-blotting with anti-Flag, anti-Myc, anti-SMAC, and anti-caspase-9 antibodies.
SK-MEL28 cells were treated with Gemcitabine (0.5 mM) and 1(0.5 mM) for 20 h in the presence of the caspase inhibitor, zVAD (10 mM). At that time the cells were lysed in NP40 lysis buffer, immunoprecipitated with the anti-Smac antibody, resolved on SDS-PAGE, and immuno-blotted with anti-ML-IAP and anti-Smac antibodies.
Cell Viability and Caspase Activation Assays
Human breast carcinoma MDA-MB-231 were obtained from ATCC. Normal human mammary epithelial cells (HMECs) were obtained from Cambrex Corp.. Cells were dissociated from tissue culture flasks by incubation with Accutase® (Innovative Cell Technology Inc.) for 5–10 minutes. Detached cells were washed with phosphate-buffered saline (PBS) and were resuspended in assay media (MDA-MB-231 cells: RPMI1640 supplemented with 10% fetal bovine serum [Sigma-Aldrich] and 2 mM L-glutamine [GlutaMAX-1; Gibco/Invitrogen Corp.]) or culture media (HMECs: MEBM® with MEGM SingleQuots® provided by Cambrex Corp.). Cells were placed in tissue culture-treated, white-wall (Corning, Inc.) or black-wall (PE Biosystems), clear-bottom, 96-well plates at 1 × 104 cells/well in a volume of 50 μL. The plates were incubated at 37°C and 5% CO2 overnight, the media was removed, and 1 or it's enantiomer were added in assay media. Cells cultured in white-wall, clear-bottom plates were incubated at 37°C and 5% CO2 for 3 days before cell viability was measured using the CellTiter-Glo® luminescent cell viability assay kit (Promega Corp.) according to the manufacturer's instructions. Cells seeded in black-wall, clear-bottom plates were incubated at 37°C and 5% CO2 for 3–24 hours before caspase-3 and -7 homogeneous activities were assessed using the Apo-ONE® caspase-3/7 assay kit (Promega Corp.) according to the manufacturer's instructions.
Tumor Xenograft Study
All procedures involving animals were performed in accordance with the guidelines of the Genentech Institutional Animal Care and Use Committee. Human breast cancer MDA-MB-231 cells were obtained from American Type Culture Collection (Manassas, VA). Cells were resuspended in PBS and the cell suspension was mixed 1:1 with Matrigel (BD Biosciences). The cells (1.5 × 107) were then implanted subcutaneously into the right flank of 130 female nude mice (Charles River Laboratories, Hollister, CA) aged 6–8 weeks. Tumor volumes were calculated using the mean diameter measured with vernier calipers using the formula v = 0.5 × a × b2, were a and b are the largest and smallest perpendicular tumor diameters, respectively. Ten mice with the appropriate mean tumor volume were assigned randomly to each of six groups. The mean tumor volume ± the standard error of the mean (SEM) for all six groups was 168 ± 3 mm3 at the initiation of treatment (Day 0). Mice were dosed 1 or vehicle (PBS) by oral gavage with a dose volume of 4.0 mL/kg, according to the schedule indicated in Figure 6. The mice were observed on each day of the study, and tumor volumes and body weights were measured twice each week. Percent tumor growth inhibition was calculated using the formula %TGI = 100 × (1 – Tumor Volumedose/Tumor Volumevehicle).
Pharmacokinetic Studies in Mouse, Rat, Dog and Monkey
Compound 1 was intravenously administered to Sprague-Dawley rats (n = 3 per dose level), Beagle dogs (n = 3 at 1 mg/kg and n = 2 at 20 mg/kg) and Cynomolgus monkeys (n = 3 per dose level) at doses of 1 and 20 mg/kg. Serial plasma samples were collected following dosing up to 24 hours post-dose. In nude mice (n = 27 per dose level), 1 was administered intravenously at doses of 1 and 100 mg/kg. Mice plasma samples (n = 3 per time point) were collected for up to 24 hours post-dose. Compound 1 plasma concentrations were determined using LC-MS/MS. Pharmacokinetic parameters were determined using non-comparmental analysis.59
Plasma Protein Binding
The plasma protein binding of 1 was determined at 0.1, 1, 10 and 100 μM by equilibrium dialysis using a 96-well equilibrium dialysis block (HTDialysis® LLC, Gales Ferry, CT, USA). Compound 1 (mix of unlabeled + 14C-labeled) in pooled plasma from female CD-1 mice, male rats, female rabbits, male dogs, male monkeys, and male humans (n ≥ 3 for all species). The radioactivity of the post-dialysis buffer and plasma samples were quantified by liquid scintillation counter. The percent unbound fraction in plasma was calculated by dividing the radiation counts in the post-dialysis buffer side by the radiation counts in the post-dialysis plasma side and multiplying by 100.
Blood Plasma Partitioning
Blood-plasma partitioning was assessed at total (unlabeled + 14C-labeled) 1 concentrations of 0.1, 1, 10 and 100 μM in pooled blood from female CD-1 mice, male rats, female rabbits, male dogs, male monkeys, and male humans (n ≥ 3 for all species). The blood samples were incubated at 37°C for 0.5 hours. Radioactivity in blood and plasma samples was quantified by liquid scintillation counter.
Hepatocyte Stability Assay and Hepatic Clearance Prediction
Compound 1 (1 μM) was incubated with human cryopreserved hepatocytes (0.5 × 106 cells/mL) at 37°C for 0, 1, 2, and 3 hours. Concentrations were assessed by LC/MS/MS. Intrinsic clearance based upon hepatocyte stability data was determined using a substrate depletion method and scaled to hepatic clearance using the well-stirred model.60
Allometric Scaling
Allometric scaling based on maximum life expectancy potential (MLP) and simple allometric scaling were used to predict human plasma clearance and volume of distribution, respectively.61
Human Pharmacokinetics
A Phase Ia, first-in-human, open-label, multicenter, standard dose-escalation study (3 + 3 design) of 1 was conducted in patients with locally advanced or metastatic solid malignancies or non-Hodgkin's lymphoma without leukemic phase. Patients were given 1 doses of 0.049, 0.1, 0.2, 0.28, 0.39, 0.54, 0.76, 1.06 or 1.48 mg/kg as a 0.5 hr intravenous infusion every 14 days. Serial plasma samples were collected at predetermined time points up to approximately 24 hours post-dose during the first two cycles. Compound 1 plasma concentrations were measured using LC/MS/MS. Pharmacokinetic parameters were calculated using non-compartmental analysis.62
Supplementary Material
Acknowledgment
We thank members of the DMPK and Purification groups within Genentech Small Molecule Drug Discovery for analytical support. We also thank the staff at the Advanced Light Source and Stanford Synchrotron Radiation Lightsource for their assistance with sample handling and data collection. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The Stanford Synchrotron Radiation Lightsource is a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209).
Abbreviations Used
- IAP
inhibitor of apoptosis.
- XIAP
X chromosome-linked inhibitor of apoptosis.
- ML-IAP
melanoma inhibitor of apoptosis.
- RIP
receptor interacting protein.
- Smac
second mitochondria-derived activator of caspases.
- DIABLO
Direct IAP binding protein with low pI.
- BIR
baculoviral IAP repeat domain.
- HATU
2-(1H-7-Azabenzotriazol-1-yl)--1,1,3,3-tetramethyl uronium hexafluorophosphate Methanaminium.
- DIPEA
diisopropylethylamine.
- BOP
benzotriazol-1-yloxytris(dimethylamino)-phosphonium hexafluorophosphate.
- HBTU
O-Benzotriazole-N,N,N',N'-tetramethyl-uronium-hexafluoro-phosphate.
- HOBt
N-Hydroxybenzotriazole.
- ADME
absorption, distribution, metabolism, and excretion.
- DIC
N, N'-diisopropylcarbodiimide.
- EDC
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide.
- PBS
phosphate-buffered saline.
- IV
intravenous.
- PK
pharmacokinetic.
- AUC
area under the curve.
- DFPE
2-(3,5-Dimethoxy-formylphenoxy)ethyl polystyrene.
- HOBt
N-hydroxybenzotriazole.
- DMF
Dimethylformamide.
Footnotes
Supporting Information Available. X-ray data collection and refinement statistics, summary of clinical pharmacokinetics parameters for 1 from Day 1 Cycle 1, Immunoprecipitation and immunoblotting of melanoma SK-MEL28 cells. This material is available free of charge via the Internet at http://pubs.acs.org.
PDB ID Codes. Compound 1/cXBIR3CS, 3UW4; Compound 1/MLXBIR3SG, 3UW5
References
- 1.Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–1568. doi: 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 2.Imoto I, Tsuda H, Hirasawa A, Miura M, Sakamoto M, Hirohashi S, Inazawa J. Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Research. 2002;62:4860–4866. [PubMed] [Google Scholar]
- 3.Imoto I, Yang ZQ, Pimkhaokham A, Tsuda H, Shimada Y, Imamura M, Ohki M, Inazawa J. Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Research. 2001;61:6629–6634. [PubMed] [Google Scholar]
- 4.Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, Dixit VM. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol. 2000;10:1359–1366. doi: 10.1016/s0960-9822(00)00781-8. [DOI] [PubMed] [Google Scholar]
- 5.Yang L, Cao Z, Yan H, Wood WC. Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Res. 2003;63:6815–6824. [PubMed] [Google Scholar]
- 6.Flygare JA, Vucic D. Development of novel drugs targeting inhibitors of apoptosis. Future Oncology. 2009;5(2):141–144. doi: 10.2217/14796694.5.2.141. [DOI] [PubMed] [Google Scholar]
- 7.Vucic D, Fairbrother WJ. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin Cancer Res. 2007;13:5995–6000. doi: 10.1158/1078-0432.CCR-07-0729. [DOI] [PubMed] [Google Scholar]
- 8.Fischer U, Schulze-Osthoff K. Apoptosis-based therapies and drug targets. Cell Death & Differentiation. 2005;12:942–961. doi: 10.1038/sj.cdd.4401556. [DOI] [PubMed] [Google Scholar]
- 9.Wright CW, Duckett CS. Reawakening the cellular death program in neoplasia through the therapeutic blockade of IAP function. J Clin Invest. 2005;115:2673–2678. doi: 10.1172/JCI26251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer research. 2004;64:7183–7190. doi: 10.1158/0008-5472.CAN-04-1918. [DOI] [PubMed] [Google Scholar]
- 11.Nachmias B, Ashhab Y, Ben-Yehuda D. The inhibitor of apoptosis protein family (IAPs): an emerging therapeutic target in cancer. Semin Cancer Biol. 2004;14:231–243. doi: 10.1016/j.semcancer.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Liston P, Fong WG, Korneluk RG. The inhibitors of apoptosis: there is more to life than Bcl2. Oncogene. 2003;22:8568–8580. doi: 10.1038/sj.onc.1207101. [DOI] [PubMed] [Google Scholar]
- 13.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 14.Asselin E, Mills GB, Tsang BK. XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer research. 2001;61:1862–1868. [PubMed] [Google Scholar]
- 15.Bertrand MJM, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Birkey Reffey S, Wurthner JU, Parks WT, Roberts AB, Duckett CS. X-linked inhibitor of apoptosis protein functions as a cofactor in transforming growth factor-beta signaling. J Biol Chem. 2001;276:26542–26549. doi: 10.1074/jbc.M100331200. [DOI] [PubMed] [Google Scholar]
- 17.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-κB control. Proc Natl Acad Sci USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dogan T, Harms GS, Hekman M, Karreman C, Oberoi TK, Alnemri ES, Rapp UR, Rajalingam K. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat Cell Biol. 2008;10:1447–1455. doi: 10.1038/ncb1804. [DOI] [PubMed] [Google Scholar]
- 19.Hofer-Warbinek R, Schmid JA, Stehlik C, Binder BR, Lipp J, de Martin R. Activation of NF-κB by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J Biol Chem. 2000;275:22064–22068. doi: 10.1074/jbc.M910346199. [DOI] [PubMed] [Google Scholar]
- 20.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc Natl Acad Sci USA. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanna MG, da Silva Correia J, Ducrey O, Lee J, Nomoto K, Schrantz N, Deveraux QL, Ulevitch RJ. IAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibition. Mol Cell Biol. 2002;22:1754–1766. doi: 10.1128/MCB.22.6.1754-1766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJA, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 23.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNF α)-induced NF-κB activation. Journal of Biological Chemistry. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu L, Zhu J, Hu X, Zhu H, Kim HT, LaBaer J, Goldberg A, Yuan J. c-IAP1 cooperates with Myc by acting as a ubiquitin ligase for Mad1. Mol Cell. 2007;28:914–922. doi: 10.1016/j.molcel.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y, Conze DB, Hanover JA, Ashwell JD. Tumor necrosis factor receptor 2 signaling induces selective c-IAP1-dependent ASK1 ubiquitination and terminates mitogen-activated protein kinase signaling. J Biol Chem. 2007;282:7777–7782. doi: 10.1074/jbc.M609146200. [DOI] [PubMed] [Google Scholar]
- 26.Varfolomeev E, Vucic D. (Un)expected roles of c-IAPs in apoptotic and NFκB signaling pathways. Cell Cycle. 2008;7:1511–1521. doi: 10.4161/cc.7.11.5959. [DOI] [PubMed] [Google Scholar]
- 27.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 28.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 29.Flygare J, Fairbrother W. Small-molecule pan-IAP antagonists: a patent review. Expert Opinion on Therapeutic Patents. 2010;20:251–267. doi: 10.1517/13543770903567077. [DOI] [PubMed] [Google Scholar]
- 30.Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;(2):519–527. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 32.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. A conserved XIAP-interaction motif in caspase-9***** and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 33.Sun C, Cai M, Meadows RP, Xu N, Gunasekera AH, Herrmann J, Wu JC, Fesik SW. NMR structure and mutagenesis of the third Bir domain of the inhibitor of apoptosis protein XIAP. J Biol Chem. 2000;275:33777–33781. doi: 10.1074/jbc.M006226200. [DOI] [PubMed] [Google Scholar]
- 34.Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406:855–862. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 35.Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- 36.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 37.Sun C, Cai M, Gunasekera AH, Meadows RP, Wang H, Chen J, Zhang H, Wu W, Xu N, Ng SC, Fesik SW. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature. 1999;(6755):818–822. doi: 10.1038/44617. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC. A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem. 1998;273:7787–7790. doi: 10.1074/jbc.273.14.7787. [DOI] [PubMed] [Google Scholar]
- 39.Silke J, Hawkins CJ, Ekert PG, Chew J, Day CL, Pakusch M, Verhagen AM, Vaux DL. The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase 3- and caspase 9-interacting sites. J Cell Biol. 2002;157:115–124. doi: 10.1083/jcb.200108085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–1008. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 41.Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang C-Y, Miller R, Yi H, Zhang T, Sun D, Kang S, Guo M, Leopold L, Yang D, Wang S. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem. 2011;54:2714–2726. doi: 10.1021/jm101505d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFα-mediated cell death. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 43.Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem. 2004;47:4417–4426. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]
- 44.Sun H, Liu L, Lu J, Bai L, Li X, Nikolovska-Coleska Z, McEachern D, Yang C-Y, Qiu S, Yi H, Sun D, Wang S. Potent bivalent Smac mimetics: effect of the linker on binding to inhibitor of apoptosis proteins (IAPs) and anticancer activity. J Med Chem. 2011;54:3306–3318. doi: 10.1021/jm101651b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun H, Lu J, Liu L, Yi H, Qiu S, Yang CY, Deschamps JR, Wang S. Nonpeptidic and potent small-molecule inhibitors of cIAP-1/2 and XIAP proteins. J Med Chem. 2010;53:6361–6367. doi: 10.1021/jm100487z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S. Design of Small-Molecule Smac Mimetics as IAP Antagonists. Small-Molecule Inhibitors of Protein-Protein Interactions. 2011:89–113. doi: 10.1007/82_2010_111. [DOI] [PubMed] [Google Scholar]
- 47.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. Efficient Method For The Preparation of Peptoids [Oligo(N-Substituted Glycines)] by Submonomer Solid-Phase Synthesis. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 48.Cohen F, Alicke B, Elliott LO, Flygare JA, Goncharov T, Keteltas SF, Franklin MC, Frankovitz S, Stephan JP, Tsui V. Orally Bioavailable Antagonists of Inhibitor of Apoptosis Proteins Based on an Azabicyclooctane Scaffold. J Med Chem. 2009;52:1723–1730. doi: 10.1021/jm801450c. [DOI] [PubMed] [Google Scholar]
- 49.Franklin MC, Kadkhodayan S, Ackerly H, Alexandru D, Distefano MD, Elliott LO, Flygare JA, Mausisa G, Okawa DC, Ong D. Structure and function analysis of peptide antagonists of melanoma inhibitor of apoptosis (ML-IAP) Biochemistry. 2003;42:8223–8231. doi: 10.1021/bi034227t. [DOI] [PubMed] [Google Scholar]
- 50.Zobel K, Wang L, Varfolomeev E, Franklin MC, Elliott LO, Wallweber HJA, Okawa DC, Flygare JA, Vucic D, Fairbrother WJ, Deshayes K. Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem Biol. 2006;1:525–533. doi: 10.1021/cb600276q. [DOI] [PubMed] [Google Scholar]
- 51.Cohen F, Koehler MFT, Bergeron P, Elliott LO, Flygare JA, Franklin MC, Gazzard L, Keteltas SF, Lau K, Ly CQ, Tsui V, Fairbrother WJ. Antagonists of inhibitor of apoptosis proteins based on thiazole amide isosteres. Bioorg Med Chem Lett. 2010;20:2229–2233. doi: 10.1016/j.bmcl.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 52.Ndubaku C, Varfolomeev E, Wang L, Zobel K, Lau K, Elliott LO, Maurer B, Fedorova AV, Dynek JN, Koehler M, Hymowitz SG, Tsui V, Deshayes K, Fairbrother WJ, Flygare JA, Vucic D. Antagonism of c-IAP and XIAP proteins is required for efficient induction of cell death by small-molecule IAP antagonists. ACS Chem Biol. 2009;4:557–566. doi: 10.1021/cb900083m. [DOI] [PubMed] [Google Scholar]
- 53.Vucic D, Franklin MC, Wallweber HJA, Das K, Eckelman BP, Shin H, Elliott LO, Kadkhodayan S, Deshayes K, Salvesen GS, Fairbrother WJ. Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem J. 2005;385:11–20. doi: 10.1042/BJ20041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael-Tomasevic A, Phu L, Helgason E, Fairbrother WJ, Deshayes K, Kirkpatrick DS, Vucic D. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010;29:4198–4209. doi: 10.1038/emboj.2010.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS. ARP/wARP and molecular replacement. Acta Crystallographica Section D-Biological Crystallography. 2001;57:1445–1450. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- 56.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D-Biological Crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 57.Keating S, Marsters J, Beresini M, Ladner C, Zioncheck K, Clark K, Arellano F, Bodary S. Putting the pieces together: Contribution of fluorescence polarization assays to small molecule lead optimization. In-Vitro Diagnostic Instrumentation. 2000;1(7):128–137. [Google Scholar]
- 58.Dynek JN, Chan SM, Liu JF, Zha JP, Fairbrother WJ, Vucic D. Microphthalmia-associated transcription factor is a critical transcriptional regulator of melanoma inhibitor of apoptosis in melanomas. Cancer Research. 2008;68:3124–3132. doi: 10.1158/0008-5472.CAN-07-6622. [DOI] [PubMed] [Google Scholar]
- 59.Gilbaldi M, Perrier D. 2nd Edition. Marcel Dekker Inc.; New York: 1982. Vol. [Google Scholar]
- 60.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ, Wastall P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. Journal of Pharmacology and Experimental Therapeutics. 1997;283:46–58. [PubMed] [Google Scholar]
- 61.Mahmood I, Balian JD. Interspecies scaling: Predicting clearance of drugs in humans. Three different approaches. Xenobiotica. 1996;26:887–895. doi: 10.3109/00498259609052491. [DOI] [PubMed] [Google Scholar]
- 62.Perrier D, Mayersohn M. Non-Compartmental Determination of the Steady-State Volume of Distribution for Any Mode of Administration. Journal of Pharmaceutical Sciences. 1982;71(3):372–373. doi: 10.1002/jps.2600710332. [DOI] [PubMed] [Google Scholar]
- 63.Keating SM, Marsters J, Beresini M, Ladner C, Zioncheck K, Clark K, Arellano F, Bodary S. In Putting the pieces together: contribution of fluorescence polarization assays to small-molecule lead optimization. In: Cohn GE, editor. Proceedings of SPIE: In-Vitro Diagnostic Instrumentation.2000. pp. 128–137. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.