

Abstract
Very recently, FOXP1 deficiency was shown to result in a phenotype of intellectual disability with significant speech and language impairment. Behavioral abnormalities should be considered as part of the clinical spectrum. Mild craniofacial abnormalities found in half of the described patients expand the clinical spectrum associated with FOXP1 mutations.
Key Words: Developmental delay, FOXP1, Intellectual disability, Speech deficit
History of the Syndrome
Neurodevelopmental disorders that disturb speech and language are highly heritable. Rare heterozygous mutations of the FOXP2 gene have been identified as one cause of hereditary developmental dyspraxia [Lai et al., 2001]. The most closely related member of the same gene family, the forkhead box P1 gene (FOXP1, MIM 605515), has expression patterns that overlap with FOXP2 in some areas of the brain, and FOXP1 and FOXP2 have the ability to form heterodimers. This structural and functional relationship between FOXP1 and FOXP2 suggests that FOXP1 may also contribute to proper speech development. In 2009, a patient was described who presented with a 3p13–14.1 microdeletion of 4 genes, including FOXP1, resulting in speech delay, contractures, muscular hypertonia and blepharophimosis [Pariani et al., 2009]. Recently, a single deletion of FOXP1 was reported to cause severe speech delay, delayed gross motor skills, Chiari I malformation, and epileptiform discharges in a child [Carr et al., 2010]. Very recently, 3 heterozygous overlapping deletions solely affecting the FOXP1 gene in unrelated patients affected with moderate intellectual disability (ID) and significant language and speech deficits have been detected by molecular karyotyping [Horn et al., 2010]. Two different de novo heterozygous mutations in the FOXP1 gene were identified in 2 unrelated children with ID, expressive language deficits, and autistic features [Hamdan et al., 2010].
In total, 6 patients with heterozygous FOXP1 deficiency have been reported to date.
Clinical Features
Intellectual Disability
Detailed clinical investigation of these patients revealed that they present mild to moderate ID in combination with a general developmental delay (table 1). In 5 patients, the non-verbal IQ score was assessed as about 50 (3 SD below the mean) [Hamdan et al., 2010; Horn et al., 2010]. Almost all showed retarded gross-motor development with unsupported walking between 18 and 36 months [Carr et al., 2010; Hamdan et al., 2010; Horn et al., 2010]. Patients started to speak at ages of 3–6 years and used combinations of 2 words at ages between 4 and 7 years. Expressive language was more affected than receptive abilities. Expressive vocabulary was limited to relatively simple, frequent words or 2–3 word sentences in a 15-year-old and 9-year-old patient, respectively. In terms of expressive communication these 2 patients were scored at an age equivalent of 1 year 11 months at the chronological age of 9 years and about 4.5 years at the chronological age of 15 years, respectively [Hamdan et al., 2010].
Table 1.
Clinical findings in patients with FOXP1 deletions and mutations
| Carr et al. [2010] | Horn et al. [2010] Pat. 1 | Horn et al. [2010] Pat. 2 | Horn et al. [2010] Pat. 3 | Hamdan et al. [2010] Pat. A | Hamdan et al. [2010] Pat. B | ||
|---|---|---|---|---|---|---|---|
| General features | age of last assessment | 41 months | 7 years | 5.5 years | 6 years | 15 years | 9 years |
| sex | male | male | female | male | female | male | |
| occipitofrontal head | |||||||
| circumference (SD) | +0.7 | +1.2 | +0.8 | +1.3 | n.d. | n.d. | |
| weight (SD) | −0.2 | +0.5 | +2.7 | +2.5 | n.d. | n.d. | |
| facial gestalt | prominent forehead, ptosis | no dysmorphisms | prominent forehead, frontal hair upsweep | prominent forehead, frontal hair upsweep | n.d. | n.d. | |
| non-verbal | |||||||
| performance IQ | n.d. | <50 | <50 | 50 | 58 | 48 | |
| gross motor delay | n.d. | + | + | + | + | + | |
| age at walking | 16 months | 24 months | 36 months | 24 months | 18 months | 20 months | |
| Speech and language | first words at age of combined words at | 3.5 years | 3.5 years | 3.5 years | 3.5 years | 3 years | 6 years |
| development | age of | n.d. | 7 years | 5 years | 5.5 years | 4 years | n.d. |
| Behavioral anomalies | n.d. | n.d. | + | + | + | + |
Two of them showed oromotor problems including difficulties with lip protrusion. In infancy, some patients had a tendency to keep their mouth open [Horn et al., 2010].
Facial Gestalt
Mild craniofacial anomalies documented in 3 of 6 patients included broad and prominent forehead, a short nose and a frontal hair upsweep in 2 patients of this cohort (fig. 1) [Carr et al., 2010; Horn et al., 2010]. The remaining patients are not reported to show a distinctive facial phenotype [Hamdan et al., 2010]. Altogether, the facial features alone do not seem to be specific enough to establish the diagnosis.
Fig. 1.
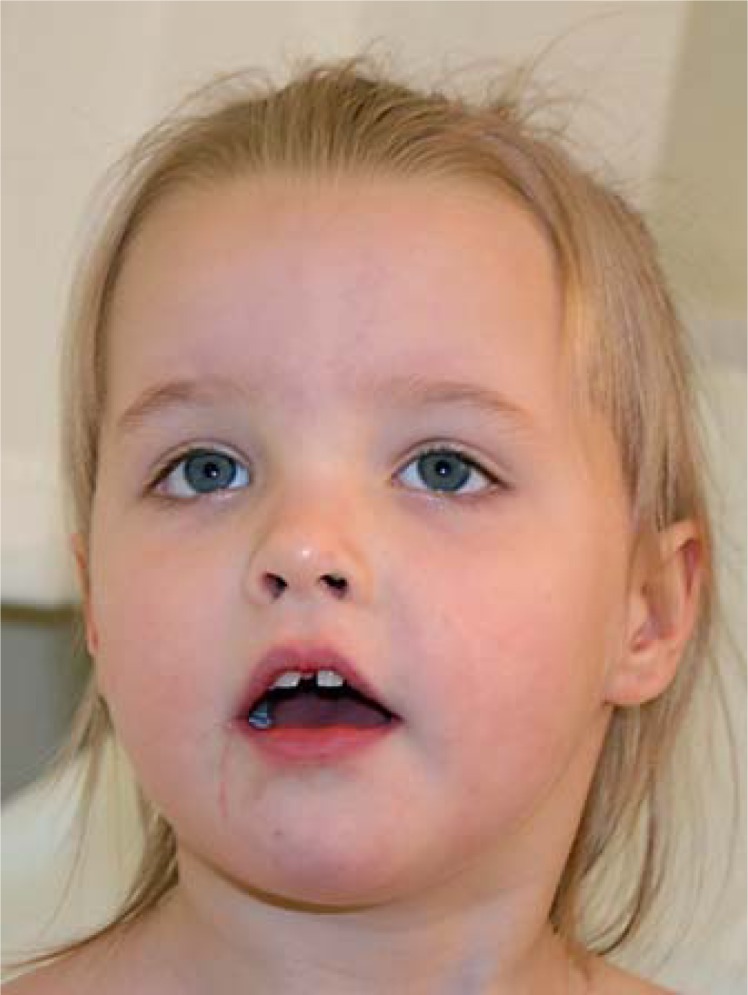
Facial features of a 4-year-old girl showing a broad and prominent forehead, frontal hair upsweep, and a short nose (patient 2 reported by Horn et al., 2010).
Growth Parameters
Patients were born after uneventful pregnancies and deliveries. Growth parameters at birth were normal in all. Also later in childhood, normal growth parameters regarding height and occipitofrontal head circumference were documented in 4 reported cases (table 1) but presence of obesity was striking in 2 patients. Regarding available data, patients show a tendency to head circumferences above mean (+0.7 to +1.3 SD) [Carr et al., 2010; Horn et al., 2010].
Behavioral Abnormalities
Four of 6 patients showed particular behavioral problems [Hamdan et al., 2010; own unpublished results]. The abnormal behaviors were characterized by both internalizing (e.g., anxiety) and externalizing (e.g., mood lability, temper tantrums, impulsivity, physical aggression) behavioral problems. Irritability, stereotypy, hyperactivity and autistic features were rated as the most significant problems on the aberrant behavior checklist [Hamdan et al., 2010]. A number of specific obsessions and compulsions as well as ritualistic behavior (e.g. rigid routines, strong attachment to specific objects) were described by the parents. Self-injurious behavior was also reported for 1 patient [Hamdan et al., 2010]. Common behavioral characteristics in 4 of 6 patients were mood lability, autistic features, and hyperactivity.
Other Findings
Brain magnetic resonance imaging and electroencephalography did not reveal any abnormalities in 3 patients but in 1 further patient a dysmorphic corpus callosum, mild hypoplasia of the cerebellar vermis, and a Chiari I malformation were reported which required suboccipital decompression of this malformation at an age of 30 months [Carr et al., 2010]. An electroencephalogram in this patient disclosed epileptiform discharges in different brain regions [Carr et al., 2010]. Another patient with a FOXP1 mutation was operated on for atresia of the midjejunum and midileum gut during the neonatal period [Hamdan et al., 2010].
Natural History
In infancy, gross-motor development is delayed in most cases [Carr et al., 2010; Hamdan et al., 2010; Horn et al., 2010]. Speech and language development is significantly retarded with first words spoken after 3 years of age. Expressive vocabulary remains limited to simple and single words. ID is of mild to moderate degree. Behavioral problems, including mood lability and hyperactivity, tend to be frequent in childhood.
Recommendations for Management
Not much information about treatment is available. In the reported patient with epileptiform discharges, oxacarbazepine therapy was initiated [Carr et al., 2010].
Genetics
Information about the FOXP1 Gene, the Corresponding Protein, and Relationship to FOXP2
FOXP1 consists of 16 coding exons spanning about 600 kb on chromosome 3p14.1. De novo deletions identified by copy number analysis included all coding exons of FOXP1 in 3 patients and are intragenic deletions in 2 reported patients [Carr et al., 2010; Hamdan et al., 2010; Horn et al., 2010]. By sequencing of the longest FOXP1 isoform, a de novo nonsense mutation was found in a further patient [Hamdan et al., 2010]. This mutation was predicted to abolish the last 152 residues of the protein, including part of the forkhead domain. Expression studies showed that this mutant impaired the transcriptional repression ability of FOXP1, consistent with a loss of function [Hamdan et al., 2010].
FOXP1 belongs to a functionally diverse family of forkhead box (FOX) transcription factors that are all characterized by a highly conserved FOX domain. FOX genes have been shown to play important roles in diverse cellular functions including metabolic and developmental processes [Carlsson and Mahlapuu, 2002]. While the function of FOXP1 during blood cell, lung and heart formation has already been addressed [Shu et al., 2001; Shi et al., 2004; Wang et al., 2004; Hu et al., 2006], the role of FOXP1 in neuronal processes, in particular brain development, is still unclear. In a mouse mutant model, Foxp1 has recently been shown to be an important accessory factor in Hox transcriptional output, thus regulating motor neuron diversification and connectivity to target muscles [Dasen et al., 2008; Rousso et al., 2008]. These findings are of particular interest with regard to the gross-motor deficits seen in patients with FOXP1 deletions and mutations.
With regard to structure and expression profiles, the most interesting gene related to FOXP1 is FOXP2 (MIM 605317). Rare mutations of FOXP2 have been described in individuals having expressive and receptive language and speech deficits generally described as developmental verbal dyspraxia [Hurst et al., 1990; Lai et al., 2001; MacDermot et al., 2005; Feuk et al., 2006]. A functional relationship between Foxp1 and Foxp2 has been previously demonstrated in mouse, as they form homo- and heterodimers necessary for efficient DNA binding [Li et al., 2004]. FOXP1 and FOXP2 also show overlapping expression patterns within brains of zebra finches and fetal human brains, particularly in subcortical regions that play important roles in sensorimotor integration and coordinated movements important for vocalization and speech, respectively [Teramitsu et al., 2004].
Non-verbal intellectual impairment could not be considered characteristic of the phenotype associated with FOXP2 mutations [Lai et al., 2001]. This is in contrast to the clinical findings in patients with FOXP1 mutations who, in addition to their speech and language disabilities, exhibit significantly reduced non-verbal IQ scores. This may indicate that additional distinct functions of FOXP1 exist, presumably acting on a more global level in neuronal development compared to FOXP2. Thus, haploinsufficiency of FOXP1 leads to abnormal development of neural structures that coordinate general cognitive and psychomotor as well as verbal abilities.
Mode of Inheritance
Up to now, all reported patients are sporadic cases, resulting from a de novo heterozygous deletion or mutation of FOXP1. Therefore, the FOXP1 related phenotype is inherited in an autosomal dominant manner.
Frequency of FOXP1 Mutations and Deletions
The frequency of these mutations has not been estimated, but these mutations seem to be a rare cause of ID and speech and language deficits. In a cohort of about 1,500 patients with ID, 3 cases with FOXP1 deletions were identified by copy number analysis [Horn et al., 2010]. Search for point mutations in 883 patients with ID did not discover a clear pathogenic variant, but a few coding variants were detected which have to be further investigated to assess their neurobiological significance [Horn et al., 2010]. Using array based genomic hybridization, 1 intragenic deletion of FOXP1 was found in a cohort of 30 patients with nonsyndromic ID and 80 patients with autism spectrum disorders [Hamdan et al., 2010]. Sequencing of FOXP1 in 110 sporadic cases with nonsyndromic ID and 135 cases with autism spectrum disorders revealed the presence of one de novo nonsense mutation [Hamdan et al., 2010].
References
- Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002;250:1–23. doi: 10.1006/dbio.2002.0780. [DOI] [PubMed] [Google Scholar]
- Carr CW, Moreno-De-Luca D, Parker C, Zimmerman HH, Ledbetter N, et al. Chiari I malformation, delayed gross motor skills, severe speech delay, and epileptiform discharges in a child with FOXP1 haploinsufficiency. Eur J Hum Genet. 2010;18:1216–1220. doi: 10.1038/ejhg.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasen JS, De Camilli A, Wang B, Tucker PW, Jessell TM. Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell. 2008;134:304–316. doi: 10.1016/j.cell.2008.06.019. [DOI] [PubMed] [Google Scholar]
- Feuk L, Kalervo A, Lipsanen-Nyman M, Skaug J, Nakabayashi K, et al. Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. Am J Hum Genet. 2006;79:965–972. doi: 10.1086/508902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. 2010;87:671–678. doi: 10.1016/j.ajhg.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn D, Kapeller J, Rivera-Brugues N, Moog U, Lorenz-Depiereux B, et al. Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat. 2010;31:E1851–E1860. doi: 10.1002/humu.21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Wang B, Borde M, Nardone J, Maika S, et al. Foxp1 is an essential transcriptional regulator of B cell development. Nat Immunol. 2006;7:819–826. doi: 10.1038/ni1358. [DOI] [PubMed] [Google Scholar]
- Hurst JA, Baraitser M, Auger E, Graham F, Norell S. An extended family with a dominantly inherited speech disorder. Dev Med Child Neurol. 1990;32:352–355. doi: 10.1111/j.1469-8749.1990.tb16948.x. [DOI] [PubMed] [Google Scholar]
- Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–523. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- Li S, Weidenfeld J, Morrisey EE. Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol Cell Biol. 2004;24:809–822. doi: 10.1128/MCB.24.2.809-822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermot KD, Bonora E, Sykes N, Coupe AM, Lai CS, et al. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet. 2005;76:1074–1080. doi: 10.1086/430841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariani MJ, Spencer A, Graham JM, Jr, Rimoin DL. A 785kb deletion of 3p14.1p13, including the FOXP1 gene, associated with speech delay, contractures, hypertonia and blepharophimosis. Eur J Med Genet. 2009;52:123–127. doi: 10.1016/j.ejmg.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousso DL, Gaber ZB, Wellik D, Morrisey EE, Novitch BG. Coordinated actions of the forkhead protein Foxp1 and Hox proteins in the columnar organization of spinal motor neurons. Neuron. 2008;59:226–240. doi: 10.1016/j.neuron.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Zhang X, Chen Z, Sulaiman K, Feinberg MW, et al. Integrin engagement regulates monocyte differentiation through the forkhead transcription factor Foxp1. J Clin Invest. 2004;114:408–418. doi: 10.1172/JCI21100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W, Yang H, Zhang L, Lu MM, Morrisey EE. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. 2001;276:27488–27497. doi: 10.1074/jbc.M100636200. [DOI] [PubMed] [Google Scholar]
- Teramitsu I, Kudo LC, London SE, Geschwind DH, White SA. Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. J Neurosci. 2004;24:3152–3163. doi: 10.1523/JNEUROSCI.5589-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, et al. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131:4477–4487. doi: 10.1242/dev.01287. [DOI] [PubMed] [Google Scholar]