

Abstract
Painful peripheral neuropathy is a dose-limiting complication of chemotherapy. Cisplatin produces a cumulative toxic effect on peripheral nerves, and 30–40% of cancer patients receiving this agent experience pain. By modeling cisplatin-induced hyperalgesia in mice with daily injections of cisplatin (1 mg/kg, i.p.) for 7 d, we investigated the anti-hyperalgesic effects of anandamide (AEA) and cyclohexylcarbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597), an inhibitor of AEA hydrolysis. Cisplatin-induced mechanical and heat hyperalgesia were accompanied by a decrease in the level of AEA in plantar paw skin. No changes in motor activity were observed after seven injections of cisplatin. Intraplantar injection of AEA (10 μg/10 μl) or URB597 (9 μg/10 μl) transiently attenuated hyperalgesia through activation of peripheral CB1 receptors. Co-injections of URB597 (0.3 mg/kg daily, i.p.) with cisplatin decreased and delayed the development of mechanical and heat hyperalgesia. The effect of URB597 was mediated by CB1 receptors since AM281 (0.33 mg/kg daily, i.p.) blocked the effect of URB597. Co-injection of URB597 also normalized the cisplatin-induced decrease in conduction velocity of Aα/Aβ-fibers and reduced the increase of ATF-3 and TRPV1 immunoreactivity in dorsal root ganglion (DRG) neurons. Since DRGs are a primary site of toxicity by cisplatin, effects of cisplatin were studied on cultured DRG neurons. Incubation of DRG neurons with cisplatin (4 μg/ml) for 24 h decreased the total length of neurites. URB597 (100 nm) attenuated these changes through activation of CB1 receptors. Collectively, these results suggest that pharmacological facilitation of AEA signaling is a promising strategy for attenuating cisplatin-associated sensory neuropathy.
Introduction
Cisplatin is a cytotoxic platinum-based drug widely used to treat various types of cancer. Unfortunately, cisplatin is accompanied by an array of dose-limiting side effects that reduce the effectiveness of treatment and affect survival. Since cisplatin at pharmacologic doses does not cross the blood–brain barrier, it almost exclusively damages peripheral tissues, including dorsal root ganglia (DRGs) and sensory fibers (Gregg et al., 1992). The motor system does not appear to be affected (Albers et al., 2011). Damage to DRG cells results in peripheral sensory neuropathy defined clinically and experimentally as a decrease in nerve conduction velocity (Verdú et al., 1999; Carozzi et al., 2009), reduced tactile sensitivity, and hyperalgesia (for review, see Windebank and Grisold, 2008; Joseph and Levine, 2009; Ta et al., 2009). The formation of DNA-platination products is considered the main trigger for the antineoplastic activities of cisplatin (Boulikas and Vougiouka, 2003; McDonald et al., 2005; Dzagnidze et al., 2007), but the mechanisms underlying the hyperalgesia have not been resolved. Since damage to sensory neurons recovers only partially or not at all, therapies have been sought to prevent or decrease neurotoxic effects of cisplatin.
N-Arachidonoyl ethanolamine [anandamide (AEA)], an endogenous cannabinoid, acts as a partial agonist at cannabinoid type-1 receptor (CB1R) (Hillard, 2000; Pertwee and Ross, 2002). Like synthetic CB1R agonists, AEA attenuates hyperalgesia in models of neuropathic (Guindon and Beaulieu, 2006), inflammatory (Guindon et al., 2006; Potenzieri et al., 2008), and tumor (Khasabova et al., 2008) pain. Clinically, the synthetic cannabinoid agonist nabilone reduces chemotherapy-induced pain (Davis, 2008).
Since AEA is hydrolyzed by the intracellular enzyme fatty acid amide hydrolase (FAAH) (Cravatt et al., 1996), inhibiting FAAH increases the availability of endogenous AEA to activate CB1R and reduce hyperalgesia (Chang et al., 2006; Russo et al., 2007; Khasabova et al., 2008; Kinsey et al., 2009; Clapper et al., 2010). CB1R-dependent neuroprotective effects of FAAH inhibitors include attenuation of cytoskeletal damage (Karanian et al., 2005) and reduction of hippocampal neuronal activity in models of excitotoxicity (Coomber et al., 2008). These effects parallel the ability of CB1R agonists to protect neurons against injuries created by ischemia and oxidative stress (for review, see Svízenská et al., 2008; Fowler et al., 2010).
It is not known whether changes in AEA signaling contribute to pain associated with cisplatin treatment and whether increasing AEA signaling reduces hyperalgesia and peripheral neurotoxicity associated with cisplatin. Therefore, the aim of the present study was to determine effects of AEA and URB597 on cisplatin-induced hyperalgesia and test their ability to reduce symptoms of neurotoxicity. Neurotoxicity was defined as an upregulation of activating transcription factor-3 (ATF-3), a marker of cellular injury (Seijffers et al., 2006), and biochemical and functional deficits in peripheral nerves. Our results support the strategy of inhibiting AEA hydrolysis to decrease the neurotoxicity and sensory symptoms associated with cisplatin treatment.
Materials and Methods
Animals.
Adult, male C3H/HeN mice (25–32 g; National Cancer Institute) were used in these studies. All procedures were approved by the University of Minnesota Institutional Animal Care and Use Committee.
Drugs.
The chemotherapeutic agent cisplatin [cis-diamminedichloroplatinum (II); Sigma-Aldrich] was dissolved in sterile saline to the final concentration (1 mg/ml). AEA was dissolved in Tocrisolve100. Stock solutions of the CB1R antagonist 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide (AM281) (10 mg/ml) and the CB2R antagonist 6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone (AM630) (10 mg/ml) were prepared in DMSO. AM281 has 300-fold greater affinity for the CB1 compared with the CB2 receptor (Lan et al., 1999). AM630 has 165-fold greater affinity at CB2 compared with CB1 receptors (Ross et al., 1999). AEA, AM281, AM630, and Tocrisolve were purchased from Tocris. A stock solution of the FAAH inhibitor, cyclohexylcarbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597) (Cayman Chemical), was prepared in ethanol (8 mg/ml). All drugs were diluted in saline to achieve the final dose.
Assessment of body weight.
Mice were weighed daily for the duration of the experiment before behavioral testing or drug injections. At the end of an experiment, the weight of each animal was compared with its weight at the start of the experiment, and the difference was expressed as a percentage of initial body weight.
Rotarod assay of motor function.
To evaluate an effect of cisplatin on motor performance, mice were placed on a rotarod treadmill at an initial speed of 3.75 rpm with progressive acceleration to 5 rpm. A cutoff time of 300 s was used. The time spent on the treadmill before falling off was recorded for each mouse on each day of testing. Baseline measurements were obtained for all mice on 3 consecutive days before the first injection of cisplatin or vehicle.
Measurement of mechanical and heat hyperalgesia.
Mechanical hyperalgesia was defined as an increase in the frequency of paw withdrawal responses to a standard von Frey monofilament that delivered a force of 3.7 mN. Mice were isolated under separate glass containers on a mesh platform and allowed to acclimate for 30 min before testing. The monofilament was applied to the plantar surface of each hindpaw 10 times (1–2 s each). Withdrawal frequency was determined and expressed as a percentage. Daily baseline measurements were obtained for all mice on 3 consecutive days before the first injection of cisplatin or vehicle. These data were used to screen mice for hypersensitivity, and animals that exhibited baseline withdrawal frequencies ≥50% were excluded from the study (<2%).
Sensitivity to heat was determined by measuring the latency in seconds of a withdrawal response to radiant heat applied to the center of the plantar surface of the hindpaw. Mice were placed on an elevated glass platform and allowed to habituate for 30 min. The thermal stimulus was applied three times on each paw, and consecutive applications of radiant heat were separated by at least 3 min. A cutoff time of 15 s was imposed to prevent tissue damage. The latency of response for each hindpaw was defined as the average of the three trials. A decrease in withdrawal latency was defined as heat hyperalgesia.
Mechanical and heat hyperalgesia were monitored in separate groups of mice that received intraperitoneal injections of cisplatin or vehicle (saline) once daily for 7 consecutive days. Behavioral testing occurred ∼23 h after the previous cisplatin injection. To determine the acute, local anti-hyperalgesic effect of cannabinoids, mice were given an intraplantar (i.pl.) drug injection (10 μl volume) into one hindpaw. Withdrawal responses evoked by the monofilament or radiant heat were then measured in each hindpaw before and every 30 min after drug injection for at least 2.5 h. To determine whether URB597 attenuated the development of cisplatin-induced hyperalgesia, cisplatin-treated mice also were given an injection of URB597 (9 μg, i.p., once daily for 7 d). The intraperitoneal route for repeated injections of URB597 was selected to avoid damage to the hindpaw. The experimenter was blinded to all treatments.
Measurement of compound action potential.
Mice were initially anesthetized with injection of sodium pentobarbital (Nembutal; 50 mg/kg, i.p.); supplemental doses (15 mg/kg) were given as needed to maintain areflexia during the experiment. The tibial nerve was exposed between the heel and the knee joint. The sciatic nerve was isolated from the distal portion of the femur to the point of entry into the pelvic girdle. The sheathing was removed from the distal portion of the tibial nerve and from the proximal end of the sciatic nerve. The length of the isolated nerve (20–24 mm) was measured to the nearest millimeter between a unipolar recording electrode on the distal end of the tibial nerve and a proximally located bipolar stimulating electrode on the sciatic nerve. The nerve and surrounding tissues were kept moist during the experiment by repeated application of saline. The sciatic nerve was stimulated using a constant current stimulator and stimulus isolator (World Precision Instruments). Stimulation pulses were 200 μs in duration and were delivered every 5 s. Current was gradually increased from 10 to 1000 μA to obtain electrical thresholds for activation of Aα/Aβ- and C-fibers. The compound action potential (CAP) evoked by stimulation of the sciatic nerve at 1.5× the threshold to activate C-fibers was used for analysis. The CAP was amplified, displayed on an oscilloscope, and stored on a computer for off-line analyses using Spike2 software program (Cambridge Electronic Design). The Aα/Aβ component of the CAP was defined by fibers with conduction velocities >20 m/s; the C component was defined by fibers with conduction velocities <2 m/s.
Western blot analysis.
Samples of tibial nerve (∼1 cm each pooled from two mice) from naive and cisplatin-treated mice were sonicated in single-detergent lysis buffer [50 mm Tris-HCl, pH 8.0, with 1% Triton X-100, 150 mm NaCl, 0.02% Na azide, 100 μg/ml PMSF, and 1 μg/ml protease inhibitor mixture (Sigma-Aldrich)], and the particulate fraction of the supernatant was obtained after serial centrifugation at 800 × g for 10 min and 14,000 × g for 25 min. Western blot analysis was performed on 10 μg of protein, which was loaded onto a 10% SDS-PAGE gel, subjected to electrophoresis, and then transferred onto polyvinylidene difluoride membranes (Bio-Rad Laboratories). Nonspecific binding to membranes was blocked by incubation in Tris-buffered saline with 3% defatted dry milk for 1 h at room temperature. The membranes were probed with a mouse anti-phosphorylated 200 kDa neurofilament (p-NF) protein antibody (clone RT97; 1/2000; Boehringer Mannheim) overnight at 4°C. The primary antibody was detected with a peroxidase conjugate of goat anti-mouse IgG (1:20,000; GE Healthcare). Immunoreactivity (ir) was visualized using the enhanced chemifluorescence detection reagent (Thermo Fisher Scientific). Loading controls were performed with a rabbit anti-actin antibody (1:1000; Sigma-Aldrich). The amount of p-NF protein was defined as the ratio of RT97-ir to actin-ir within the same sample.
Immunohistochemistry.
One day after the seventh injection of cisplatin alone, cisplatin with URB597, or vehicle mice were tested behaviorally to confirm the occurrence of mechanical hyperalgesia and were then prepared for immunohistochemical analysis of DRG samples. Animals were anesthetized deeply with sodium pentobarbital (50 mg/kg, i.p.) and perfused intracardially with 20 ml of PBS, pH 7.35, followed by 100 ml of 4% (w/v) paraformaldehyde in phosphate buffer, pH 6.9. DRGs (L3–L5) were removed, postfixed for 12 h in the perfusion fixative, and cryoprotected by immersion for 24 h in 30% sucrose at 4°C. DRGs were embedded in Tissue-Tek embedding medium (Bayer), rapidly frozen on dry ice, and processed for immunohistochemistry. The DRGs were cut into 15 μm sections on a cryostat and thaw mounted on gelatin-coated slides. Sections were preincubated in a blocking solution of 3% normal donkey serum (Jackson ImmunoResearch Laboratories) with 0.3% Triton X-100 and 0.1% sodium azide for 1 h at room temperature. Approximately every sixth section was incubated with a pool of antibodies for detection of two antigens: rabbit anti-ATF-3 (C-19; 1:500; Santa Cruz Biotechnology) plus mouse anti-neuron-specific nuclear protein antibody (NeuN; 1:100; Millipore Bioscience Research Reagents), or guinea pig anti-TRPV1 (1:500; VR1 C terminus; Neuromics) plus mouse anti-β-tubulin III (1:200; Sigma-Aldrich). Colabeling for NeuN- or β-tubulin III-ir was used to identify neurons (Moskowitz and Oblinger, 1995; Kim et al., 2009). Sections were incubated with primary antibodies overnight at room temperature. After rinses with PBS, tissue sections were incubated for 1 h with a combination of Alexa Fluor 555-conjugated donkey anti-rabbit IgG (1:1000) or DyLight 594-conjugated donkey anti-guinea pig (1:800) with Alexa Fluor 488 donkey anti-mouse IgG (1:1000; Invitrogen) antibodies. Finally, the sections were rinsed in PBS, protected with a medium of glycerol/PBS (5:1 v/v), containing 0.1% p-phenylenediamide, and coverslipped. Immunoreactivity was visualized with an Olympus BX50 fluorescence microscope (Olympus Corporation) using a 10× fluorite objective, and digitized images were obtained with an SCION1394 camera. Specificity of the ATF-3 (C-19) antibody was confirmed with an absorption control. When the diluted antiserum was incubated with the ATF-3 (C-19) blocking peptide (1:1 v/v; Santa Cruz Biotechnology), immunoreactivity was not observed. The specificity of the TRPV1 antibody for TRPV1 protein was previously described (Guo et al., 1999). MetaMorph software (Molecular Devices) was used to define somal area and intensity of immunofluorescence. A neuron was scored “immunopositive” if its average intensity was >2 SDs greater than the mean intensity of neurons labeled with the secondary antibody alone. Only neurons with a visible nucleus were counted.
Immunocytochemistry.
Primary cultures of mouse DRG neurons were prepared from dorsal root ganglia dissected from all levels of the spinal cord as described previously (Khasabova et al., 2007). Cisplatin, URB597, or cannabinoid receptor antagonists were dissolved in medium, and drug treatments were initiated 1 h after plating. Effects of drug treatments on β-tubulin III- or NeuN-ir were determined after 24 h in vitro. Following a wash in HEPES buffer (in mm: 25 HEPES, 135 NaCl, 2.5 CaCl2, 3.5 KCl, 1 MgCl2, and 3.3 glucose, pH 7.4 and 335–340 mOsm), cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and then rinsed with PBS. Cultures were incubated for 1 h in PBS containing 0.1% Na azide, 0.3% Triton X-100, 5% normal donkey serum (Jackson ImmunoResearch Laboratories), and then incubated with mouse anti-β-tubulin III antibody (1:200; Sigma-Aldrich) that specifically recognizes an epitope located on isotype III of β-tubulin. Mouse anti-NeuN-ir was used to determine neuronal survival. Specimens were incubated with primary antibodies overnight at room temperature. After rinses with PBS, samples were incubated for 1 h in FITC-donkey anti-rabbit IgG (1:100; Jackson ImmunoResearch Laboratories) combined with 7-amino-4-methylcoumarin-3-acetic acid-labeled donkey anti-mouse IgG (1:100; Jackson ImmunoResearch Laboratories). Finally, samples were rinsed in PBS, and bound fluorophores were protected by mounting as described above. No immunofluorescence was seen in the absence of primary antibodies. Fluorophores were visualized on a Nikon E400 fluorescence microscope using 10 or 20× fluorite objectives, and digitized images were obtained with a MTI camera. MetaMorph software was used to measure the total length of neurites/neuron. The data were collected by an observer who was blinded to the treatment of the samples.
Measurement of endogenous AEA, 2-arachidonoyl glycerol, and palmitoylethanolamide.
Samples of plantar paw skin were dissected and homogenized with 500 μl of chloroform. Samples of cultured DRG cells were scraped from the bottom of the plate with a rubber policeman, sonicated in HEPES. All samples were mixed with 5 vol of chloroform containing 5 pmol of d8-AEA, 100 pmol of d8-2-arachidonoyl glycerol (d8-2-AG), and 100 pmol of d4-palmitoylethanolamide (d4-PEA) (Cayman Chemical) as internal standards. Extraction of lipids occurred by incubation at 4°C overnight. Mixtures were then homogenized with an equal volume of methanol/Tris-HCl (50 mm) (1:1). Homogenates were centrifuged at 3000 × g for 15 min (4°C). The organic phase was evaporated with a gentle stream of nitrogen gas. Targeted isotope-dilution HPLC/atmospheric pressure chemical ionization/mass spectrometry was conducted on each sample as described previously (Khasabova et al., 2008). The AEA, 2-AG, and PEA levels in experimental samples were estimated from the ratio of the area of deuterated compounds and AEA (0.02–20 pmol), 2-AG (2–2000 pmol), or PEA (2–2000 pmol) standards and were expressed as picomoles per gram of tissue or picomoles per sample (DRG).
Statistical analyses.
Data are presented as the mean ± SEM or the median with the 25th and 75th percentile range. Data were analyzed by one- and two-way ANOVAs (with repeated measures when applicable) followed by post hoc comparisons between groups. Levels of endogenous AEA were analyzed by Student's t test. The occurrence of ATF-3-ir and the length of tubulin-ir neurites were analyzed by Kruskal–Wallis ANOVA on ranks test. For all statistical analyses, a probability value of <0.05 was considered significant.
Results
Assessment of a model of cisplatin-induced hyperalgesia
A range of doses of cisplatin (0.5, 1, and 3 mg/kg, i.p.; daily for 7 d) was used to optimize the development of mechanical hyperalgesia in mice without producing deterioration in general health status. Weight loss (16%) was observed only for mice given the highest dose of cisplatin daily (3 mg/kg; p < 0.05, two-way ANOVA with Bonferroni's t test; n = 6–8 mice/treatment group; Fig. 1A).
Figure 1.
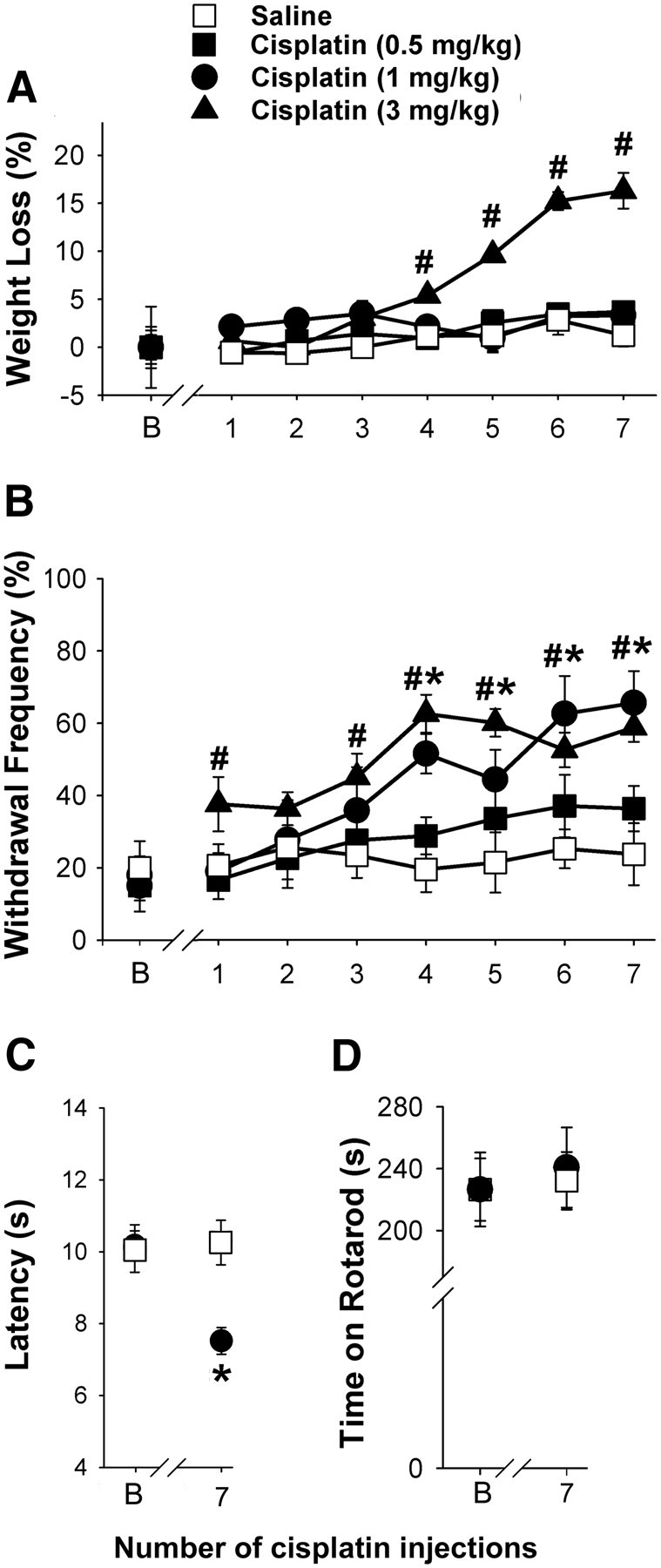
Assessment of a model of cisplatin-induced mechanical hyperalgesia. A, Weight loss was observed only in mice given the highest daily dose of cisplatin (3 mg/kg). At doses of 0.5 or 1 mg/kg, cisplatin did not produce weight loss (B, baseline). B, Mechanical hyperalgesia occurred following repeated injections of cisplatin (1 and 3 mg/kg of body weight, daily for 7 d, i.p.). The withdrawal frequency to a mechanical stimulus (3.7 mN) increased with the increased cumulative dose compared with responses before the first injection of cisplatin. C, The latency to withdrawal from a radiant heat stimulus was shorter following 7 d of treatment with cisplatin (1 mg/kg). D, In the rotarod test, mice that received cisplatin (1 mg/kg) performed as well as the vehicle-treated group. #Significantly different from baseline in the group treated with 3 mg/kg cisplatin; *significantly different from baseline in the group treated with 1 mg/kg cisplatin (p < 0.05, two-way ANOVA with Bonferroni's test; n = 6–15 mice/treatment). Error bars indicate SEM.
Cisplatin produced a dose-dependent bilateral increase in the frequency of withdrawal evoked by the von Frey monofilament (3.7 mN) applied to the hindpaw of mice (Fig. 1B). Withdrawal frequencies increased following doses of 1 and 3 mg/kg. Withdrawal frequency was ∼20% before cisplatin treatments and increased to ∼60% within 7 d of treatment with the 1 or 3 mg/kg doses.
Since repeated injections of 1 mg/kg cisplatin produced robust mechanical hyperalgesia with no weight loss and no obvious deterioration in general health, this dose of cisplatin was chosen for subsequent studies. Mice that received this dose of cisplatin also developed heat hyperalgesia over the 7 d of cisplatin injections (Fig. 1C). In contrast to changes in nociceptive responses, motor function measured with the rotarod assay was not changed by cisplatin treatment (p = 0.841, two-way ANOVA with Bonferroni's t test; n = 4–8 mice/group; Fig. 1D). Therefore, it is not likely that the measure of withdrawal of the cisplatin-treated mice to either the mechanical or thermal stimulus was confounded by a change in motor function.
To assess the contribution of endogenous AEA to cisplatin-evoked mechanical hyperalgesia, AEA was measured in plantar skin from the hindpaws of saline- and cisplatin-treated mice. To exclude the involvement other lipids with similar physiological activity, levels of 2-AG and PEA were determined in the same samples. The level of AEA in cisplatin-treated mice decreased compared with saline-treated mice (Table 1). This change was specific to AEA as the level of 2-AG was the same between treatment groups while the level of PEA increased in plantar skin from cisplatin-treated mice.
Table 1.
Effect of cisplatin on levels of endocannabinoids and PEA in plantar paw skin
| Treatment | AEA (pmol) | PEA (nmol) | 2-AG (nmol) |
|---|---|---|---|
| Saline | 34.9 ± 2.57 (9) | 7.9 ± 0.89 (9) | 1.6 ± 0.25 (9) |
| Cisplatin | 23.8 ± 1.87* (7) | 14.6 ± 2.61* (7) | 1.8 ± 0.17 (7) |
Mice were treated with saline or cisplatin (1 mg/kg) for 7 d prior to tissue collection. Data are expressed as picomoles per gram of tissue or nanomoles per gram of tissue.
*Different from saline within the same group at p < 0.05 (Student's t test). Numbers in parentheses represent the sample size.
AEA attenuated hyperalgesia in mice treated with cisplatin
Given that a decrease in AEA in plantar paw skin was associated with hyperalgesia 1 d after the seventh injection of cisplatin (1 mg/kg, i.p.), we determined whether a single injection of AEA (10 μg, i.pl.) into one hindpaw would attenuate mechanical hyperalgesia in the injected paw. Only mice exhibiting mechanical hyperalgesia (≥50% withdrawal frequency for at least one hindpaw; ∼60% of cisplatin-treated mice) were included in these experiments. Thus, before intraplantar injection of AEA, the mean withdrawal frequency of cisplatin-treated mice was 60.0 ± 5.0%. The withdrawal frequency to the mechanical stimulus decreased by 60 min after AEA injection in the paw ipsilateral to the injection, and the greatest decrease in withdrawal frequency, to 30.0 ± 5.0%, occurred at 90 min after injection (p < 0.01, two-way ANOVA with Bonferroni's test; n = 4–7 mice/treatment group; Fig. 2A). The withdrawal frequency returned to predrug values by 120 min. There was no evidence of anti-hyperalgesia in the paw contralateral to injection. The mean withdrawal frequency for the contralateral paw was 56 ± 2.4% before injection and 50 ± 3.2% at 90 min after administration of AEA into the contralateral paw (n = 4–7; p = 0.879, two-way ANOVA), indicating that the anti-hyperalgesia produced by AEA was mediated locally.
Figure 2.

Administration of AEA or URB597 decreased mechanical hyperalgesia produced by cisplatin. A, A single intraplantar injection of AEA (10 μg) into a hindpaw decreased withdrawal responses to a mechanical stimulus ipsilateral to the injection in cisplatin-treated mice (PD, predrug). Co-injection of AM281 (10 μg, i.pl.), the CB1R antagonist, but not AM630 (4 μg, i.pl.), the CB2R antagonist, blocked the effect of AEA. Neither antagonist had an effect when injected alone. B, Intraplantar injection of URB597 (9 μg) into a hindpaw attenuated mechanical hyperalgesia ipsilateral to the injection in cisplatin-treated mice. The effect of URB597 was blocked by co-injection of the CB1R antagonist AM281 (10 μg, i.pl.); AM630 (4 μg, i.pl.) had no effect. *Significantly different from vehicle; #significantly different from AEA or URB597 (p < 0.05, two-way ANOVA with Bonferroni's test; n = 4–8 mice/treatment). Error bars indicate SEM.
To determine whether the anti-hyperalgesic effect of AEA was mediated through CB1 or CB2 receptors, selective cannabinoid receptor antagonists were coadministered with AEA. AM281 (10 μg), a selective CB1R antagonist, but not AM630 (4 μg), a CB2R antagonist, blocked the anti-hyperalgesic effect of AEA when it was co-injected into the same paw (Fig. 2A). The same dose of AM630 effectively blocked the anti-hyperalgesic effect of 2-AG in a murine model of cancer pain (Khasabova et al., 2011). Injection of AM281 into the hindpaw contralateral to the AEA injection did not block the effect of AEA (55 ± 3.1%; p = 0.773, two-way ANOVA). Neither antagonist altered the mechanical sensitivity of cisplatin-treated mice when given alone (n = 5 mice/group; p = 0.653, two-way ANOVA).
Systemic as well as local injection of URB597 increases tissue levels of AEA (Lichtman et al., 2004; Fegley et al., 2005). In the present study, a single intraplantar injection of URB597 (9 μg) reduced cisplatin-evoked mechanical hyperalgesia (Fig. 2B; p < 0.001, two-way ANOVA with Bonferroni's test; n = 4–7 mice/treatment group). The maximum decrease in the withdrawal frequency occurred at 90 min after injection, and the magnitude of the effect was comparable with that produced by intraplantar injection of AEA (from 57.1 ± 1.8% before injection to 31.4 ± 4.6% after). Similarly, a single injection of URB597 reversed heat hyperalgesia in cisplatin-treated mice (6.8 ± 0.47 s cisplatin vs 9.0 ± 0.27 cisplatin plus URB597 at 60 min after injection; p < 0.001, one-way ANOVA, with Bonferroni's test; n = 7–8 mice/group). The anti-hyperalgesia produced by URB597 was mediated by peripheral CB1R because coadministration of the CB1R antagonist, AM281, blocked the effect of URB597 on mechanical hyperalgesia. The CB2R antagonist, AM630, did not alter the anti-hyperalgesic effect of URB597 (n = 5; p = 0.372; Fig. 2B). URB597 did not produce anti-hyperalgesia in the paw contralateral to injection (59.3 ± 2.0% before and 55.6 ± 1.6% at 90 min after injection; n = 4–7; p = 0.397, two-way ANOVA). Together, these data support the conclusion that intraplantar injection of URB597 decreased cisplatin-induced mechanical hyperalgesia by increasing the level of endogenous AEA locally, and this effect was mediated by peripheral CB1R.
Repeated administration of URB597 attenuated the development of hyperalgesia produced by cisplatin
The development of hyperalgesia was delayed and its magnitude was reduced when daily injections of cisplatin were accompanied by URB597 (9 μg, i.p.; Fig. 3). Mechanical hyperalgesia following cisplatin alone appeared by day 2, and withdrawal frequency reached 74.2 ± 3.4% after 7 d of treatment (Fig. 3A); however, mechanical hyperalgesia did not develop until day 4 in mice treated with cisplatin and URB597. When cisplatin was accompanied by URB597, mechanical hyperalgesia was reduced by ∼35% after 7 d of treatment. The anti-hyperalgesia produced by URB597 was mediated by CB1 receptors because coadministration with AM281 (10 μg, i.p.) abolished the anti-hyperalgesic effect of URB597. Repeated injections of AM281 alone had no effect on the development of cisplatin-evoked mechanical hyperalgesia.
Figure 3.

Repeated systemic injections of URB597 (0.3 mg/kg daily, i.p.) reduced the development of cisplatin-evoked hyperalgesia in cisplatin-treated mice. A, When cisplatin was co-injected with URB597, the development of mechanical hyperalgesia was delayed, and it was reduced in magnitude (B, baseline). The effect of URB597 was blocked by daily co-injection of the CB1 receptor antagonist AM281 (0.33 mg/kg, i.p.). B, Cotreatment with URB597 blocked the development of heat hyperalgesia. C, Acute treatment with AM281 (10 μg, i.p.) reversed the anti-hyperalgesic effect of repeated URB597 administration. The y-axis in C is the same as in B. D, Repeated administration of URB597 for 7 d had no effect on motor function as measured by the rotarod assay. *Significantly different from vehicle; #significantly different from URB597 (p < 0.05, two-way ANOVA with Bonferroni's test; n = 4–8 mice/treatment). Error bars indicate SEM.
The development of cisplatin-induced heat hyperalgesia was also reduced by daily coadministration of URB597. Heat hyperalgesia was evident following the fourth injection of cisplatin (the first day of data collection), and URB597 blocked the occurrence of heat hyperalgesia on all days of testing (Fig. 3B). Because URB597 binds to FAAH with high affinity and slow dissociation kinetics, leading to its description as an “irreversible” FAAH inhibitor (Kathuria et al., 2003), we determined whether the anti-hyperalgesic effect of URB597 in this experimental paradigm was due to the activation of CB1R by an endogenous ligand. After determination of heat sensitivity following seven injections of cisplatin, the mice received an additional treatment of cisplatin and URB597. On the following day, the mice were tested for sensitivity to heat and then given an injection of the CB1R antagonist AM281 (intraperitoneally). Treatment with AM281 decreased the withdrawal latency in URB597-treated mice (Fig. 3C), indicating that the reduction in heat hyperalgesia was mediated by activation of CB1R by an endogenous ligand. However, the residual heat hyperalgesia in the presence of AM281 indicated that thermal dysfunction was still present. This conclusion is supported by evidence that the same dose of AM281 did not affect heat sensitivity in naive mice (p = 0.28, two-way ANOVA; 3–4 mice/group).
Collectively, these data support the conclusion that the low level of AEA in the plantar paw skin contributed to the development of cisplatin-evoked mechanical and heat hyperalgesia through a CB1R-dependent mechanism. Importantly, inhibition of AEA hydrolysis by treatment with URB597 for 7 d did not alter the performance of the mice on the rotarod (p = 0.729, one-way ANOVA; Fig. 3D). Therefore, the inhibitory effect of URB597 on the withdrawal responses to either the mechanical or thermal stimulus was not due to a general depression in motor function.
Cisplatin increased the expression of TRPV1 in sensory neurons
Evidence that a CB1R antagonist reversed the anti-hyperalgesic effect of daily cotreatments with URB597 suggested that the effect of cisplatin on the structural underpinning of heat hyperalgesia was not altered by cotreatment with URB597. Because TRPV1 contributes to heat transduction (Caterina MJ et al., 2000), TRPV1-ir was visualized in L3–L5 DRGs from mice treated with vehicle, cisplatin, or cisplatin plus URB597 (Fig. 4A). Quantification of the proportion of TRPV1-ir neurons in DRGs of mice across treatment groups indicated that cisplatin injections increased the occurrence of TRPV1-ir neurons (Fig. 4B). It is noteworthy that the proportion of labeled neurons was decreased by cotreatment with URB597, but the effect of cisplatin was not reduced to the level of immunoreactive neurons in the vehicle control group (each treatment group was different from each other at p < 0.05, one-way ANOVA with Student–Newman–Keuls test; four mice per group). The mean diameter of TRPV1-ir neurons did not change (p = 0.24 across treatment groups, one-way ANOVA). These data indicate that daily treatment with URB597 reduced the neurochemical effect of cisplatin on the expression of TRPV1.
Figure 4.
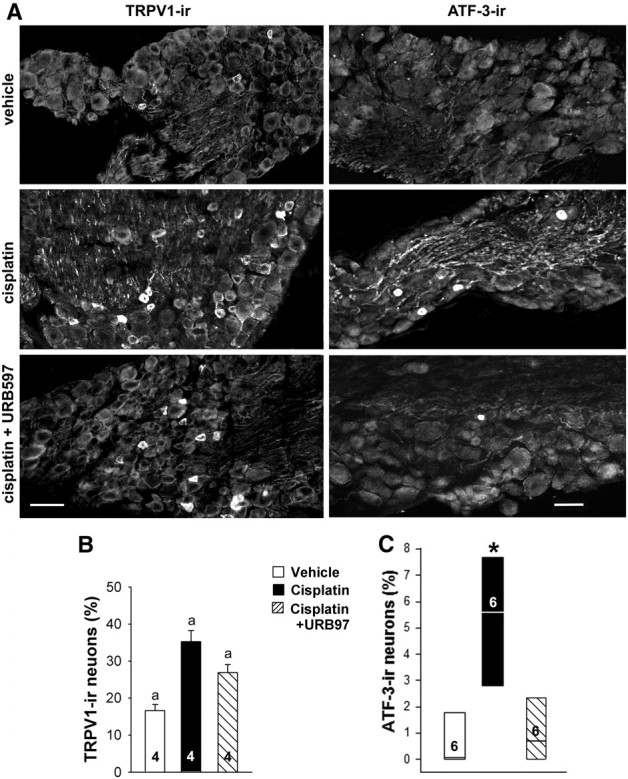
URB597 attenuated effects of cisplatin on protein expression in DRGs. A, TRPV1- and ATF-3-ir were detected by immunofluorescence in L3–L5 DRGs from mice treated with vehicle, cisplatin, or cisplatin plus URB597. Cisplatin (1 mg/kg of body weight, daily for 7 d, i.p.) increased the occurrence of TRPV1- and ATF3-ir in neurons. Co-injection of URB597 (0.3 mg/kg daily, i.p.) with cisplatin attenuated the effect cisplatin on protein-ir. Scale bars: 10 μm (for images within each antigen). B, Quantitative summary of the effect of treatments on TRPV1-ir in neurons. Data are expressed as the mean ± SEM. aSignificantly different from each other group (p < 0.05, one-way ANOVA with Student–Newman–Keuls test; n = 4 mice/treatment). C, Quantitative summary of the effect of treatments on ATF-ir in neurons. Data are expressed as the median and 25th and 75th percentile range. *Significantly different from vehicle control and cisplatin plus URB597 groups (n = 6 mice/treatment; p < 0.001, Kruskal–Wallis ANOVA on ranks test).
In comparison, URB597 reversed the effect of cisplatin on a general marker of neurotoxicity. An increase in the number of ATF-3-ir neurons was noted in DRGs of cisplatin-treated mice when compared with saline-treated mice (n = 6 mice/group, p < 0.001, Kruskal–Wallis ANOVA on ranks test). The somal area of ATF-ir neurons was 1256 ± 80.6 μm2, which falls outside the range of murine DRG neurons described as nociceptors (Dirajlal et al., 2003). Co-injection of URB597 with cisplatin for 7 d blocked the expression of ATF-3 following cisplatin (Fig. 4C).
URB597 blocked effects of cisplatin on large myelinated fibers
Electrophysiology was used to assess the effect of cisplatin on conduction velocity of large-diameter fibers in the sciatic nerve. In all groups of mice, electrical stimulation of the sciatic nerve evoked a well defined Aα/Aβ component of the CAP (Fig. 5A). The conduction velocity of the Aα/Aβ-fibers was slower in mice treated with cisplatin compared with vehicle- and cisplatin-plus-URB597-treated groups (n = 5; p < 0.05, one-way ANOVA with Bonferroni's t tests; Fig. 5B). Conduction velocity of Aα/Aβ-fibers was 26.6 ± 3.39 m/s in the cisplatin-treated group in contrast to measures in vehicle- and cisplatin-plus-URB597-treated groups of 36.7 ± 1.76 and 35.7 ± 2.69 m/s, respectively.
Figure 5.

URB597 attenuated effects of cisplatin in nerve. A, Representative examples of Aα/Aβ components of the CAP from mice treated with vehicle, cisplatin, and cisplatin plus URB597. Electrical stimuli are indicated by arrows, and latencies of spike initiations are shown by dashed lines. Latencies corresponded to conductance velocities of 37.2 m/s (vehicle), 26.9 m/s (cisplatin), and 36.1 m/s (cisplatin plus URB597). B, Quantitative summary showing that treatment with cisplatin (1 mg/kg, daily for 7 d, i.p.) reduced the conduction velocity of Aα/Aβ-fibers. Cotreatment with URB597 (0.3 mg/kg daily, i.p.) blocked the effect of cisplatin. C, Western blots of p-NF and actin protein in tibial nerves of mice treated with (1) vehicle, (2) cisplatin alone, or (3) cisplatin plus URB597. D, Quantitative summary of the amount of p-NF protein defined as the ratio of RT97-ir to actin-ir within the same sample. The decrease in p-NF was protected by coadministration of URB597. *Significantly different (p < 0.05, one-way ANOVA with Bonferroni's test). Values inside the bars represent the sample size. Error bars indicate SEM.
The decrease in conduction velocity of large myelinated fibers was associated with a decrease in expression of p-NF protein, a marker of myelinated axons (Perry et al., 1991; Sann et al., 1995). Western blots were prepared of p-NF protein in tibial nerves of mice treated with vehicle, cisplatin, or cisplatin with URB597 (Fig. 5C). Densitometric analyses of immunoblots demonstrated a decrease in p-NF in cisplatin-treated mice compared with saline-treated mice. The effect of cisplatin was blocked in mice cotreated with URB597 (Fig. 5D). Thus, daily administration of URB597 with cisplatin reduced neurochemical and conductive abnormalities in large myelinated peripheral nerve fibers produced by cisplatin alone.
URB597 attenuated a neurotoxic effect of cisplatin on DRG neurons in vitro
To further evaluate the neuroprotective function of URB597, we determined whether URB597 decreased direct effects of cisplatin on DRG neurons in vitro. Since cisplatin decreases the process of tubulin assembly/disassembly (Tulub and Stefanov, 2001), and polymerization of β-tubulin III is essential for neurite extension in regenerating axons (Moskowitz and Oblinger, 1995), β-tubulin III-ir was used to evaluate effects of cisplatin (4 μg/ml) and URB597 (100 nm) on DRG neurons after 24 h of treatment. The concentration and duration of cisplatin treatment were selected on the basis of previous studies (McDonald et al., 2005; Klein et al., 2007). The clinical relevance of in vitro cisplatin treatment was confirmed by the decreased survival of fibrosarcoma cells (Fig. 6). In addition, we used HPLC/MS analysis to confirm that treatment with URB597 (100 nm) increased the level of AEA in cultured DRG cells without altering the levels of 2-AG and PEA (n = 3 cultures/group; p < 0.05, Student's t test; Table 2). The number of NeuN-ir neurons in DRG cultures was not altered following cisplatin treatment, indicating that cisplatin did not affect neuronal survival (n = 4131–5038 neurons/group across 4 different culture preparations; p = 0.546, Student's t test).
Figure 6.
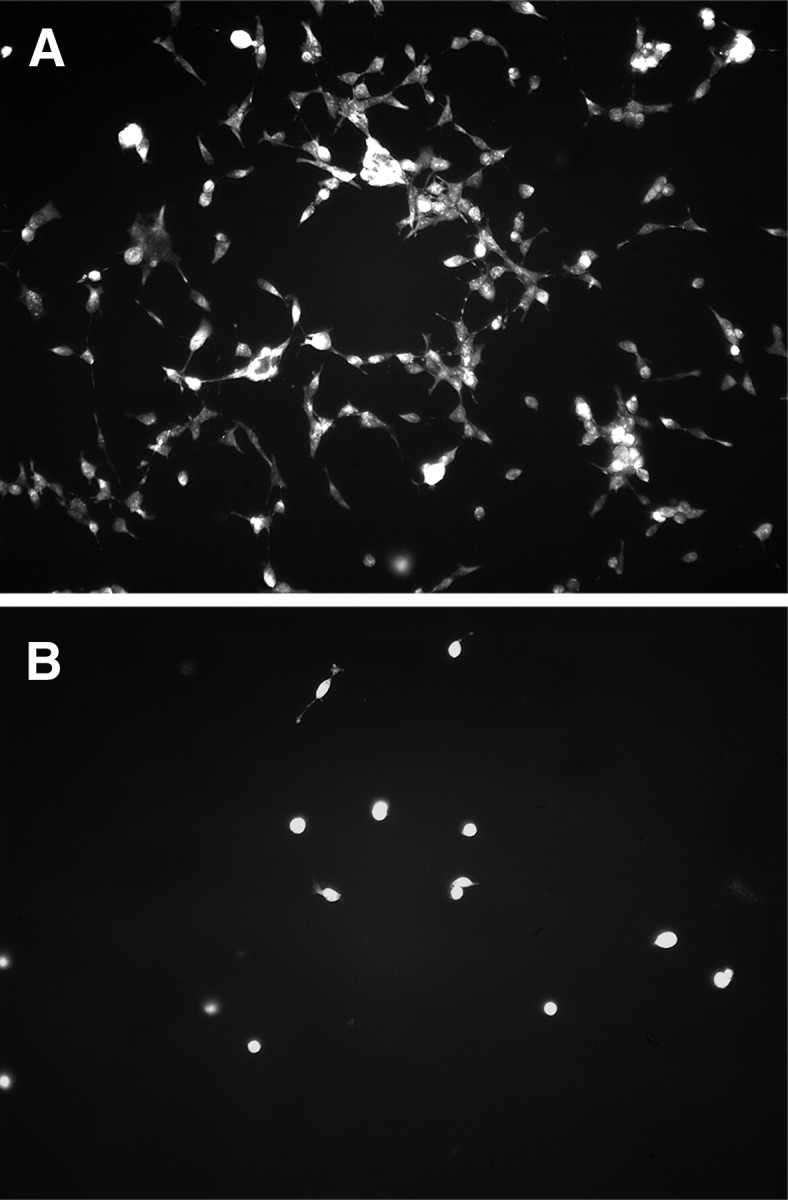
Cytotoxicity of cisplatin in vitro was confirmed on cultured fibrosarcoma cells. In comparison to the vehicle-treated culture (A), treatment with cisplatin (4 μg/ml for 24 h; B) decreased the number of viable fibrosarcoma cells. Cultures were incubated in acridine orange (0.67 μm for 5 min) to visualize live cells (Bank, 1987).
Table 2.
Effect of URB597 on levels of endocannabinoids and PEA in primary cultures of DRGs
| Treatment | AEA (pmol) | PEA (pmol) | 2AG (pmol) |
|---|---|---|---|
| Vehicle | 0.56 ± 0.12 | 25.9 ± 4.76 | 12.9 ± 2.36 |
| URB597 | 1.38 ± 0.03* | 27.5 ± 5.93 | 10.3 ± 2.09 |
Primary cultures of DRGs were treated with vehicle or URB597 (100 nm) for 15 min after 1 d in vitro. Each sample was equivalent to five to six DRGs. Data are expressed as picomoles per sample.
*Different from vehicle-treated within the same endocannabinoid group at p < 0.05 (n = 3 cultures/treatment, Student's t test).
Under control conditions, cultured DRG neurons extended neurites within 24 h after plating. Although cisplatin did not affect the number of neurons in culture, the total neurite length of DRG neurons was reduced following cisplatin treatment (median of 2170 μm compared with 3843 μm in vehicle-treated neurons). Coadministration of cisplatin with URB597 prevented the effect of cisplatin on total neurite length, producing a result comparable with vehicle-treated neurons (median of 3688 μm; Fig. 7). The effect of URB597 on neurite growth was mediated by CB1 receptors because AM281 (1 μm), but not AM630 (1 μm), blocked the increase in neurite length following treatment with URB597 (p < 0.001, Kruskal–Wallis ANOVA on ranks; n = 3 different culture preparations). Application of URB597 alone to DRG neurons had no effect on neurite extension (Fig. 7D). Together, these data indicate that an increase in AEA following URB597 protected isolated DRG neurons from the decrease in neurite growth following cisplatin, and this occurred through CB1 receptors.
Figure 7.
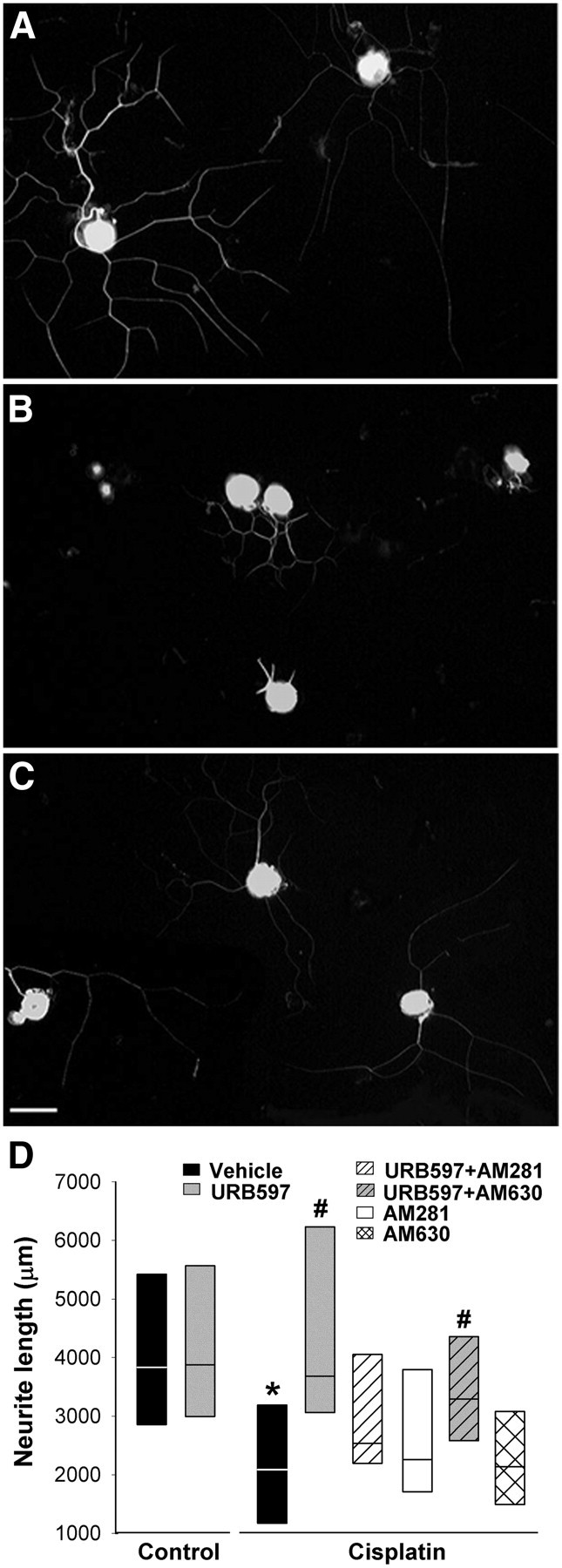
Adult DRG neurons treated with cisplatin in vitro exhibited reduced neurite growth, which was prevented by cotreatment with URB597. A, Dissociated DRG neurons cultured in the control conditions exhibited long, branched neurites with strong tubulin-ir. B, DRG neurons treated with cisplatin (4 μg/ml) for 24 h had fewer and shorter tubulin-ir neurites compared with neurons in the control condition. C, Cotreatment with URB597 (100 nm) reversed the effect of cisplatin. Scale bar, 50 μm. All images are the same magnification. D, Quantitative summary of the effect of treatments on neurite length. Data are expressed as the median and 25th and 75th percentile range. The effect of URB597 was blocked by cotreatment with the CB1R antagonist AM281 (1 μm). The CB2R antagonist AM630 (1 μm) did not block the effect of URB597. Neither antagonist alone had effect on cisplatin-treated DRG neurons. URB597 had no effect on the length of tubulin-ir neurites in the control conditions. *Significantly different from vehicle in the control condition. #Significantly different from vehicle, URB597 plus AM281, or URB597 plus AM630 groups in cisplatin-treated conditions (n = 143–191 neurons/treatment in 3 different culture preparations; p < 0.001, Kruskal–Wallis ANOVA on ranks).
Discussion
The present study demonstrates for the first time that facilitation of AEA signaling may protect sensory neurons from cisplatin-induced injury as well as reduce the pain associated with the neuropathy. An acute, local increase in AEA in the periphery transiently attenuated cisplatin-evoked hyperalgesia via activation of CB1R. More importantly, long-term facilitation of AEA signaling by inhibition of AEA hydrolysis with URB597 delayed the onset and decreased the magnitude of mechanical and heat hyperalgesia caused by repeated administration of cisplatin. The behavioral effect of coadministration of URB597 with cisplatin was accompanied by a reduction in biochemical and functional aberrations of DRG neurons in mice treated with cisplatin alone. Following an earlier example that used primary cultures of adult DRG neurons to characterize the neurotoxic effects of cisplatin (Malgrange et al., 1994), the present study used this model to uncover a neuroprotective effect of URB597 against cisplatin in DRG neurons.
General characteristics of a murine model of cisplatin-evoked mechanical hyperalgesia
Consistent with earlier clinical (Sleijfer et al., 1985; Cooley et al., 1994) and experimental (Tredici et al., 1998; Joseph and Levine, 2009; Ta et al., 2009) reports, mechanical and heat hyperalgesia occurred in the majority of mice treated with cisplatin in the present study. Because cisplatin does not cross the blood–brain barrier (Gregg et al., 1992) and no obvious deterioration in general health or motor activity occurred during the 7 d course of treatment, the hyperalgesia observed in cisplatin-treated mice is most likely due to changes in sensory neurons. This conclusion is supported by the pattern of neurochemical and functional changes that occurred in somatosensory neurons in vivo, including the increased occurrence of ATF-3- and TRPV1-ir in DRG neurons, a decrease in the expression of p-NF protein in peripheral axons, and the slowing of the conduction velocity observed in the electrically evoked sensory CAP.
The expression of ATF-3 in DRG neurons is a specific marker of axonal injury. ATF-3 is a transcription factor that is synthesized de novo in sensory neurons in response to injury of peripheral axons (Hai and Hartman, 2001); noxious stimuli in the absence of nerve injury do not induce its expression (Tsujino et al., 2000). Thus, the occurrence of ATF-3 in DRG neurons in the present study is most likely secondary to axonal damage caused by cisplatin. Other antimitotic drugs share this effect on ATF-3 (Jimenez-Andrade et al., 2006; Jamieson et al., 2007; Peters et al., 2007).
The increased expression of TRPV1-ir in cisplatin-treated mice is consistent with an earlier report of increased TRPV1 mRNA in trigeminal ganglia from cisplatin-treated mice (Ta et al., 2010). However, the previous report did not detect a change in the proportion of TRVP1-ir neurons in the ganglia. The difference in results may be due to the sensitivity of the immunohistochemical protocols or a difference in the ganglia sampled. Most noteworthy, TRPV1 was shown to be required for heat hyperalgesia in cisplatin-treated mice (Ta et al., 2010).
The decrease in expression of p-NF protein in peripheral nerve of cisplatin-treated mice complements a previous study of cisplatin-treated rats that showed a decrease in the number of pNP-ir cell bodies in lumbar DRGs (Jamieson et al., 2009). A decrease in the expression of neurofilament proteins in sensory neurons is associated with peripheral neuropathy in humans treated with cisplatin (Walsh et al., 1982; Gastaut and Pellissier, 1985). Phosphorylated neurofilaments are major cytoskeletal proteins of myelinated nerve fibers, and the amount of pNP-ir is positively correlated to conduction velocity in Aα/Aβ-fibers (Lawson and Waddell, 1991). The reduction in conduction velocity we observed following cisplatin treatment is consistent with a clinical report (Yamamoto et al., 1997) and other preclinical studies (Bárdos et al., 2003; Yoon et al., 2009). Together, these behavioral, biochemical, and functional changes support the use of short-term cisplatin treatment in mice as a model for studying effects of repeated cisplatin administration on sensory neurons and the development of new anti-hyperalgesic and neuroprotective approaches to chemotherapy-induced neuropathy.
Anandamide signaling and hyperalgesia
Because AEA is an agonist of the CB1R (Hillard, 2000; Pertwee and Ross, 2002) and CB1R tone in nociceptors modulates the threshold for their activation (Agarwal et al., 2007), we speculate that the reduction in the AEA content of plantar skin in cisplatin-treated mice contributed to the behavioral hypersensitivity observed in these mice. Similarly, a decrease in CB1R activity may contribute to the observed increase in spontaneous activity of Aδ- and C-fibers observed in rats treated with a related compound, oxaliplatin (Xiao et al., 2012). This assertion is supported by evidence that local injection of AEA or URB597, an inhibitor of AEA hydrolysis, acutely reduced the hyperalgesia in cisplatin-treated mice. Evidence that the effects of AEA and URB597 were reversed by a CB1 but not CB2 receptor antagonist confirmed that the acute effects of each were mediated by CB1R. Thus, the acute anti-hyperalgesic effect of AEA in cisplatin-treated mice is consistent with its effects in models of inflammatory, neuropathic, and cancer pain (Calignano et al., 1998; Guindon and Beaulieu, 2006; Guindon et al., 2006; Khasabova et al., 2008). The mechanism underlying the reduction in tissue content of AEA remains to be resolved.
URB597 delayed and decreased the development of hyperalgesia
An important observation of the present study is that repeated systemic administration of URB597 delayed and decreased the severity of cisplatin-induced mechanical and heat hypersensitivity through activation of CB1 receptors. URB597 is a highly selective FAAH inhibitor (Cravatt et al., 1996) that penetrates the brain and elevates endogenous AEA (Lichtman et al., 2004; Hohmann et al., 2005). Although URB597 is considered an effective inhibitor of FAAH, other amides such as N-PEA and N-oleoylethanolamide are also substrates for FAAH (Cravatt et al., 2001) and have anti-hyperalgesic properties (LoVerme et al., 2006; Suardíaz et al., 2007). However, these fatty acid amides do not activate CB1R (Lambert and Di Marzo, 1999). Instead, CB2R or PPARα mediate their anti-hyperalgesic effects in other models of peripheral injury (Sagar et al., 2008; Kinsey et al., 2009). In cisplatin-treated mice, both the acute and long-term effects of URB597 on hyperalgesia were reversed by a CB1R antagonist. Importantly, repeated systemic administration of URB597 did not impair motor function. In addition, unlike other CB1R agonists, repeated administration of URB597 does not result in physical dependence (Schlosburg et al., 2009), thereby increasing the potential clinical utility of FAAH inhibitors.
In addition to decreasing hyperalgesia, repeated administrated of URB597 considerably decreased the severity of cisplatin-induced peripheral neurotoxicity in vivo. When URB597 was co-injected with cisplatin, the effects of cisplatin on the biochemical markers used to characterize its neurotoxicity (ATF-3 and p-NF) were reversed along with the functional changes in subpopulations of A-fibers. Although we cannot exclude the possibility that other ethanolamides have a role in modulating these diverse changes, if these markers are accepted as markers of neuronal stress, it is interesting to consider the effect of URB597 on neurite growth in vitro and the extent to which CB1R may contribute to the fundamental health of DRG neurons.
Anandamide signaling and neuronal neurotoxicity
The primary antineoplastic activity of cisplatin is attributed to its platination of nuclear DNA. Recent evidence indicates that sensory changes associated with cisplatin treatment may be due to mitochondrial dysfunction that results from platination of mitochondrial DNA (Flatters and Bennett, 2006; Podratz et al., 2011; Xiao et al., 2011). Alternatively, because cisplatin interferes with the polymerization of microtubules (Tulub and Stefanov, 2001; Goshima et al., 2010), the scaffold for axonal transport, impaired axonal transport may also contribute to the sensory changes following cisplatin treatment. The total length of β-tubulin-ir neurites was used as a measure of cisplatin neurotoxicity in vitro (Malgrange et al., 1994). It is noteworthy that URB597 reversed the inhibitory effect of cisplatin on neurite extension by DRG neurons through a CB1R-dependent mechanism. Biochemical measures confirmed that URB597 increased the level of AEA in the cultures. These data are consistent with CB1R-mediated stimulation of neurite outgrowth in an in vitro model of diabetic neuropathy (Zhang et al., 2009); however, the mechanism by which CB1R signaling blocks cisplatin-mediated disruption of neurite extension remains to be determined. We speculate that signal transducer and activator of transcription 3 (STAT3) plays a key role in this process. Phosphorylation of STAT3 is a downstream target of CB1R signaling, and phosphorylated STAT3 also binds to tubulin to promote polymerization and neurite extension (He et al., 2006; Ng et al., 2006; Bromberg et al., 2008). Given the fundamental role of microtubules in axonal transport and the beneficial effect of neurotrophic factors in vivo in cisplatin-induced peripheral neurotoxicity (Wongtawatchai et al., 2009), maintaining the retrograde transport of neurotrophic factors may attenuate neurotoxic effects of cisplatin in vivo.
Conclusion
We have shown that cisplatin produces hyperalgesia and toxicity to sensory neurons as indicated by neurochemical, morphological, and functional measures. Increasing AEA signaling at CB1 receptors not only reduced the hyperalgesia but reduced the neurotoxicity of cisplatin as well. Although the mechanisms by which AEA reduce neurotoxicity remain to be resolved, the present studies underscore the dual utility in exploiting the endocannabinoid system for management of neuropathic pain produced by chemotherapy.
Footnotes
This work was supported by National Institute of Drug Abuse Grant DA011471 (D.A.S.) and National Cancer Institute Grant CA138684 (V.S.S.). We are grateful to P. Villalta and B. Mater at the University of Minnesota Cancer Center for assistance in the quantification of endocannabinoids and to L. G. Coicou, P. Cortez, C. E. McShane, M. Tracy, and D. Vetter for technical assistance.
The authors declare no competing financial interests.
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ, Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, Kuner R. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers JW, Chaudhry V, Cavaletti G, Donehower RC. Interventions for preventing neuropathy caused by cisplatin and related compounds. Cochrane Database Syst Rev. 2011;2011:CD005228. doi: 10.1002/14651858.CD005228.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank HL. Assessment of islet cell viability using fluorescent dyes. Diabetologia. 1987;30:812–816. doi: 10.1007/BF00275748. [DOI] [PubMed] [Google Scholar]
- Bárdos G, Móricz K, Jaszlits L, Rabloczky G, Tory K, Rácz I, Bernáth S, Sümegi B, Farkas B, Literáti-Nagy B, Literáti-Nagy P. BGP-15, a hydroximic acid derivative, protects against cisplatin- or taxol-induced peripheral neuropathy in rats. Toxicol Appl Pharmacol. 2003;190:9–16. doi: 10.1016/s0041-008x(03)00155-8. [DOI] [PubMed] [Google Scholar]
- Boulikas T, Vougiouka M. Cisplatin and platinum drugs at the molecular level. Oncol Rep. 2003;10:1663–1682. [PubMed] [Google Scholar]
- Bromberg KD, Ma'ayan A, Neves SR, Iyengar R. Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science. 2008;320:903–909. doi: 10.1126/science.1152662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Carozzi V, Chiorazzi A, Canta A, Oggioni N, Gilardini A, Rodriguez-Menendez V, Avezza F, Crippa L, Ceresa C, Nicolini G, Bossi M, Cavaletti G. Effect of the chronic combined administration of cisplatin and paclitaxel in a rat model of peripheral neurotoxicity. Eur J Cancer. 2009;45:656–665. doi: 10.1016/j.ejca.2008.10.038. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breitenbucher JG, Chaplan SR, Webb M. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley ME, Davis L, Abrahm J. Cisplatin: a clinical review. Part II—Nursing assessment and management of side effects of cisplatin. Cancer Nurs. 1994;17:283–293. [PubMed] [Google Scholar]
- Coomber B, O'Donoghue MF, Mason R. Inhibition of endocannabinoid metabolism attenuates enhanced hippocampal neuronal activity induced by kainic acid. Synapse. 2008;62:746–755. doi: 10.1002/syn.20547. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MP. Oral nabilone capsules in the treatment of chemotherapy-induced nausea and vomiting and pain. Expert Opin Investig Drugs. 2008;17:85–95. doi: 10.1517/13543784.17.1.85. [DOI] [PubMed] [Google Scholar]
- Dirajlal S, Pauers LE, Stucky CL. Differential response properties of IB4-positive and -negative unmyelinated sensory neurons to protons and capsaicin. J Neurophysiol. 2003;89:513–524. doi: 10.1152/jn.00371.2002. [DOI] [PubMed] [Google Scholar]
- Dzagnidze A, Katsarava Z, Makhalova J, Liedert B, Yoon MS, Kaube H, Limmroth V, Thomale J. Repair capacity for platinum-DNA adducts determines the severity of cisplatin-induced peripheral neuropathy. J Neurosci. 2007;27:9451–9457. doi: 10.1523/JNEUROSCI.0523-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122:245–257. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Rojo ML, Rodriguez-Gaztelumendi A. Modulation of the endocannabinoid system: neuroprotection or neurotoxicity? Exp Neurol. 2010;224:37–47. doi: 10.1016/j.expneurol.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Gastaut JL, Pellissier JF. Neuropathy caused by cisplatin. Clinical, electrophysiological and morphological study (in French) Rev Neurol (Paris) 1985;141:614–626. [PubMed] [Google Scholar]
- Goshima Y, Usui H, Shiozawa T, Hida T, Kuraoka S, Takeshita S, Yamashita N, Ichikawa Y, Kamiya Y, Gotoh T, Gotoh T. Computational analysis of the effects of antineoplastic agents on axonal transport. J Pharmacol Sci. 2010;114:168–179. doi: 10.1254/jphs.09352fp. [DOI] [PubMed] [Google Scholar]
- Gregg RW, Molepo JM, Monpetit VJ, Mikael NZ, Redmond D, Gadia M, Stewart DJ. Cisplatin neurotoxicity: the relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. J Clin Oncol. 1992;10:795–803. doi: 10.1200/JCO.1992.10.5.795. [DOI] [PubMed] [Google Scholar]
- Guindon J, Beaulieu P. Antihyperalgesic effects of local injections of anandamide, ibuprofen, rofecoxib and their combinations in a model of neuropathic pain. Neuropharmacology. 2006;50:814–823. doi: 10.1016/j.neuropharm.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Guindon J, De Léan A, Beaulieu P. Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain. 2006;121:85–93. doi: 10.1016/j.pain.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- He JC, Neves SR, Jordan JD, Iyengar R. Role of the Go/i signaling network in the regulation of neurite outgrowth. Can J Physiol Pharmacol. 2006;84:687–694. doi: 10.1139/y06-025. [DOI] [PubMed] [Google Scholar]
- Hillard CJ. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000;61:3–18. doi: 10.1016/s0090-6980(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Jamieson SM, Liu JJ, Connor B, Dragunow M, McKeage MJ. Nucleolar enlargement, nuclear eccentricity and altered cell body immunostaining characteristics of large-sized sensory neurons following treatment of rats with paclitaxel. Neurotoxicology. 2007;28:1092–1098. doi: 10.1016/j.neuro.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Jamieson SM, Subramaniam J, Liu JJ, Jong NN, Ip V, Connor B, McKeage MJ. Oxaliplatin-induced loss of phosphorylated heavy neurofilament subunit neuronal immunoreactivity in rat DRG tissue. Mol Pain. 2009;5:66. doi: 10.1186/1744-8069-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Peters CM, Mejia NA, Ghilardi JR, Kuskowski MA, Mantyh PW. Sensory neurons and their supporting cells located in the trigeminal, thoracic and lumbar ganglia differentially express markers of injury following intravenous administration of paclitaxel in the rat. Neurosci Lett. 2006;405:62–67. doi: 10.1016/j.neulet.2006.06.043. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Comparison of oxaliplatin- and cisplatin-induced painful peripheral neuropathy in the rat. J Pain. 2009;10:534–541. doi: 10.1016/j.jpain.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanian DA, Brown QB, Makriyannis A, Kosten TA, Bahr BA. Dual modulation of endocannabinoid transport and fatty acid amide hydrolase protects against excitotoxicity. J Neurosci. 2005;25:7813–7820. doi: 10.1523/JNEUROSCI.2347-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Stucky CL, Harding-Rose C, Eikmeier L, Beitz AJ, Coicou LG, Hanson AE, Simone DA, Seybold VS. Chemical interactions between fibrosarcoma cancer cells and sensory neurons contribute to cancer pain. J Neurosci. 2007;27:10289–10298. doi: 10.1523/JNEUROSCI.2851-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Khasabov SG, Harding-Rose C, Coicou LG, Seybold BA, Lindberg AE, Steevens CD, Simone DA, Seybold VS. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. J Neurosci. 2008;28:11141–11152. doi: 10.1523/JNEUROSCI.2847-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Chandiramani A, Harding-Rose C, Simone DA, Seybold VS. Increasing 2-arachidonoyl glycerol signaling in the periphery attenuates mechanical hyperalgesia in a model of bone cancer pain. Pharmacol Res. 2011;64:60–67. doi: 10.1016/j.phrs.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Adelstein RS, Kawamoto S. Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J Biol Chem. 2009;284:31052–31061. doi: 10.1074/jbc.M109.052969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Brown D, Turnley AM. Phenoxodiol protects against cisplatin induced neurite toxicity in a PC-12 cell model. BMC Neurosci. 2007;8:61. doi: 10.1186/1471-2202-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DM, Di Marzo V. The palmitoylethanolamide and oleamide enigmas: are these two fatty acid amides cannabimimetic? Curr Med Chem. 1999;6:757–773. [PubMed] [Google Scholar]
- Lan R, Gatley J, Lu Q, Fan P, Fernando SR, Volkow ND, Pertwee R, Makriyannis A. Design and synthesis of the CB1 selective cannabinoid antagonist AM281: a potential human SPECT ligand. AAPS PharmSci. 1999;1:E4. doi: 10.1208/ps010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson SN, Waddell PJ. Soma neurofilament immunoreactivity is related to cell size and fibre conduction velocity in rat primary sensory neurons. J Physiol. 1991;435:41–63. doi: 10.1113/jphysiol.1991.sp018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- Malgrange B, Delrée P, Rigo JM, Baron H, Moonen G. Image analysis of neuritic regeneration by adult rat dorsal root ganglion neurons in culture: quantification of the neurotoxicity of anticancer agents and of its prevention by nerve growth factor or basic fibroblast growth factor but not brain-derived neurotrophic factor or neurotrophin-3. J Neurosci Methods. 1994;53:111–122. doi: 10.1016/0165-0270(94)90151-1. [DOI] [PubMed] [Google Scholar]
- McDonald ES, Randon KR, Knight A, Windebank AJ. Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: a potential mechanism for neurotoxicity. Neurobiol Dis. 2005;18:305–313. doi: 10.1016/j.nbd.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Moskowitz PF, Oblinger MM. Sensory neurons selectively upregulate synthesis and transport of the βIII-tubulin protein during axonal regeneration. J Neurosci. 1995;15:1545–1555. doi: 10.1523/JNEUROSCI.15-02-01545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DC, Chan SF, Kok KH, Yam JW, Ching YP, Ng IO, Jin DY. Mitochondrial targeting of growth suppressor protein DLC2 through the START domain. FEBS Lett. 2006;580:191–198. doi: 10.1016/j.febslet.2005.11.073. [DOI] [PubMed] [Google Scholar]
- Perry MJ, Lawson SN, Robertson J. Neurofilament immunoreactivity in populations of rat primary afferent neurons: a quantitative study of phosphorylated and non-phosphorylated subunits. J Neurocytol. 1991;20:746–758. doi: 10.1007/BF01187848. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids. 2002;66:101–121. doi: 10.1054/plef.2001.0341. [DOI] [PubMed] [Google Scholar]
- Peters CM, Jimenez-Andrade JM, Kuskowski MA, Ghilardi JR, Mantyh PW. An evolving cellular pathology occurs in dorsal root ganglia, peripheral nerve and spinal cord following intravenous administration of paclitaxel in the rat. Brain Res. 2007;1168:46–59. doi: 10.1016/j.brainres.2007.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podratz JL, Knight AM, Ta LE, Staff NP, Gass JM, Genelin K, Schlattau A, Lathroum L, Windebank AJ. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol Dis. 2011;41:661–668. doi: 10.1016/j.nbd.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenzieri C, Brink TS, Pacharinsak C, Simone DA. Cannabinoid modulation of cutaneous Adelta nociceptors during inflammation. J Neurophysiol. 2008;100:2794–2806. doi: 10.1152/jn.90809.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, Pertwee RG. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo R, Loverme J, La Rana G, Compton TR, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- Sagar DR, Kendall DA, Chapman V. Inhibition of fatty acid amide hydrolase produces PPAR-alpha-mediated analgesia in a rat model of inflammatory pain. Br J Pharmacol. 2008;155:1297–1306. doi: 10.1038/bjp.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sann H, McCarthy PW, Jancsó G, Pierau FK. RT97: a marker for capsaicin-insensitive sensory endings in the rat skin. Cell Tissue Res. 1995;282:155–161. doi: 10.1007/BF00319142. [DOI] [PubMed] [Google Scholar]
- Schlosburg JE, Kinsey SG, Lichtman AH. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J. 2009;11:39–44. doi: 10.1208/s12248-008-9075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seijffers R, Allchorne AJ, Woolf CJ. The transcription factor ATF-3 promotes neurite outgrowth. Mol Cell Neurosci. 2006;32:143–154. doi: 10.1016/j.mcn.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Sleijfer DT, Meijer S, Mulder NH. Cisplatin: a review of clinical applications and renal toxicity. Pharm Weekbl Sci. 1985;7:237–244. doi: 10.1007/BF01959196. [DOI] [PubMed] [Google Scholar]
- Suardíaz M, Estivill-Torrús G, Goicoechea C, Bilbao A, Rodríguez de Fonseca F. Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain. 2007;133:99–110. doi: 10.1016/j.pain.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Svízenská I, Dubový P, Sulcová A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol Biochem Behav. 2008;90:501–511. doi: 10.1016/j.pbb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Ta LE, Low PA, Windebank AJ. Mice with cisplatin and oxaliplatin-induced painful neuropathy develop distinct early responses to thermal stimuli. Mol Pain. 2009;5:9. doi: 10.1186/1744-8069-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta LE, Bieber AJ, Carlton SM, Loprinzi CL, Low PA, Windebank AJ. Transient receptor potential vanilloid 1 is essential for cisplatin-induced heat hyperalgesia in mice. Mol Pain. 2010;6:15. doi: 10.1186/1744-8069-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tredici G, Tredici S, Fabbrica D, Minoia C, Cavaletti G. Experimental cisplatin neuronopathy in rats and the effect of retinoic acid administration. J Neurooncol. 1998;36:31–40. doi: 10.1023/a:1005756023082. [DOI] [PubMed] [Google Scholar]
- Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K, Yonenobu K, Ochi T, Noguchi K. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol Cell Neurosci. 2000;15:170–182. doi: 10.1006/mcne.1999.0814. [DOI] [PubMed] [Google Scholar]
- Tulub AA, Stefanov VE. Cisplatin stops tubulin assembly into microtubules. A new insight into the mechanism of antitumor activity of platinum complexes. Int J Biol Macromol. 2001;28:191–198. doi: 10.1016/s0141-8130(00)00159-8. [DOI] [PubMed] [Google Scholar]
- Verdú E, Vilches JJ, Rodríguez FJ, Ceballos D, Valero A, Navarro X. Physiological and immunohistochemical characterization of cisplatin-induced neuropathy in mice. Muscle Nerve. 1999;22:329–340. doi: 10.1002/(sici)1097-4598(199903)22:3<329::aid-mus5>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Walsh TJ, Clark AW, Parhad IM, Green WR. Neurotoxic effects of cisplatin therapy. Arch Neurol. 1982;39:719–720. doi: 10.1001/archneur.1982.00510230045013. [DOI] [PubMed] [Google Scholar]
- Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13:27–46. doi: 10.1111/j.1529-8027.2008.00156.x. [DOI] [PubMed] [Google Scholar]
- Wongtawatchai T, Agthong S, Kaewsema A, Chentanez V. Sex-related differences in cisplatin-induced neuropathy in rats. J Med Assoc Thai. 2009;92:1485–1491. [PubMed] [Google Scholar]
- Xiao WH, Zheng H, Zheng FY, Nuydens R, Meert TF, Bennett GJ. Mitochondrial abnormality in sensory, but not motor, axons in paclitaxel-evoked painful peripheral neuropathy in the rat. Neuroscience. 2011;199:461–469. doi: 10.1016/j.neuroscience.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao WH, Zheng H, Bennett GJ. Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience. 2012;203:194–206. doi: 10.1016/j.neuroscience.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kachi T, Yamada T, Nagamatsu M, Sobue G. Sensory conduction study of cisplatin neuropathy: preservation of small myelinated fibers. Intern Med. 1997;36:829–833. doi: 10.2169/internalmedicine.36.829. [DOI] [PubMed] [Google Scholar]
- Yoon MS, Katsarava Z, Obermann M, Schäfers M, Liedert B, Dzagnidze A, Kribben A, Egensperger R, Limmroth V, Diener HC, Thomale J. Erythropoietin overrides the triggering effect of DNA platination products in a mouse model of cisplatin-induced neuropathy. BMC Neurosci. 2009;10:77. doi: 10.1186/1471-2202-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Challapalli SC, Smith PJ. Cannabinoid CB1 receptor activation stimulates neurite outgrowth and inhibits capsaicin-induced Ca2+ influx in an in vitro model of diabetic neuropathy. Neuropharmacology. 2009;57:88–96. doi: 10.1016/j.neuropharm.2009.04.017. [DOI] [PubMed] [Google Scholar]