

Background: Dodecin prevents riboflavin from autodegradation and exerting photo-induced cellular stress.
Results: Ultrafast depopulation of excited states and ground state recovery is observed by transient spectroscopy.
Conclusion: Ultrafast electron transfer in combination with proton transfer is responsible for deactivation of flavin excited states.
Significance: A comprehensive study of parameters in the binding pocket affecting the riboflavin quenching mechanism is given.
Keywords: Electron Transfer, Flavin, Flavoproteins, Protein Dynamics, Spectroscopy, Flavin-binding Protein, Proton Transfer, Ultrafast Spectroscopy
Abstract
Dodecins, a group of flavin-binding proteins with a dodecameric quaternary structure, are able to incorporate two flavins within each of their six identical binding pockets building an aromatic tetrade with two tryptophan residues. Dodecin from the archaeal Halobacterium salinarum is a riboflavin storage device. We demonstrate that unwanted side reactions induced by reactive riboflavin species and degradation of riboflavin are avoided by ultrafast depopulation of the reactive excited state of riboflavin. Intriguingly, in this process, the staggered riboflavin dimers do not interact in ground and photoexcited states. Rather, within the tetrade assembly, each riboflavin is kept under the control of the respective adjacent tryptophan, which suggests that the stacked arrangement is a matter of optimizing the flavin load. We further identify an electron transfer in combination with a proton transfer as a central element of the effective excited state depopulation mechanism. Structural and functional comparisons of the archaeal dodecin with bacterial homologs reveal diverging evolution. Bacterial dodecins bind the flavin FMN instead of riboflavin and exhibit a clearly different binding pocket design with inverse incorporations of flavin dimers. The different adoption of flavin changes photochemical properties, making bacterial dodecin a comparably less efficient quencher of flavins. This supports a functional role different for bacterial and archaeal dodecins.
Introduction
Riboflavin-binding proteins (RfbPs)3 have been characterized in eukaryotes as proteins for storage and transport of riboflavin. RfbPs are high affinity binders, and reduce the photochemical reactivity of the highly reactive riboflavins by efficiently quenching light-induced excited states. Dodecin has recently been identified as a RfbP in archaea, representing the first protein of this function in prokaryotes (1, 2). With one ligand per 68 residues, dodecin represents the flavoprotein with highest flavin load known to date. Dodecin has also been found in bacteria where it was characterized as a binder of FMN (2–4).
Dodecins have been characterized structurally by x-ray crystallography from the archaeon Halobacterium salinarum (HsDodA) and from the bacteria, Thermus thermophilus (TtDodB), Halorhodospira halophila (HhDodB), and Mycobacterium tuberculosis (MtDodB). Structures revealed a dodecameric fold, providing six identical binding pockets, which each enables binding of two antiparallel arranged flavins between two tryptophan residues building an aromatic tetrade arrangement (Fig. 1A) (1–7).
FIGURE 1.

Riboflavin (yellow) embedded into the binding pocket of HsDodA (A) and HhDodB (B). Shown are the amino acids responsible for the flavin binding, respectively. Water molecules coordinated to a Mg2+ at the bottom of the binding pocket are hydrogen-bonded to the flavin (see supplemental Figs. S3 and S4). For both proteins the tryptophan-flavin distance is 3.3 Å, the flavin-flavin distance is 3.5 Å.
Although structurally similar, archaeal and bacterial dodecins differ in their ligand binding spectrum and in the binding site architecture (Fig. 1). Although both share the incorporation of antiparallel stacked flavin dimers, the bacterial dodecin incorporates si-si-stacked pairs of FMN, whereas archaeal dodecin binds re-re-stacked pairs of riboflavin. An alignment of approximately 100 dodecins reflects the separation of bacterial and archaeal dodecin on a sequence level (2). The physiological function of the bacterial dodecins is still not known. Coenzyme A bound to a TtDodB encouraged speculations toward a bifunctional dodecin storage protein (4), whereas due to similarities to the recently identified EmoB, also binding dimers of FMN, it is tempting to speculate that the bacterial dodecin might be able to deliver FMNH2 for redox reactions in partner proteins (8).
A broad in vitro and in vivo analysis of HsDodA revealed that archaeal dodecin is a riboflavin storage protein for buffering FMN and FAD concentration during the H. salinarum life cycle. Cellular flavin concentrations and the dodecin expression profile show that dodecin sequesters riboflavin under growth-limiting concentrations and releases riboflavin into the biosynthesis of the physiologically important flavin derivatives FMN and FAD when favorable growth conditions induce metabolic activity (2).
Storage of micromolar amounts of the flavin requires efficient neutralization of the versatile flavin chemical reactivity to not affect the cellular integrity. Interaction with potent reaction partners is prevented by the encapsulation of riboflavins in sealed binding pockets. Preventing flavin photoreactivity is a more difficult task. Whereas primary light-induced activation to excited flavin states cannot be prevented by the protein environment, fast quenching of light-induced excited states has been identified as a strategy to suppress uncontrolled photochemical reactivity. Eukaryotic RfbPs typically bind riboflavin between the aromatic residues of mostly tryptophan- and tyrosine-built triads of stacked aromatic rings. Mechanistically, fast electron transfer from the aromatic moiety to the riboflavin has been proposed to quench the excited state of riboflavin by building a couple of a positively charged amino acid radical and a negatively charged flavin semiquinone (2, 9). Ultrafast electron transfer mechanisms from an aromatic moiety to a photoexcited flavin are not only observed for RfbP, but for other flavoproteins, like for BLUF (blue light sensing using FAD) domains, cryptochromes, and DNA photolyases (10–13).
Whereas the fast quenching of the riboflavin excited states in dodecin has been suggested to involve electron transfer from the adjacent tryptophan, similar to what has been reported for the eukaryotic RfbPs, the unique dodecin binding site also raises the question of whether the photocycle of dodecin might involve other or additional relaxation channels.
What role does tetrade-π-stacking play for the quenching mechanism? Does the ribityl chain affect the quenching mechanism? Do riboflavin-riboflavin-interactions contribute in the quenching mechanism? Do hydrogen bonds to the flavin, e.g. from surrounding water molecules, have an effect on the mechanism? Does the design of the binding pocket have any effect? To address these important questions, we initiated a broad systematic photophysical analysis with several wild-type and mutant dodecins in complex with several flavin ligands, in deuterated and nondeuterated solvents. With the broad set of samples, we achieve a combinatorial approach for identifying the key elements in the HsDodA quenching mechanism. For example, the influence of the ribityl chain was studied with riboflavin versus lumiflavin bound to dodecin; the influence of the staggered arrangement of isoalloxazines with riboflavin versus the single binder FAD; the influence of proton transfer on the quenching reaction with H2O versus D2O buffer solutions; and the influence of magnesium, coordinated at the bottom of the binding pocket, with wild-type versus magnesium-free mutant dodecins. The method of choice to observe fast photophysical processes on a subnanosecond time scale is the femtosecond pump-probe technique, where a short laser pulse starts the reaction and a second time-delayed pulse interrogates the system.
Our data show effective fluorescence quenching of riboflavin when embedded into HsDodA, indicating a fast depopulation of the reactive excited state. Because the concept of ultrafast electron transfer between flavins and closely positioned aromates like adenine, tryptophan, and tyrosine is well established in flavin photochemistry (9, 14–20), a similar process involving electron transfer from the adjacent tryptophan to the photoexcited flavin can be assumed for dodecin. However, in this contribution, the role of the unique dimeric arrangement of the isoalloxazines, the possibility of riboflavin fluorescence self-quenching (21), the function of other residues in the binding pocket, and a putative more complex quenching reaction comprising electron and proton transfer (20) are evaluated on its impact on the HsDodA photophysical properties.
EXPERIMENTAL PROCEDURES
Sample Preparation
The dodecin proteins are prepared as described by Grininger et al. (5). Wild-type and mutant dodecin from H. salinarum with different flavin cofactors and free riboflavin were measured in 20 mm Tris buffer, pH 7.5, with 1 m NaCl and 5 mm MgCl2. Dodecin from H. halophila was measured in 20 mm Tris buffer, pH 8, with 300 mm NaCl and 100 mm ectoine. The samples were concentrated to an optical density of 0.25–0.4 at 450 nm. D2O samples were prepared by successive steps of dialysis of dodecameric dodecin against D2O buffer at a pD of 7.9 (22).
Transient Visible-pump/Visible-probe Spectroscopy
The experimental setup for the pump/probe spectroscopy in the visible spectral range was described in detail elsewhere (23). In brief, the pulse source for these experiments was a Clark CPA 2001 femtosecond laser system (central wavelength 775 nm, pulse length 170 fs, pulse energy 800 μJ, repetition rate1 kHz). Excitation pulses were generated by frequency doubling of the laser fundamental in a BBO crystal (388 nm) or by using a noncollinear optical parametric amplifier (NOPA) (475 nm) (24, 25). Pulses with a central wavelengths of 345 and 440 nm were generated by sum frequency mixing of 775 nm and NOPA pulses in a type I BBO crystal (θ = 36° and 26°, respectively). The energy of the pump pulse was between 25 and 100 nJ. Probe pulses in a spectral range of 430–750-nm pulses were generated in a sapphire window as single filament white light. The cross-correlation width was below 250 fs for the entire investigated spectral range. The continuum pulses were dispersed by two spectrometers (sample and reference) and recorded with two 42-segment diode arrays (multichannel detection), resulting in a spectral resolution of 8 nm. Data acquisition was performed in single shot detection mode as balanced and referenced measurement providing signal-to-noise ratios up to 104. To guarantee nonexcited sample for successive laser shots, the sample was moved in two dimensions. Pure solution was measured as reference to correct for the coherent artifact. UV/visible absorption spectra were taken before and after the time-resolved experiments to ensure that no long lived photoproducts were accumulated.
Data Analysis
Prior to further analysis, the transient data were corrected for the coherent signals and group velocity dispersion. The coherent signal of the solution can be subtracted from the sample signal (26). The delay time zero was determined for all wavelengths employing a procedure (27, 28), which uses the temporal evolution of the coherent signal and a least squares fit algorithm. For the quantitative data analysis we employed a kinetic model, which describes the data as sum of exponential decays. A Marquardt downhill algorithm optimizes n global time constants (τi) for all wavelengths simultaneously with wavelength-dependent amplitudes Ai(λ) for each component. The model function assumes Gaussian pump and probe pulses with a (1/e) cross-correlation width tcc.
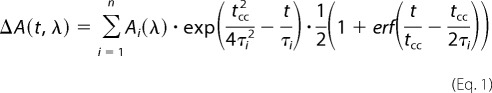 |
The wavelength-dependent fit amplitudes Ai(λ) represent the decay-associated difference spectrum (DADS) for each decay and thus represent the part of the difference spectrum that decays with the corresponding time constant (29). In this definition, an infinite time constant is equal to a time-independent offset in the transient absorbance changes at positive delay times, and therefore it mainly represents the absorbance change that remains at the maximum delay time in our experiments.
RESULTS AND DISCUSSION
Even though the positions of the riboflavin absorption bands are very sensitive to the environment (30), only small shifts of the absorption maxima of oxidized flavins have been observed for flavin dimers in solution (21) and a number of other flavoproteins (31, 32). Accordingly, steady-state spectroscopy for dodecin shows that the riboflavin absorption band at 445 nm (S0→S1 transition is only marginally blue-shifted compared with free riboflavin in aqueous solution, and the band at 373 nm (S0→S2 transition) shows no shift of the absorption maximum (Fig. 2). Furthermore, the relation of the two absorption bands changes if riboflavin is bound to HsDodA. Both absorption bands are attributed to π→π* transitions (33). The shoulder around 480 nm for bound riboflavin is a common phenomenon observed also for other flavoproteins and probably results from interactions between the π-systems or hydrogen bonding in the binding pocket (32, 34).
FIGURE 2.

Absorption spectra of riboflavin in aqueous buffer solution at pH 7.5 (gray) and incorporated into HsDodA (black), as well as riboflavin incorporated into HhDodB (gray, dotted). Spectra are normalized to their absorbance at 372 nm. The arrows denote the used excitation wavelengths in transient spectroscopy.
The kinetic measurements of riboflavin in aqueous solution and incorporated into diverse dodecin proteins were carried out for different excitation wavelengths to induce distinct electronic transitions and to provide varying excess vibrational energies. Additionally to the excitation of the S0→S1 transition of riboflavin at 476 nm, the low energy wing, and at 440, 388, and 345 nm was chosen to excite the S0→S2 transition at the low and at the high energy wing, respectively. Because transient absorbance changes for the different excitation wavelengths did not show any significant differences, we suggest that the S2→S1 transition is faster than the time resolution of the experiment for bound as well as for free riboflavin. This finding is in agreement with experiments by Stanley and McFarlane, where an internal conversion from S2 to S1 in less than 100 fs for flavins was demonstrated (17, 35). Thus, the discussion of photodynamics is restricted to the 388 nm excitation because the best signal-to-noise ratio was obtained in this experiment.
The transient absorbance changes of riboflavin in aqueous solution (17, 35, 36) and bound to HsDodA revealed entirely different spectral and dynamic behavior (Fig. 3, A and B, and Fig. 4). The excited state of free riboflavin displayed a lifetime longer than the maximum delay time of the experiment (1.5 ns), consistent with the reported lifetime of about 5 ns (37). In contrast, bound riboflavin showed lifetimes of its difference signals clearly shortened to only a few picoseconds, e.g. the ground state bleach (negative absorbance change at 450 nm). Furthermore, the pattern of positive and negative absorbance changes shows distinctive differences: the intensive negative signal of the stimulated emission at 520–600 nm observed for free riboflavin in aqueous solution is missing for HsDodA-bound riboflavin (2), and the positive signal does not reach that far into the long wavelength region above 700 nm. A discussed mechanism for the fast quenching of the excited state, on the basis of previous investigations on flavoproteins (9, 18, 19), is an ultrafast electron transfer from a tryptophan residue to the excited flavin. If such an electron transfer occurs, the spectrum should show the absorption characteristics of a cationic tryptophan radical, expected around 560 nm (38, 39) and of an anionic flavosemiquinone around 510 nm (40–42). For RfbP from chicken egg white, an electron-transferred state consisting of a cationic tryptophan radical and a flavosemiquinone has already been identified which shows spectral characteristics as observed for dodecin (7, 17). However, a remarkable difference between the reported data on chicken RfbP is the positive signal around 500 nm, which is not synchronized with the positive signal at longer wavelengths, and decays more slowly.
FIGURE 3.

Transient absorbance changes of riboflavin in aqueous solution (A) and bound to HsDodA (B) and HhDodB (C). Positive absorbance changes are depicted in red, negative in blue. The scale is linear from −1 ps to +1 ps and logarithmic for longer delay times.
FIGURE 4.

Transient absorbance changes of free (○), HsDodA (▴), and HhDodB (□) bound riboflavin at different wavelengths. The transient absorbance changes were normalized with respect to their absorption in the steady-state spectra at 388 nm. Symbols represent measured data points, and solid lines show results of the global fitting procedure.
Knowing the intermolecular distances in the dodecin binding pocket (3.3 Å between the isoalloxazine and the tryptophan for HsDodA) (2) as well as the net ΔG0 of 0.4 eV (9), the electron transfer rate, and the time constant of a potential electron transfer from a tryptophan residue to an electronically excited flavin molecule can be estimated (43). Accordingly, a time constant of a few hundred femtoseconds can be derived, which is close to the time resolution of the experiment, and therefore difficult to resolve.
Data of HsDodA occupied with riboflavin were successfully approximated in a global fit analysis with four time constants (Fig. 4 and supplemental Table 1). The fastest time constant τ1, close to the instrumental response time, is necessary to describe the rise of the signal. Another time constant, τ4, longer than the maximum delay time of the experiment, is used to include residual signals, which are here nearly 0. Two further time constants, τ2 and τ3, describe the decay of the difference signals: a fast time constant τ2 of 800 ± 100 fs and a slower one τ3 of 4 ± 1 ps. The DADS denotes a high positive amplitude for τ2 between 500 and 650 nm, indicating the decay of a positive absorption change in this region (Fig. 5). The spectral signature of the DADS of τ2 is in good agreement with the spectrum of the cationic tryptophan radical as well as of the neutral flavin semiquinone (38–42). Below 475 nm, the negative amplitude indicates the recovery of the ground state. The slower time constant τ3 also contributes to the ground state recovery, but the DADS indicates a spectral signature different from the one seen for τ2. τ3 exhibits a considerable amplitude at 500 nm, where the amplitude of τ2 crosses zero, and a lower amplitude than τ2 for wavelengths longer than 550 nm. These results suggest that two different quenching pathways with different time constants exist for the photoexcited flavin in the dodecin binding pocket, or that two different species, generated after photoexcitation, decay on different time scales. The specific characteristic of dodecin to relax excited states at different time scales might be based on the unique incorporation of dimeric riboflavin or on a proton neutralizing an anionic semiquinone. To get further insight into the complexity of the quenching pathway, also with regard to a proton transfer process, we initiated experiments with a set of wild-type and mutant HsDodA flavin complexes with various ligands, as well with a HsDodA-riboflavin complex in D2O.
FIGURE 5.

DADS of τ2 (HsDodA, 800 fs; HhDodB, 900 fs) and τ3 (HsDodA, 4 ps; HhDodB, 7 ps) of riboflavin incorporated into dodecin proteins (▴ HsDodA, □ HhDodB).
For evaluation of the role of the ribityl chain in the quenching mechanism, we used the capability of HsDodA to incorporate not only riboflavin, but also other flavins such as lumiflavin with a methyl group replacing the ribityl chain. Steady-state spectra of HsDodA with lumiflavin, essentially positioned similarly in the dodecin binding pocket as riboflavin, showed the typical flavin absorption bands as described for riboflavin and only a marginal shift in the absorption bands if it is embedded into dodecin (supplemental Fig. S1). Ultrafast spectroscopy revealed identical spectral and dynamic behavior (supplemental Fig. S2), indicating that the ribityl chain and the slightly different position do not play a major role for the excited state depopulation mechanism. For evaluation of the involvement of the stacked flavin in the quenching pathway, we used the properties of HsDodA to incorporate only one FAD molecule per binding pocket. In solution, fluorescence self-quenching of riboflavin of high concentrations due to dimerization is a common phenomenon (21, 30, 44). The distance between the two isoalloxazine moieties in the HsDodA binding pocket is only 3.2 Å, and self-quenching effects of dodecin bound riboflavin similar to riboflavin dimers in solution might be involved in dodecin photochemistry. FAD allows studying the effect of dimer stacking, as dodecin traps FAD from solution in a closed conformation, forming a tryptophan-isoalloxazine-adenine-tryptophan instead of tryptophan-isoalloxazine-isoalloxazine-tryptophan aromatic tetrade (6). Steady-state and transient spectroscopy of FAD in solution demonstrated a quenching of the excited state of the flavin by interactions with the adenine subunit in the closed conformation (17, 45–47). If for the quenching mechanism the second aromatic moiety is important, one should notice a difference in the transient data for the different bound cofactors riboflavin and FAD, respectively. Steady-state spectra of FAD show a shoulder at 480 nm and a small red shift of the absorption band at 350 nm if it is embedded into HsDodA (supplemental Fig. S1). Transient data show similar spectral and dynamic behavior as found for riboflavin (supplemental Fig. S2), suggesting a faster quenching of the exited state by the amino acid residues in the HsDodA binding pocket than by the molecule occupying the second flavin position. This also implies that the second flavin in the binding pocket does not influence the main excited state depopulation pathway.
Literature also reports a potential quenching of excited flavins by ultrafast electron and proton transfer (20). Therefore, the influence of hydrogen-bonded water molecules in the binding pocket was studied by replacement of H2O by D2O. The CW spectra show a blue shift of the S0→S2 absorption, whereas the S0→S1 band shows no shift if deuterated solvent is used (supplemental Fig. S2). Transient data show a slower decay of all signals for the samples in D2O (Fig. 6) and indicate that a proton transfer from a solvent molecule is part of the studied mechanism. A global fit analysis of the data of D2O and H2O samples taken under identical conditions (excitation wavelength, excitation energy, spatial and temporal overlap of pump and probe, etc.) showed that the data could successfully be approximated with four time constants. A fast time constant (τ1) and one longer than the maximum delay time of the experiment (τ4) were necessary for reasons described above. The time constant τ2 showed similar DADS profiles for both solvents but was longer for dodecin in D2O (τ2(D2O) 1.2 ± 0.2 ps versus τ2(H2O) 800 ± 100 fs) (Fig. 7). The time constant τ3 was somewhat slower for the D2O sample (τ3(D2O) 6 ± 2 ps versus τ3(H2O) 5 ± 1 ps), and the DADS showed spectral differences, especially around 600 nm. Variation of the starting conditions of the global fit analysis indicate that the time constant τ3 is inaccurate, and therefore the results are handled with care. However, global fit analysis indicates that the proton transfer plays a role at least for the faster component (τ2). The DADS of the time constant τ3 shows a significant amplitude at 500 nm and is therefore assigned to the decay of the difference signal at this wavelength. A similar signal is not seen for other flavin-binding proteins (9). A definitive assignment of this difference signal cannot be given, yet. However, some processes can be excluded: (i) single proton or hydrogen transfer without an electrontransfer, excluded because such processes should also occur in water and other flavin-binding proteins; (ii) a single electron transfer process without proton transfer because of the missing contribution of the tryptophan radical in the DADS of τ3; (iii) attendance of the second flavin in the binding pocket due to an unchanged signature for τ3 when FAD is binding. The spectral signature of the DADS indicates that a neutral tryptophan radical and an anionic riboflavin semiquinone may be intermediates of the slower pathway (38–42). It should be mentioned that τ3 could also describe cooling effects.
FIGURE 6.

Transient absorbance changes of riboflavin incorporated into HsDodA in H2O (▴) and D2O (○) buffer, respectively, at selected wavelengths after excitation with 388-nm pulses. The transient absorbance changes were normalized with respect to their absorption in the steady-state spectra at 388 nm.
FIGURE 7.

DADS of τ2 (HsDodA in H2O (800 fs), HsDodA in D2O (1.2 ps)) and τ3 (HsDodA in H2O (5 ps), HsDodA in D2O (6 ps)) of riboflavin incorporated into dodecin in H2O (▴) and D2O (○) buffer.
For the faster process, a proton donor transfer to the flavin is part of the mechanism. The tryptophan nitrogen is hydrogen bonding to Glu-38 and can be ruled out as the acid in this process. Instead, water molecules were identified as putative proton donors. These water molecules are in close vicinity to the riboflavin N5 position, which had been reported as good proton acceptor in riboflavin excited states (48). Especially, Mg2+-coordinated waters close to the N5 atom were regarded as putative proton donors, as Mg2+ coordination of waters facilitates deprotonation (49). To investigate the role of the Mg2+ in the quenching mechanism, we studied the mutated HsDodA samples D41S which do not bind a Mg2+ at that position as was studied by x-ray crystallography. Transient data show results identical to the wild-type HsDodA, and therefore we conclude that the water coordinated by the Mg2+ does not play a significant role (supplemental Fig. S5). It cannot be ruled out, however, that hydrogen-bonded water molecules act as proton donors even in the absence of the Mg2+.
Dodecin proteins are not only found in H. salinarum, but also in numerous other species for example M. tuberculosis, Legionella pneumophila, or H. halophila (2, 3). Adjacent to some smaller variations in the protein structure, the dodecin proteins show remarkable differences in the design of their binding pockets, resulting in a varying alignment of incorporated flavins. Whereas in archaeal dodecin, represented by structural data on HsDodA, the flavin dimers are in re-re-orientation, bacterial dodecin have si-si-orientated flavins as shown for HhDodB, TtDodB, and MtDodB (Fig. 1). The effect of the binding pocket design and the orientation of the flavin on the excited state quenching of riboflavin in HhDodB were also investigated by transient spectroscopy (Fig. 3C). The lifetime of the difference signals is clearly shorter than for free riboflavin, but compared with HsDodA, the decay of the difference signals is considerably slower. The stimulated emission cancels the positive absorbance change at 530 nm, thus the quenching of the excited state is less effective. Furthermore, the positive signal extends more into the long wavelength region, similar as for free riboflavin (Fig. 3, A and C, and Fig. 4). This leads to the conclusion that, compared with HsDodA, a slower recovery of the initial ground state is achieved by either slower transfer and back transfer of an electron, or another slower, less effective quenching mechanism.
The data of HhDodB were also successfully fitted with four time constants. According to the HsDodA data analysis, τ1 and τ4 are assigned as stated above. Time constants of τ2 = 900 ± 100 fs and τ3 = 7 ± 2 ps were found and describe the decay of the signals (the time constants for HsDodA were τ2 = 800 ± 100 fs and τ3 = 4 ± 1 ps). Both time constants are slower for HhDodB.
The ground state recovery for HsDodA and HhDodB gives the best comparison of the quenching dynamics. It shows that HhDodB has strongly pronounced amplitudes in the slower process (τ3), whereas in the fast process (τ2) the amplitudes are similar. Although different in ground state recovery, the DADS of the slow time constant is similar in both species and indicates that the respective reaction pathway is similar for both species. The DADS for the shorter time constant clearly shows a different spectral signature for λ > 500 nm and indicates significant differences in their deactivation pathway (Fig. 5). These dynamic differences are nicely reflected in the structural differences of both dodecins (Fig. 1): Most evident is the divergent alignment of the flavin residues in the binding pocket and a less overlapping alignment of the aromatic tetrade because the tryptophan residues are tilted and the flavins are shifted against each other. Also, the polar interactions holding the flavins are different in both binding pockets.
CONCLUSION
Our studies on dodecin proteins show that the lifetime of photoexcited riboflavin is reduced several orders of magnitude in an ultrafast depopulation mechanism. On the basis of results on other flavin-binding proteins, we expect an electron transfer from a tryptophan residue to the photoexcited flavin to play a central role for the quenching mechanism. This idea is supported by the spectral profile of the difference spectrum that fits well with the cationic tryptophan radical. The broad flavin tolerance of dodecin allows studying the role of the ribityl chain and the second flavin in the binding pocket in the quenching mechanism. Our results show that these elements are not relevant for the mechanism. Thus, the benefit of dimeric incorporation of riboflavin is to maximize the flavin load while still keeping the flavin photochemistry under the control of an adjacent aromatic amino acid, similar as is found in the well characterized chicken RfbP. Proton transfer from a surrounding water molecule as part of excited state quenching was detected by a longer time constant when H2O is replaced by D2O. From our data we can conclude that an electron transfer and back-transfer coupled with a proton transfer is responsible for the ultrafast depopulation of the flavin excited state. The most reasonable mechanism is illustrated in Fig. 8. Another explanation for the solvent dependence of τ2 might be a proton transfer-coupled electron back-transfer. A second process, which occurs on a slightly slower time scale, cannot be definitely assigned, yet. Therefore, we postulate the following quenching mechanism: a fast electron transfer from a tryptophan to the photoexcited riboflavin occurs faster than the time resolution of our experiment (250 fs) and a back-transfer with a time constant of 800 fs. This electron transfer process is coupled to a proton transfer from surrounding water molecules to the N5 of the flavin. A second simultaneous mechanism with a intermediate absorbing at 500 nm and decaying with a 4-ps time constant is not finally identified. The maximum in the DADS at 500 nm indicates that a neutral tryptophan radical or an anionic flavin radical might be part of that mechanism (38, 40). As mentioned before, hot riboflavin molecules might also be responsible for this signal. A contribution of the second flavin in the binding pocket to the difference signals is excluded, due to the results of dodecin-bound FAD.
FIGURE 8.
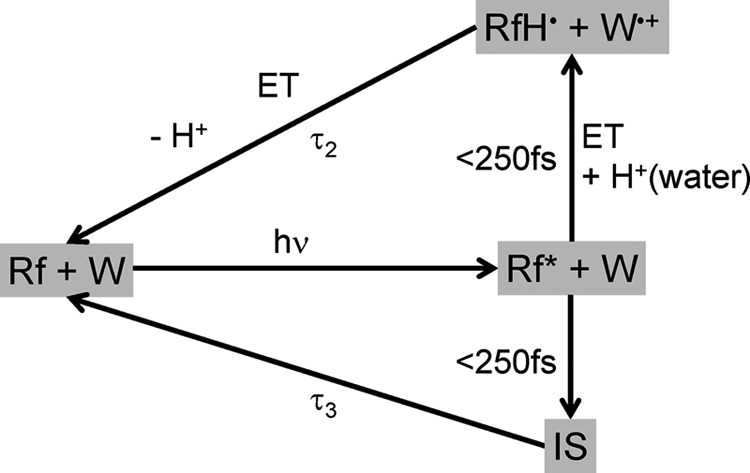
Proposed reaction pathway of HsDodA leading to the fast depopulation of the excited state of riboflavin (Rf). After the photoexcitation, an electron transfer from a tryptophan residue (W) to riboflavin and a proton transfer from a surrounding water to N5 of riboflavin occurs faster than the time resolution of the instrumental setup. Electron- and proton back-transfer with the time constant τ2 result in the initial ground state. In a second parallel reaction pathway, depopulation of the excited state of the flavin results in an intermediate (IS) with yet unknown identity. It decays with the time constant τ3 to the initial ground state.
In addition, we show the impact of the binding pocket design on excited state quenching by comparing ultrafast spectral behavior of archaeal and bacterial dodecin. Dodecins from the different domains of life show entirely different behavior in their transient data, thus demonstrating the conspicuous effect of the different binding pockets which were manifested by evolutionary divergence. Most prominent is the considerably faster decay of the difference signals for the archaeal HsDodA as key mechanism for faster quenching of bound riboflavin (2). It is striking that the DADS of the slower time constant shows nearly identical spectral features for HsDodA and HsDodB, thus describing the decay of similar states for both species.
Supplementary Material
Acknowledgment
We thank Prof. Dr. Hanna Grajek for helpful discussions concerning flavin dimers.
This work was supported by Collaborative Research Center 807 and Cluster of Excellence Frankfurt “Macromolecular Complexes.”

This article contains supplemental Figs. S1–S5 and Table S1.
- RfbP
- riboflavin-binding protein
- DADS
- decay-associated difference spectrum
- HhDodB
- bacterial H. halophila dodecin (ligand composition not specified)
- HsDodA
- archaeal H. salinarum dodecin (ligand composition not specified)
- MtDodB
- bacterial M. tuberculosis dodecin
- TdDodB
- bacterial T. thermophilus dodecin.
REFERENCES
- 1. Bieger B., Essen L. O., Oesterhelt D. (2003) Crystal structure of halophilic dodecin: a novel, dodecameric flavin-binding protein from Halobacterium salinarum. Structure 11, 375–385 [DOI] [PubMed] [Google Scholar]
- 2. Grininger M., Staudt H., Johansson P., Wachtveitl J., Oesterhelt D. (2009) Dodecin is the key player in flavin homeostasis of archaea. J. Biol. Chem. 284, 13068–13076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu F., Xiong J., Kumar S., Yang C., Ge S., Li S., Xia N., Swaminathan K. (2011) Structural and biophysical characterization of Mycobacterium tuberculosis dodecin Rv1498A. J. Struct. Biol. 175, 31–38 [DOI] [PubMed] [Google Scholar]
- 4. Meissner B., Schleicher E., Weber S., Essen L. O. (2007) The dodecin from Thermus thermophilus, a bifunctional cofactor storage protein. J. Biol. Chem. 282, 33142–33154 [DOI] [PubMed] [Google Scholar]
- 5. Grininger M., Zeth K., Oesterhelt D. (2006) Dodecins: a family of lumichrome-binding proteins. J. Mol. Biol. 357, 842–857 [DOI] [PubMed] [Google Scholar]
- 6. Grininger M., Seiler F., Zeth K., Oesterhelt D. (2006) Dodecin sequesters FAD in closed conformation from the aqueous solution. J. Mol. Biol. 364, 561–566 [DOI] [PubMed] [Google Scholar]
- 7. Vinzenz X., Grosse W., Linne U., Meissner B., Essen L. O. (2011) Chemical engineering of Mycobacterium tuberculosis dodecin hybrids. Chem. Commun. 47, 11071–11073 [DOI] [PubMed] [Google Scholar]
- 8. Nissen M. S., Youn B., Knowles B. D., Ballinger J. W., Jun S. Y., Belchik S. M., Xun L., Kang C. (2008) Crystal structures of NADH:FMN oxidoreductase (EmoB) at different stages of catalysis. J. Biol. Chem. 283, 28710–28720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhong D., Zewail A. H. (2001) Femtosecond dynamics of flavoproteins: charge separation and recombination in riboflavin (vitamin B2)-binding protein and in glucose oxidase enzyme. Proc. Natl. Acad. Sci. U.S.A. 98, 11867–11872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Z., Tan C., Guo X., Kao Y. T., Li J., Wang L., Sancar A., Zhong D. (2011) Dynamics and mechanism of cyclobutane pyrimidine dimer repair by DNA photolyase. Proc. Natl. Acad. Sci. U.S.A. 108, 14831–14836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gauden M., van Stokkum I. H., Key J. M., Lührs D. Ch., Van Grondelle R., Hegemann P., Kennis J. T. (2006) Hydrogen-bond switching through a radical pair mechanism in a flavin-binding photoreceptor. Proc. Natl. Acad. Sci. U.S.A. 103, 10895–10900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gauden M., Grinstead J. S., Laan W., van Stokkum I. H., Avila-Perez M., Toh K. C., Boelens R., Kaptein R., van Grondelle R., Hellingwerf K. J., Kennis J. T. (2007) On the role of aromatic side chains in the photoactivation of BLUF domains. Biochemistry 46, 7405–7415 [DOI] [PubMed] [Google Scholar]
- 13. Chaves I., Pokorny R., Byrdin M., Hoang N., Ritz T., Brettel K., Essen L. O., van der Horst G. T., Batschauer A., Ahmad M. (2011) The cryptochromes: blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 62, 335–364 [DOI] [PubMed] [Google Scholar]
- 14. Tanaka F., Chosrowjan H., Taniguchi S., Mataga N., Sato K., Nishina Y., Shiga K. (2007) Donor-acceptor distance-dependence of photoinduced electron-transfer rate in flavoproteins. J. Phys. Chem. B 111, 5694–5699 [DOI] [PubMed] [Google Scholar]
- 15. Nakabayashi T., Islam M. S., Ohta N. (2010) Fluorescence decay dynamics of flavin adenine dinucleotide in a mixture of alcohol and water in the femtosecond and nanosecond time range. J. Phys. Chem. B 114, 15254–15260 [DOI] [PubMed] [Google Scholar]
- 16. Shirdel J., Penzkofer A., Prochazka R., Shen Z., Strauss J., Daub J. (2007) Absorption and emission spectroscopic characterisation of a pyrene-flavin dyad. Chem. Phys. 331, 427–437 [Google Scholar]
- 17. Stanley R. J., MacFarlane A. W. (2000) Ultrafast excited state dynamics of oxidized flavins: direct observations of quenching by purines. J. Phys. Chem. A 104, 6899–6906 [Google Scholar]
- 18. Mataga N., Chosrowjan H., Taniguchi S., Tanaka F., Kido N., Kitamura M. (2002) Femtosecond fluorescence dynamics of flavoproteins: comparative studies on flavodoxin, its site-directed mutants, and riboflavin-binding protein regarding ultrafast electron transfer in protein nanospaces. J. Phys. Chem. B 106, 8917–8920 [Google Scholar]
- 19. Mataga N., Chosrowjan H., Shibata Y., Tanaka F., Nishina Y., Shiga K. (2000) Dynamics and mechanisms of ultrafast fluorescence quenching reactions of flavin chromophores in protein nanospace. J. Phys. Chem. B 104, 10667–10677 [Google Scholar]
- 20. Mataga N., Chosrowjan H., Shibata Y., Tanaka F. (1998) Ultrafast fluorescence quenching dynamics of flavin chromophores in protein nanospace. J. Phys. Chem. B 102, 7081–7084 [Google Scholar]
- 21. Grajek H., Gryczynski I., Bojarski P., Gryczynski Z., Bharill S., Kulak L. (2007) Flavin mononucleotide fluorescence intensity decay in concentrated aqueous solutions. Chem. Phys. Lett. 439, 151–156 [Google Scholar]
- 22. Glasoe P. K., Long F. A. (1960) Use of glass electrodes to measure acidities in deuterium oxide 1,2. J. Phys. Chem. 64, 188–190 [Google Scholar]
- 23. Huber R., Köhler T., Lenz M. O., Bamberg E., Kalmbach R., Engelhard M., Wachtveitl J. (2005) pH-dependent photoisomerization of retinal in proteorhodopsin. Biochemistry 44, 1800–1806 [DOI] [PubMed] [Google Scholar]
- 24. Wilhelm T., Piel J., Riedle E. (1997) Sub-20-fs pulses tunable across the visible from a blue-pumped single-pass noncollinear parametric converter. Opt. Lett. 22, 1494–1496 [DOI] [PubMed] [Google Scholar]
- 25. Riedle E., Beutter M., Lochbrunner S., Piel J., Schenkl S., Sporlein S., Zinth W. (2000) Generation of 10- to 50-fs pulses tunable through all of the visible and the NIR. Appl. Phys. B 71, 457–465 [Google Scholar]
- 26. Lorenc M., Ziolek M., Naskrecki R., Karolczak J., Kubicki J., Maciejewski A. (2002) Artifacts in femtosecond transient absorption spectroscopy. Appl. Phys. B 74, 19–27 [Google Scholar]
- 27. Ernsting N. P., Kovalenko S. A., Senyushkina T., Saam J., Farztdinov V. (2001) Wave-packet-assisted decomposition of femtosecond transient ultraviolet-visible absorption spectra: application to excited-state intramolecular proton transfer in solution. J. Phys. Chem. A 105, 3443–3453 [Google Scholar]
- 28. Kovalenko S. A., Dobryakov A. L., Ruthmann J., Ernsting N. P. (1999) Femtosecond spectroscopy of condensed phases with chirped supercontinuum probing. Phys. Rev. A 59, 2369–2384 [Google Scholar]
- 29. van Stokkum I. H., Larsen D. S., van Grondelle R. (2004) Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta 1657, 82–104 [DOI] [PubMed] [Google Scholar]
- 30. Koziol J., Knobloch E. (1965) The solvent effect on the fluorescence and light absorption of riboflavin and lumiflavin. Biochim. Biophys. Acta 102, 289–300 [DOI] [PubMed] [Google Scholar]
- 31. Ghisla S., Massey V., Lhoste J. M., Mayhew S. G. (1974) Fluorescence and optical characteristics of reduced flavins and flavoproteins. Biochemistry 13, 589–597 [DOI] [PubMed] [Google Scholar]
- 32. Massey V., Ganther H. (1965) On the interpretation of the absorption spectra of flavoproteins with special reference to d-amino acid oxidase. Biochemistry 4, 1161–1173 [DOI] [PubMed] [Google Scholar]
- 33. Sun M., Moore T. A., Song P. S. (1972) Molecular luminescence studies of flavins. I. The excited states of flavins. J. Am. Chem. Soc. 94, 1730–1740 [DOI] [PubMed] [Google Scholar]
- 34. Harbury H. A., Foley K. A. (1958) Molecular interaction of isoalloxazine derivatives. Proc. Natl. Acad. Sci. U.S.A. 44, 662–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. MacFarlane A. W., 4th, Stanley R. J. (2001) Evidence of powerful substrate electric fields in DNA photolyase: implications for thymidine dimer repair. Biochemistry 40, 15203–15214 [DOI] [PubMed] [Google Scholar]
- 36. Weigel A., Dobryakov A. L., Veiga M., Pérez Lustres J. L. (2008) Photoinduced processes in riboflavin: superposition of pi pi*-n pi* states by vibronic coupling, transfer of vibrational coherence, and population dynamics under solvent control. J. Phys. Chem. A 112, 12054–12065 [DOI] [PubMed] [Google Scholar]
- 37. Heelis P. F. (1982) The photophysical and photochemical properties of flavins (isoalloxazines). Chem. Soc. Rev. 11, 15–39 [Google Scholar]
- 38. Solar S., Getoff N., Surdhar P. S., Armstrong D. A., Singh A. (1991) Oxidation of tryptophan and N-methylindole by N3, Br2−, and (SCN)2− radicals in light- and heavy-water solutions: a pulse radiolysis study. J. Phys. Chem. 95, 3639–3643 [Google Scholar]
- 39. Baldwin J., Krebs C., Ley B. A., Edmondson D. E., Huynh B. H., Bollinger J. M. (2000) Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical. J. Am. Chem. Soc. 122, 12195–12206 [Google Scholar]
- 40. Massey V. (2000) The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 28, 283–296 [PubMed] [Google Scholar]
- 41. Massey V., Palmer G. (1966) On the existence of spectrally distinct classes of flavoprotein semiquinones: a new method for the quantitative production of flavoprotein semiquinones. Biochemistry 5, 3181–3189 [DOI] [PubMed] [Google Scholar]
- 42. Miura R. (2001) Versatility and specificity in flavoenzymes: control mechanisms of flavin reactivity. Chem. Rec. 1, 183–194 [DOI] [PubMed] [Google Scholar]
- 43. Page C. C., Moser C. C., Chen X., Dutton P. L. (1999) Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402, 47–52 [DOI] [PubMed] [Google Scholar]
- 44. Grajek H., Zurkowska G., Kuśba J. (2005) Influence of diffusion on nonradiative energy transfer between FMN molecules in aqueous solutions. J. Photochem. Photobiol. B 80, 145–155 [DOI] [PubMed] [Google Scholar]
- 45. Islam S. D., Susdorf T., Penzkofer A., Hegemann P. (2003) Fluorescence quenching of flavin adenine dinucleotide in aqueous solution by pH-dependent isomerisation and photo-induced electron transfer. Chem. Phys. 295, 137–149 [Google Scholar]
- 46. Weber G. (1950) Fluorescence of riboflavin and flavin-adenine dinucleotide. Biochem. J. 47, 114–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chosrowjan H., Taniguchi S., Mataga N., Tanaka F., Visser A. J. W. G. (2003) The stacked flavin adenine dinucleotide conformation in water is fluorescent on picosecond timescale. Chem. Phys. Lett. 378, 354–358 [Google Scholar]
- 48. Song P. S. (1968) On the basicity of the excited state of flavins. Photochem. Photobiol. 7, 311–313 [DOI] [PubMed] [Google Scholar]
- 49. Katz A. K., Glusker J. P., Markham G. D., Bock C. W. (1998) Deprotonation of water in the presence of carboxylate and magnesium ions. J. Phys. Chem. B 102, 6342–6350 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.