

Abstract

The synthesis of two complex subunits en route to spirolide C is described. A key alkyllithium addition to an aldehyde joins the fragments, which are advanced in order to investigate a ring-closing metathesis to form the 23-membered all-carbon macrocyclic framework.
The cyclic imine group of marine toxins is an emerging class of natural products with global distribution in marine environments.1 After the discovery of the first member of this family in 1995, the group has grown to include more than 30 members with complex chemical structures, including pinnatoxins,2 pteriatoxins,3 spirolides,4 gymnodimines,5 and spiro-prorocentrimine.6 Initially, the mode of action for spirolides, pinnatoxins, and gymnodimine was attributed to calcium-channel activation,7 which has been subsequently revised to a potent and selective inhibition of nicotinic acetylcholine receptors (nAChRs), where the cyclic imines have been found to be among the most powerful nonpeptidic antagonists of nAChRs known to date.8
The structure of the spirolides combines the most challenging aspects within the cyclic imine group of toxins: the characteristic spiroimine subunit, the tetrasubstituted alkene carrying a butenolide fragment, the 5,5,6-bispiroketal, and a highly functionalized 23-membered all-carbon macrocyclic framework.9 Previously, the groups of Brimble10 and Ishihara11 described their approaches to solving selected problems posed by the complexity of spirolides, concentrating on the synthesis of the 5,5,6-spirobicyclic ring system and the construction of the spiroimine subunit. Herein, we disclose the progress of our own work,12 with a focus on the formation of the 23-membered macrocyclic ring by ring-closing metathesis (RCM).
An early strategic decision in our plan was to assemble the 5,5,6-bispiroketal after the construction of the macro-cyclic framework to ensure the desired stereocontrol in the ketalization, which would be difficult to achieve in the acyclic system, in particular with regard to the formation of the 5,5-ketal, where no anomeric stabilization can be relied upon to control stereochemistry (Scheme 1).10a,11 An example of the desired macrocyclic product is compound 2, which could be accessed in a convergent manner from organolithium reagent 3 and aldehyde 4 followed by RCM (disconnections along the highlighted bonds at C13−C14 and C27–C28). Aldehyde 5, previously prepared by a key diastereoselective Ireland–Claisen rearrangement, serves as a precursor to 4.13
Scheme 1.
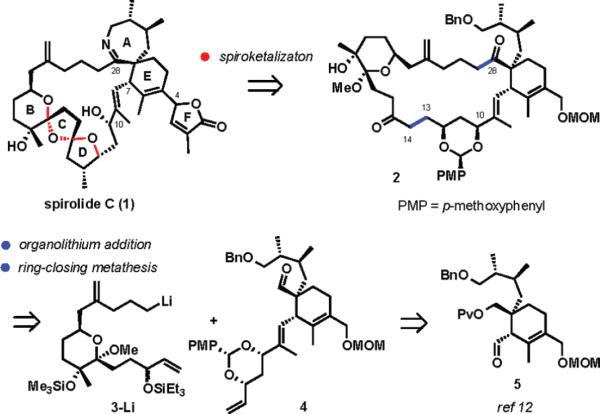
Synthesis Plan for Spirolide C
The synthesis of intermediate 4 began with a three-step preparation of thioester 6 from aldehyde 5 (Scheme 2A). Aldol addition of S-ethyl thiopropionate followed by mesylation with methanesulfonic anhydride and elimination with DBU afforded 6 as a single stereoisomer in 93% overall yield. Thioester 6 was reduced to the corresponding α,β-unsaturated aldehyde under conditions reported by Fukuyama.14 Soderquist asymmetric allylation was reproducible on varying scales and provided homoallylic alcohol 7 in 76% isolated yield with 5:1 diastereo-selectivity.15–17 Formation of the tert-butyl carbonate (LiN(SiMe3)2, Boc2O) followed by iodolactonization (NIS, (CF3)2CHOH)18 and treatment with methanolic potassium carbonate delivered epoxy alcohol 9 in 69% yield over the three steps. Treatment of epoxy alcohol 9 with the sulfur ylide generated from trimethylsulfonium iodide produced the requisite allylic diol19 and was accompanied by the removal of the pivalate ester group. Protection of the 1,3-syn-diol as a p-methoxybenzylidene acetal and oxidation of the primary hydroxy group under Swern conditions20 completed the synthesis of intermediate 4.
Scheme 2.

Synthesis of Intermediate 4
During the development of the synthetic route to 4 described above, we explored the synthesis of the desired 1,3-syn-diol acetal based on the recently developed Recatalyzed transposition of allylic alcohols directed by hydroxy groups.21 To this end, diol 10 was prepared from 7 by cross-metathesis with (Z)-2-butene-1,4-diol diacetate followed by methanolysis (97% over two steps, Scheme 2B).22 However, only a small amount of 11 could be detected, and generally decomposition was observed under a variety of conditions.
The synthesis of the bispiroketal precursor fragment (3) was initiated by methylation of lithium acetylide generated from 1223 followed by hydrozirconation–odination24 to form the corresponding (E)-iodoalkene. Palladium-catalyzed cross-coupling of the iodide and the alkylzinc halide 13 afforded trisubstituted alkene 14 (57% over three steps).25 Hydrolysis of the acetonide, epoxide formation,26 and alkylation of lithiated sulfone 15 with the epoxide afforded 16 in excellent yield. Introduction of the exomethylene group was accomplished by a Julia-type process after protection of the secondary hydroxyl group as a triethylsilyl ether.27
Sharpless asymmetric dihydroxylation28 of diene 17 took place at the trisubstituted double bond, selectively affording the desired diol in 57% yield (85% based on recovered 17). Oxidation by the Parikh–Doering method29 cleanly afforded hydroxy ketone 18. Removal of the triethylsilyl ether with concomitant ketalization afforded the sensitive methyl ketal 19 in 91% yield. Attempts to form the triethylsilyl ether at the tertiary hydroxy group (TESOSO2CF3, 2,6-lutidine; TESCl, ImH, DMF, 40 °C, 12 h) were thwarted by a facile elimination of the ketal methoxy group, affording the exocyclic vinyl ether. On the other hand, silylation of 19 with chlorotrimethylsilane in DMF at 40 °C efficiently delivered the desired monosilylated product after treatment with potassium carbonate in methanol. Iododehydroxylation of the primary alcohol delivered 20 in 89% yield over three steps.
Careful oxidative removal of the p-methoxybenzyl ether with DDQ followed by a Swern oxidation of the resulting primary hydroxy group, addition of vinylmagnesium bromide generated an inconsequential ~1:1 mixture of diastereomers. Protection of the resulting secondary alcohol as its triethylsilyl ether completed the synthesis of the key intermediate 3 (Scheme 3).
Scheme 3.

Synthesis of Intermediate 3
The fragment coupling was accomplished as planned by a direct addition of the functionalized alkyllithium reagent 3-Li generated by lithum-iodine exchange from iodide 3 with aldehyde 4 in 98% yield (Scheme 4).30 Dess–Martin oxidation and desilylation delivered substrate 22 for key RCM studies.31
Scheme 4.

Fragment Coupling
We carried out extensive studies on ring-closing metathesis of tetraene 22, the results of which are summarized in Table 1. In all cases, the HGII catalyst provided cleaner reactions than the GII catalyst. Although the formation of the 23-membered all-carbon macrocycle was found to be rather challenging, we succeeded in forming the desired product, albeit in low yield (entries 1–4). NMR studies have shown conclusively that the RCM process is initiated at the C14 terminal alkene selectively. The main byproduct was the C15 methyl ketone, presumably arising by fragmentation of the intermediate ruthenium carbene at C14.32 Various attempts to minimize side reactions using additives such as 1,4-benzoquinone33 or 2,6-lutidine34 proved to be unproductive. To our delight, in no case was a RCM observed between the olefins at C13 or C14 and C24.35
Table 1.
RCM Studies of 22 and Its Derivativesa
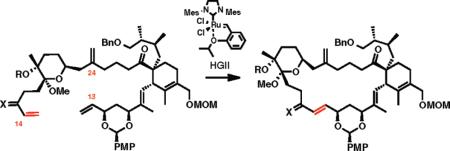
| entry | R; X | solvent | time (h), temp (°C) | conversion, % |
|---|---|---|---|---|
| 1 | H; H, OH | OH2Cl2 | 1, 40 | 10 |
| 2 | H; H, OH | CH2Cl2 | 18, 40 | 10 |
| 3 | H; H, OH | CH2Cl2 | 18, 20 | 20 |
| 4 | H; H, OH | CH2Cl2 | 12, 40 | 17b |
| 5 | H; O | CH2Cl2 | 7 d, 40 | 5 (95 brsm) |
| 6 | H; O | C2H4Cl2 | 24, 85 | 0 |
| 7 | SiMe3; H, OSiEt3 | CH2Cl2 | 24, 40 | 0 |
| 8 | SiMe3; H, OSiEt3 | PhMe | 24, 90 | 10 (90 brsm) |
| 9 | SiMe3; H, OSiEt3 | C2H4Cl2 | 24, 90 | 0 |
All reactions were carried out under an inert atmosphere of argon in degassed solvents. Reactions were performed on scales of 1–2 mg with a concentration of 0.005 M. Percent conversions were determined by NMR spectroscpy.
Isolated yield.
Ring-closing metathesis with the C13 enone, prepared by oxidation of the C13 hydroxyl, was substantially cleaner, although slower (~5% conversion after 7 days), and pre vented formation of C13 methyl ketone (entry 5). Increasing the reaction temperature from 40 to 85 °C resulted in complete decomposition (entry 6). Finally, fully silylated diol 22 was also more stable and resistant to the methyl ketone formation, but also exhibited low reactivity (entry 8).
To conclude our study on the key formation of the 23-membered macrocyclic framework en route to the spirolides, we tested the hypothesis that a more compact substrate such as spiroketal 23 would be a superior substrate for RCM (Scheme 5). Intermediate 23 could be readily prepared from diol 22 in one step in a nearly quantitative yield. Indeed, when 23 was subjected to optimized conditions for RCM (GII 30 mol %, 1,2-dichloroethane, 60 °C, 24 h), a clean reaction ensued affording the anticipated macrocyclic product (25% isolated yield) and the recovered starting material (54% isolated yield).
Scheme 5.

RCM with Spiroketal 23
In closing, we report the successful fragment coupling and extensive studies on RCM reactions of the 23-membered all-carbon macrocyclic ring system en route to the spirolides. While these studies revealed that the RCM is no doubt challenging, the powerful reaction was successful in forming the expected compounds and provided sufficient amounts of the macrocyclic products to enable key spiroketalization studies central to our approach in the synthesis of spirolides.
Supplementary Material
Acknowledgment
Financial support was provided by the National Institutes of Health, National Institute of General Medical Sciences (R01 GM077379), and kind gifts from Amgen and Eli Lilly. C.E.S. is supported by a Tobacco-Related Disease Research Program Dissertation Award and the UCSB Graduate Research Mentorship Fellowship. We are grateful to Dr. Hongjun Zhou (UC Santa Barbara) for continued assistance with NMR spectroscopy.
Footnotes
Supporting Information Available. Experimental procedures and copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- (1).(a) Cembella A, Krock B. In: Seafood and Freshwater Toxins. 2nd ed. Botana LM, editor. CRC Press; Boca Raton: 2008. pp. 561–580. [Google Scholar]; (b) Gueret SM, Brimble MA. Nat. Prod. Rep. 2010;27:1350–1366. doi: 10.1039/c005400n. [DOI] [PubMed] [Google Scholar]
- (2).(a) Uemura D, Chuo T, Haino T, Nagatsu A, Fukuzawa S, Zheng S, Chen H. J. Am. Chem. Soc. 1995;117:1155–1156. [Google Scholar]; (b) Chou T, Kamo O, Uemura D. Tetrahedron Lett. 1996;37:4023–4026. [Google Scholar]; (c) Chou T, Haino T, Kuramoto M, Uemura D. Tetrahedron Lett. 1996;37:4027–4030. [Google Scholar]
- (3).(a) Takada N, Umemura N, Suenaga K, Chou T, Nagatsu A, Haino T, Yamada K, Uemura D. Tetrahedron Lett. 2001;42:3491–3494. [Google Scholar]; (b) Takada N, Umemura N, Suenaga K, Uemura D. Tetrahedron Lett. 2001;42:3495–3497. [Google Scholar]
- (4).(a) Hu T, Curtis JM, Oshima Y, Quilliam MA, Walter JA, Watson-Wright WM, Wright JLC. J. Chem. Soc., Chem. Commun. 1995:2159–2161. [Google Scholar]; (b) Hu T, Curtis JM, Walter JA, Wright JLC. Tetrahedron Lett. 1996;37:7671–7674. [Google Scholar]; (c) Falk M, Burton IW, Hu T, Walter JA, Wright JLC. Tetrahedron. 2001;57:8659–8665. [Google Scholar]; (d) Hu T, Burton IW, Cemella AD, Curtis JM, Quilliam MA, Walter JA, Wright JLC. J. Nat. Prod. 2001;64:308–312. doi: 10.1021/np000416q. [DOI] [PubMed] [Google Scholar]
- (5).Seki T, Satake M, Mackenzie L, Kaspar H, Yasumoto T. Tetrahedron Lett. 1995;36:7093–7096.. Miles CO, Wilkins AL, Stirling DJ, MacKenzie AL. J. Agric. Food Chem. 2000;48:1373–1376. doi: 10.1021/jf991031k.. Miles CO, Wilkins AL, Stirling DJ, MacKenzie AL. J. Agric. Food Chem. 2003;51:4838–4840. doi: 10.1021/jf030101r.. For total synthesis, see: Kong K, Romo D, Lee C. Angew. Chem., Int. Ed. 2009;48:7402–7405. doi: 10.1002/anie.200903432.. Kong K, Moussa Z, Lee C, Romo D. J. Am. Chem. Soc. 2011;133:19844–19856. doi: 10.1021/ja207385y..
- (6).Lu C-K, Lee G-H, Huang R, Chou H-N. Tetrahedron Lett. 2001;42:1713–1716. [Google Scholar]
- (7).See, for example: (a) References 2a and 4a. Gill S, Murphy M, Clausen J, Richard D, Quilliam M, MacKinnon S, LaBlanc P, Mueller R, Pulido O. Neurotoxicology. 2003;24:593–604. doi: 10.1016/S0161-813X(03)00014-7..
- (8).(a) Bourne Y, Radic Z, Araoz R, Talley TT, Benoit E, Servent D, Taylor P, Molgo J, Marchot P. Proc. Natl. Acad. Sci. 2010;107:6076–6081. doi: 10.1073/pnas.0912372107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Araoz R, Servent D, Molgo J, Iorga BI, Fruchart-Gaillard C, Benoit E, Gu Z, Stivala CE, Zakarian A. J. Am. Chem. Soc. 2011;133:10499–10511. doi: 10.1021/ja201254c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For reviews on the synthesis of cyclic imine toxins, see: Beaumont S, Ilardi EA, Tappin N, Zakarian A. Eur. J. Org. Chem. 2010;30:5743–5765. doi: 10.1002/ejoc.201000842.. O'Connor PD, Brimble MA. Nat. Prod. Rep. 2007;24:869–885. doi: 10.1039/b700307m..
- (10).(a) Meilert K, Brimble MA. Org. Lett. 2005;7:3497–3500. doi: 10.1021/ol051260u. [DOI] [PubMed] [Google Scholar]; (b) Furkert DP, Brimble MA. Org. Lett. 2002;4:3655–3658. doi: 10.1021/ol026605c. [DOI] [PubMed] [Google Scholar]; (c) Meilert K, Brimble M,A. Org. Biomol. Chem. 2006;4:2184–2192. doi: 10.1039/b604334h. [DOI] [PubMed] [Google Scholar]; (d) Brimble MA, Caprio V, Furert DP. Tetrahedron. 2001;57:4023–4034. [Google Scholar]; (e) Brimble MA, Fares FA, Turner P. Aust. J. Chem. 2000;53:845–851. [Google Scholar]; (f) Zhang YC, Furkert DP, Gueret SM, Lombard F, Brimble MA. Tetrahedron Lett. 2011;52:4896–4898. [Google Scholar]
- (11).Ishihara J, Tshizaka T, Suzuki T, Hatakeyama S. Tetrahedron Lett. 2004;45:7855–7858. [Google Scholar]
- (12).Stivala CE, Zakarian A. Org. Lett. 2009;11:839–842. doi: 10.1021/ol8027797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Qin Y.-c., Stivala CE, Zakarian A. Angew. Chem., Int. Ed. 2007;46:7466–7469. doi: 10.1002/anie.200702142. [DOI] [PubMed] [Google Scholar]; (b) Gu Z, Herrmann AT, Stivala CE, Zakarian A. Synlett. 2010:1717–1722. [Google Scholar]
- (14).(a) Fukuyama T, Lin S-C, Li L. J. Am. Chem. Soc. 1990;112:7050–7051. [Google Scholar]; (b) Fukuyama T, Tokuyama H. Aldrichim. Acta. 2004;37:87–96. [Google Scholar]
- (15).(a) Burgos CH, Canales E, Matos K, Soderquist JA. J. Am. Chem. Soc. 2005;127:8044–8049. doi: 10.1021/ja043612i. [DOI] [PubMed] [Google Scholar]; (b) Canales E, Prasad KG, Soderquist JA. J. Am. Chem. Soc. 2005;127:11572–11573. doi: 10.1021/ja053865r. [DOI] [PubMed] [Google Scholar]; (c) Soto-Cairoli B, Soderquist JA. Org. Lett. 2009;11:401–404. doi: 10.1021/ol802685e. [DOI] [PubMed] [Google Scholar]
- (16).Brown asymmetric allylation generated homoallylic alcohol 7 in 91% yield as a 10:1 mixture of diastereomers; however, this result was irreproducible. Jadhav PK, Bhat KS, Perumal T, Brown HC. J. Org. Chem. 1986;51:432–439..
- (17).The separable minor diastereomer could be recycled through an oxidation–reduction sequence, providing an additional 7–10% yield of 7.
- (18).Ilardi EA, Stivala CE, Zakarian A. Org. Lett. 2008;10:1727–1730. doi: 10.1021/ol800341z. [DOI] [PubMed] [Google Scholar]
- (19).Alcaraz L, Hartnett JJ, Mioskowski C, Martel JP, Le Gall T, Shin D-S, Falck JR. Tetrahedron Lett. 1994;35:5449–5452. [Google Scholar]
- (20).Omura K, Swern D. Tetrahedron. 1978;34:1651–1660. [Google Scholar]
- (21).Herrmann AT, Saito T, Stivala CE, Tom J, Zakarian A. J. Am. Chem. Soc. 2010;132:5962–5963. doi: 10.1021/ja101673v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cluzeau J, Capdevielle P, Cossy J. Tetrahedron Lett. 2005;46:6945–6948. [Google Scholar]
- (23).Toro A, Lemelin C-A, Preville P, Belanger G, Deslongchamps P. Tetrahedron. 1999;55:4655–4684. [Google Scholar]
- (24).(a) Huang Z, Negishi E-I. Org. Lett. 2006;8:3675–3678. doi: 10.1021/ol061202o. [DOI] [PubMed] [Google Scholar]; (b) Baba S, Negishi E. J. Am. Chem. Soc. 1976;98:6729–6731. [Google Scholar]
- (25).(a) Negishi E, Okukado N, King AO, Van Horn DE, Spiegel BI. J. Am. Chem. Soc. 1978;100:2254–2256. [Google Scholar]; (b) Hayashi T, Konishi M, Kobori Y, Kumada M, Higichi T, Hirosu K. J. Am. Chem. Soc. 1984;106:158–163. [Google Scholar]
- (26).Cink RD, Forsyth CJ. J. Org. Chem. 1995;60:8122–8123. [Google Scholar]
- (27).(a) Smith AB, Doughty VA, Lin Q, Zhuang L, McBriar MD, Boldi AM, Moser WH, Murase N, Nakayama K, Sobukawa M. Angew. Chem., Int. Ed. 2001;40:191–195. doi: 10.1002/1521-3773(20010105)40:1<191::AID-ANIE191>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; (b) Julia M, Paris JM. Tetrahedron Lett. 1973:4833–4836. [Google Scholar]
- (28).Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483–2547. [Google Scholar]
- (29).Parikh JR, Doering W. v. E. J. Am. Chem. Soc. 1967;89:5505–5507. [Google Scholar]
- (30).Bailey WF, Brubaker JD, Jordan KP. J. Organomet. Chem. 2003;681:210–214. [Google Scholar]
- (31).(a) Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]; (b) Boeckman RK, Shao P, Mullins J. J. Org. Synth. 2000;77:141–152. [Google Scholar]
- (32).See the Supporting Information for the structure of the ketone. Gurjar MK, Yakambram P. Tetrahedron Lett. 2001;42:3633–3636.. Renzulli ML, Rocheblave L, Avramova SI, Galletti E, Castagnolo D, Tafi A, Corelli F, Botta M. Tetrahedron. 2007;63:497–509.. Hoye TR, Zhao H. Org. Lett. 1999;1:169–171. doi: 10.1021/ol9906514..
- (33).Hong SH, Sanders DP, Lee CW, Grubbs RH. J. Am. Chem. Soc. 2005;127:17160–17161. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- (34).2,6-Lutidine was used to avoid possible fragmentation of the sensitive methyl ketal.
- (35).For a similar problen in the synthesis of pinnatoxins, see: Stivala CE, Zakarian A. J. Am. Chem. Soc. 2008;130:3774–3776. doi: 10.1021/ja800435j.. (b) Reference 8b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.